

Abstract
In plants, small-interfering RNAs (siRNAs) mediate epigenetic silencing via the RNA-directed DNA methylation (RdDM) pathway, which is particularly prominent during reproduction and seed development. However, there is limited understanding of the origins and dynamics of reproductive siRNAs acting in different cellular and developmental contexts. Here, we used the RNaseIII-like protein RTL1 to suppress siRNA biogenesis in Arabidopsis pollen, and found distinct siRNA subsets produced during pollen development. We demonstrate that RTL1 expression in the late microspore and vegetative cell strongly impairs epigenetic silencing, and resembles RdDM mutants in their ability to bypass interploidy hybridization barriers in the seed. However, germline-specific RTL1 expression did not impact transgenerational inheritance of triploid seed lethality. These results reveal the existence of multiple siRNA subsets accumulated in mature pollen, and suggest that mobile siRNAs involved in the triploid block are produced in germline precursor cells after meiosis, or in the vegetative cell during pollen mitosis.
Similar content being viewed by others

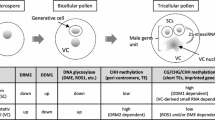

Introduction
Plant small RNA pathways have been extensively studied based on their important roles in plant development and reproduction1,2,3,4. They are mostly produced as 20- to 24-nucleotides (nt) RNA molecules that are classified as microRNAs (miRNA) and small-interfering RNAs (siRNAs) depending on their precursor transcript and processing machinery5,6. The expansion of plant siRNA pathways was propelled by the appearance of RNA polymerases solely dedicated to the production of siRNAs and their targets5,6, such as Pol IV and Pol V that mediate transcriptional silencing via the RNA-directed DNA methylation (RdDM) pathway7.
In plants, cytosine methylation is widespread and occurs in the CG, CHG,and CHH contexts (where H is A, C, or T), which are all initiated by RdDM. This pathway is essential for normal development and fertility in many plant species8,9,10, but has limited phenotypic impact in the model plant Arabidopsis thaliana, which allows studying siRNA biogenesis and activity in different developmental contexts. However, most studies have used genetic mutations that disrupt siRNA biogenesis and activity throughout the entire life cycle, which prevents the identification of siRNA subsets produced in specific tissues and cell types. For example, in the Arabidopsis male gametophyte, three haploid cells differentiate after meiosis to form a mature pollen grain11. After the first pollen mitosis, the vegetative cell nucleus exits cell cycle and undergoes epigenetic reprogramming leading to transcriptional activation of transposable elements (TEs) that are rapidly targeted by miRNAs and RdDM12,13,14,15,16. In contrast, the generative cell undergoes a second mitosis to generate two sperm cells that progress through S-phase prior to double fertilization, when RdDM pathways are transiently switched off12,17. For this reason, epigenetic reprogramming in the male germline has been largely associated to the activity of mobile siRNAs originated from the vegetative cell14,18,19 and anther tissues during meiosis20, but the biological significance of this mechanism remains poorly understood.
In order to investigate the origins, dynamics and function of siRNAs accumulated in A. thaliana pollen, we developed a method to suppress siRNA biogenesis specifically in pollen by using the antiviral RNaseIII-like protein RTL1 that is able to suppress siRNA biogenesis when expressed by strong constitutive promoters21. By using two pollen-specific promoters with very different expression dynamics, we were able to identify distinct TE-derived siRNA subsets produced during pollen development, and demonstrate that RTL1 expression in pollen impairs epigenetic silencing to promote triploid seed viability in the next generation.
Results
RTL1-mediated suppression of siRNA biogenesis in Arabidopsis pollen
Ectopic expression of RTL1 in pollen of Col-0 background was achieved by using the well-known LAT52 and MGH3 promoters that are differentially expressed throughout pollen development22. While the LAT52 promoter is expressed in the late microspore stage before becoming preferentially expressed in the vegetative cell during pollen mitosis, the MGH3 promoter is strongly expressed in the male germline after the first pollen mitosis22,23. Ectopic expression of RTL1 in mature pollen was confirmed in two independent transgenic lines of each construct (Supplementary Fig. 1a), which were subsequently used to perform small RNA sequencing and differential expression analysis (Fig. 1a and Supplementary Fig. 2). Comparative analysis using wild-type pollen (WT Col-0) as control revealed 817 loci with significantly reduced siRNA levels in pLAT52::RTL1 and 247 in pMGH3::RTL1 pollen, which were mostly non-overlapping (Fig. 1b, c, Supplementary Fig. 2a, and Supplementary Data 1). A few hundred loci showing enriched small RNA levels were also detected in both pLAT52::RTL1 and pMGH3::RTL1 samples (Supplementary Fig. 2b), but were considered to be indirect normalization effects resulting from the strong loss of siRNA in these lines. Therefore, we focused our analysis on down-regulated siRNA loci that included protein-coding genes targeted by 21/22-nt siRNAs in WT Col-0 pollen. By re-analyzing the transcriptomes of WT Col-0 pollen cell types that were already available24, we observed that siRNA depletion in pLAT52::RTL1 pollen corresponded to genes preferentially expressed in the vegetative cell, while reduced 21/22-nt siRNA levels in pMGH3::RTL1 pollen corresponded to genes preferentially expressed in sperm cells (Fig. 1d and Supplementary Fig. 1c). Taken together, these results confirmed that RTL1 is able to suppress siRNA biogenesis during pollen development, and suggested that the two pollen cell types produce different siRNA subsets.

a Schematic representation of RTL1 expression in mature pollen driven by the pLAT52 and pMGH3 promoters that are preferentially expressed in the vegetative cell and sperm cells, respectively. VCN is vegetative cell nucleus, and SC is sperm cell. b Principal component analysis after variance-stabilizing transformations shows reproducibility of small RNA sequencing experiments for two independent transgenic lines of each construct and wild-type Col-0, and highlights variance along PC1 and PC2 between all sample types. c Heatmap representation of siRNA depletion in pLAT52::RTL1 and pMGH3::RTL1 pollen (vs WT Col-0) shows that distinct loci are affected in the two transgenic lines. d Barplots present examples of different protein-coding genes showing reduced levels of 21/22-nt siRNA in pLAT52::RTL1 or in pMGH3::RTL1 pollen. Dots and bars represent expression level of individual replicates and mean values (n = 2), respectively, error bars represent the standard error, and RPM is reads per million. Source data are provided as a Source Data file.
RTL1 strongly impairs biogenesis of TE-derived siRNAs in pollen
Interestingly, siRNA depletion in pLAT52::RTL1 and pMGH3::RTL1 pollen occurred primarily at TEs that seem to be differentially impacted in the two transgenic lines (Fig. 2a–c). This includes ATGP1, ATHILA4, VANDAL3 and ATLANTYS2 families that lost siRNAs mainly in pLAT52::RTL1 pollen, while siRNAs from ATGP2, ATGP2N, VANDAL6 and ATCOPIA36 families were primarily depleted in pMGH3::RTL1 pollen (Fig. 2b, c and Supplementary Fig. 3). Depletion of 24-nt siRNAs in pMGH3::RTL1 pollen was particularly surprising, given that downstream RdDM components are not expressed in sperm cells12,25. To confirm that these siRNAs are indeed produced within the canonical RdDM pathway during pollen development, we took advantage of a Fluorescence Tagged Line (FTL1230)26 carrying a pollen-expressed DsRed transgene genetically linked to NRPD1a (largest subunit of Pol IV) (Supplementary Fig. 4a). This FTL line was crossed with the nrpd1a-3 mutant to obtain double heterozygous FTL1230/+;nrpd1a-3/+ plants, which allowed purification of segregating wild-type (DsRed positive, WTFTL) and mutant (DsRed negative, nrpd1a-3 FTL) pollen by fluorescence-activated cell sorting (FACS) (Supplementary Fig. 4b, c). This experiment showed that most TEs within the LTR/Gypsy superfamily lost siRNA in nrpd1a-3FTL pollen, including the ATGP1 and ATGP2N families that were the most affected in pLAT52::RTL1 and pMGH3::RTL1 pollen, respectively (Fig. 2c, d and Supplementary Fig. 4d). These results demonstrate that the multiple TE-derived siRNA subsets depleted in pLAT52::RTL1 and pMGH3::RTL1 pollen are at least partially dependent on the gametophytic activity of Pol IV, which is the main pathway for siRNA biogenesis in Arabidopsis pollen15,27,28.

a Comparative small RNA analysis between transgenic and WT Col-0 pollen shows that siRNA depletion in pLAT52::RTL1 and pMGH3::RTL1 pollen occurs primarily at TEs. Among all TEs annotated in the Arabidopsis TAIR10 genome, the LTR/Gypsy superfamily was found over-represented in the lists of differentially expressed siRNA loci. b Different members of the LTR/Gypsy superfamily showed significantly reduced siRNA levels in pLAT52::RTL1 or pMGH3::RTL1 pollen. The bar plots show the fraction of TE superfamilies in the two datasets. c Many retrotransposons from the ATGP1 family lost siRNAs specifically in pLAT52::RTL1 pollen, while ATGP2 and ATGP2N lost siRNAs specifically in pMGH3::RTL1 pollen. The bar plots show the fraction of TE families in the two datasets. d siRNAs depleted in pLAT52::RTL1 or pMGH3::RTL1 pollen are dependent on Pol IV activity. Dots and bars represent expression level of individual replicates and mean values (n = 2), respectively, error bars represent the standard error, and RPM is reads per million. Source data are provided as a Source Data file.
RTL1 expression impacts the pollen epigenome
RTL1-mediated suppression of siRNA biogenesis specifically in pollen provided an opportunity to investigate the role of gametophytic siRNAs in germline reprogramming and epigenetic inheritance. Therefore, we profiled DNA methylation in pLAT52::RTL1 and pMGH3::RTL1 pollen and seedlings by whole-genome bisulfite sequencing (WGBS), which was compared to WT Col-0, FACS-purified WTFTL and nrpd1a-3FTL pollen, as well as nrpd1a and p35S::RTL1 seedlings as controls (Supplemental Data 5). The analysis of differentially methylated regions (DMRs) in pLAT52::RTL1 and pMGH3::RTL1 pollen detected 2965 and 1552 hypomethylated DMRs in the CHH context, respectively (Fig. 3a), which mapped mainly to TEs and intergenic regions that were similarly hypomethylated in nrpd1a-3FTL pollen (Fig. 3b, Supplementary Fig. 6a, and Supplementary Data 2). Combined, the methylomes of pLAT52::RTL1 and pMGH3::RTL1 pollen resembled that of nrpd1a-3FTL to a large extent (Fig. 3b and Supplementary Fig. 5), although differences were expected given that RTL1 targets all siRNA types21. In contrast, the methylomes of pLAT52::RTL1 and pMGH3::RTL1 seedlings showed only a small number of hypo- and hypermethylated CHH DMRs as compared to p35S::RTL1 and nrpd1a seedlings (Supplementary Figs. 6a and 7), suggesting that the loss of pollen siRNAs has a limited impact on RdDM activity in the sporophyte. Importantly, the striking differences in siRNA depletion between pLAT52::RTL1 and pMGH3::RTL1 pollen were not reflected in their methylomes, as we found a significant overlap between hypomethylated regions in the two datasets (Fig. 3b and Supplementary Fig. 6b, c). This could be partially explained by the poor representation ATGP2 and ATGP2N families in the list of hypomethylated DMRs (Supplementary Fig. 6c), while these two families represent the vast majority of TEs that lost siRNA in pMGH3::RTL1 and nrpd1a-3FTL pollen (Fig. 2b and Supplementary Fig. 4d). This result suggests that ATGP2 and ATGP2N siRNAs are not actively involved in RdDM in pollen. Indeed, transcriptome analysis by mRNA sequencing showed that TEs remained transcriptionally repressed in pMGH3::RTL1 pollen, as only six genes (including RTL1) were significantly deregulated (Supplementary Fig. 8 and Supplementary Data 3). In contrast, the transcriptome of pLAT52::RTL1 pollen showed 718 up-regulated and 855 down-regulated genes and TEs (Supplementary Fig. 8 and Supplementary Data 3), including retrotransposons from the ATHILA4A, ATHILA4, and ATGP1 families that were equally up-regulated in nrpd1a-3FTL mutant pollen (Fig. 3c, d). These analyses demonstrate that RTL1 expression driven by the LAT52 promoter strongly impairs epigenetic silencing during pollen development, but has a limited impact on RdDM activity in the next generation.

a Heatmap and boxplot representation of CHH methylation levels at hypomethylated DMRs in pLAT52::RTL1 and pMGH3::RTL1 pollen (as compared to WT Col-0) shows overlap with reduced CHH levels in nrpd1a-3 FTL pollen. b Venn diagram shows the overlaps between hypomethylated regions in CHH context in nrpd1a-3FTL, pLAT52::RTL1 and pMGH3::RTL1 pollen. The statistical significance of each overlap was calculated using the R package SuperExactTest48. c Genome browser tracks show CHH methylation (green) and RNA sequencing (black) datasets at selected loci. Partial CHH hypomethylation is observed specifically in nrpd1a-3FTL and pLAT52::RTL1 pollen, leading to the transcriptional activation of retrotransposons. d RNA sequencing confirmed RTL1 expression in both pLAT52::RTL1 and pMGH3::RTL1 pollen, but up-regulation of LTR/Gypsy retroelements from the ATHILA4A, ATHILA4 and ATGP1 families occurs only in pLAT52::RTL1 and nrpd1a-3FTL pollen. Dots and bars represent expression level of individual replicates and mean values (n = 2), respectively, error bars represent the standard error, and normalized expression values were calculated by DESeq2 using the median of ratios method. Source data are provided as a Source Data file and in Supplementary Data 2.
RTL1 expression in pollen impacts triploid seed viability
Previous studies have shown that the paternal RdDM pathway triggers interploidy hybridization barriers in the seed, which is known as the “triploid block” response15,27,29,30. Therefore, to test if RTL1-mediated suppression of pollen siRNAs also allows bypassing the triploid block, we introduced pLAT52::RTL1 and pMGH3::RTL1 transgenes into the jas-3 mutant (Col-0 background) that produces 30 to 40% of unreduced diploid pollen and triploid seeds that collapse at high frequencies31,32. Strikingly, a significant decrease in triploid seed collapse was observed in three independent jas-3;pLAT52::RTL1 lines over five consecutive generations, while there was no significant effect in jas-3;pMGH3::RTL1 plants (Fig. 4a, b and Supplementary Fig. 9a). Importantly, the lower levels of triploid seed collapse in jas-3;pLAT52::RTL1 plants resembled the jas-3;nrpd1a-3 double mutant (Fig. 4b), suggesting that the suppressive effect is caused by the loss of Pol IV-dependent siRNAs that have been previously implicated in the triploid block response27,29,30. However, we cannot discard the possibility that other siRNA types are also involved, or that the suppressive effect mediated by RTL1 expression in pollen is not associated to siRNA activity. Importantly, increased viability of triploid seeds in jas-3;pLAT52::RTL1 lines coincided with down-regulation of the paternally expressed imprinted gene PHERES1 (PHE1) in developing siliques, which is an essential regulator of genomic imprinting and triploid seed lethality in Arabidopsis33 (Supplementary Fig. 9b). This demonstrates that RTL1 expression during pollen development impacts imprinted gene expression in interploid hybrid seeds with paternal excess, thus mimicking strong epigenetic suppressors of the triploid block27,34,35.

a The triploid block response in jas-3 mutants is quantified by counting the number of collapsed seeds in six siliques of selfed plants, which reflects the amount of diploid pollen produced in this mutant background (30 to 40%). Seed abortion is significantly reduced in jas-3/nrpd1a-3 double mutant plants in the F3 generation, and also across five consecutive generations of three independent jas-3;pLAT52::RTL1 lines, while jas-3;pMGH3::RTL1 remains similar to jas-3 controls. Numbers above each box represent the number of individual plants used for each genotype, and their levels of triploid seed collapse is represented by the dots on top of each box. Boxes represent the interquartile range (IQR) showing the lower (Q1) and upper (Q3) quartiles surrounding the median (central line), and whiskers represent the minimum (Q1 –1.5*IQR) and maximum (Q3 + 1.5*IQR) values. Statistically significant differences in the percentage of collapsed seeds were calculated by ANOVA with a post hoc Dunnett test, using jas-3 as the reference group (ns is not significant, * is P < 0.05, ** is P < 0.01, *** is P < 0.001). Source data are provided as a Source Data file. b Representative images of seeds show lower levels of seed abortion in suppressor lines jas-3/nrpd1a-3 and jas-3;pLAT52::RTL1, in comparison with a non-suppressor line jas-3;pMGH3::RTL1 and jas-3 controls.
Discussion
We developed a method to suppress siRNA biogenesis in Arabidopsis pollen, based on the ectopic expression of RTL1 driven by two pollen-specific promoters. Our results provide conclusive evidence that siRNA biogenesis is reinforced during pollen development, including the male germline where RdDM activity is significantly reduced12,13,25. Indeed, RTL1 targeted a completely different subset of TE siRNAs when expressed by the germline-specific promoter MGH3, perhaps reflecting the distinctive chromatin organization of this cell type12,13,36,37,38. This highlights the importance of having mobile siRNAs produced in germline precursor cells or companion vegetative cells in pollen14,18,19,20,23, and in the female gametophyte39, as it may compensate the fact that siRNA biogenesis in the germline is limited to only certain TEs. Our results provide further support to these ideas, as RTL1 expression driven by the LAT52 promoter allowed bypassing the triploid block in hybrid seeds, while germline-specific RTL1 expression did not impact triploid seed lethality. This strongly suggests the existence of mobile siRNAs originated from the late microspore or vegetative nucleus, which must then accumulate in the germline in order to reach the endosperm after fertilization, possibly loaded in some of the many Argonaute (AGO) proteins that are highly abundant in sperm cells40. AGO1 and AGO5 are good candidates in this model, as they have been both independently implicated in the triploid block response16. This hypothesis would also explain why RTL1 activity in the germline is not able to target siRNAs originated from elsewhere, as mobile siRNAs loaded into AGO proteins may be protected from RTL1 activity. Alternatively, mobile siRNAs accumulated in sperm cells could be modified in a way that prevents targeting by RTL1. Indeed, pollen siRNAs involved the triploid block were recently shown to be heavily modified by pseudouridine, which seems to be an essential feature of mobile siRNAs in both plants and mammals41.
The biological significance of active siRNA biogenesis in sperm cells remains unclear, given that hundreds of genes and TEs produce siRNAs that are seemingly not engaged in PTGS and RdDM activity in the male germline. This is particularly puzzling for ATGP2 and ATGP2N siRNAs, which combined represent a large fraction within the most abundant TE-derived siRNAs detected in wild-type Arabidopsis pollen (Supplementary Fig. 3c). As downstream components of RdDM are not expressed in sperm cells12, one possibility is that sperm-borne siRNAs are sequestered only to participate in RdDM later in germline specification during pollen tube growth, or maybe after fertilization in the early seed. A recent analysis of parental siRNA contributions in the Arabidopsis endosperm supports this idea, as the loss of paternal siRNAs resulted in reduced DNA methylation and significant changes in gene expression42. However, our results have shown that RTL1 expression in pollen had only a minor impact on RdDM activity in the sporophyte (Supplementary Fig. 6), suggesting that siRNA biogenesis in pollen is mostly dispensable for transgenerational epigenetic inheritance. This raises yet another possibility that sperm-borne small RNAs are mainly involved in translational repression, as previously shown with artificial miRNAs23. It is tempting to speculate that a subset of germline small RNAs is part of a surveillance mechanism that is primed to act only in situations of epigenetic instability caused by stress or hybridizations that lead to bursts of TE expression and activity. These outstanding questions may now be tested by using RTL1-mediated suppression of sperm siRNAs in different plant species and developmental contexts.
Methods
Plant materials, plasmid cloning, and plant transformation
The Arabidopsis mutant lines jas-3 (SAIL_813_H03) and nrpd1a-3 (SALK_128428), as well as the fluorescence-tagged line FTL123026 were used in this study, and are all in Col-0 background. Plants were grown in greenhouse long-day conditions (16 h light and 8 h dark). The pLAT52::RTL1-Myc and pMGH3::RTL1-Myc constructs were generated by PCR amplification of the respective promoters from genomic DNA (primers listed in Supplementary Table 1), and cloned into the p35S::RTL1-Myc binary plasmid21. The 35S promoter was replaced using HindIII and XbaI restriction sites, and each plasmid was transformed into Arabidopsis Col-0 and jas-3 mutants by floral dipping43. Transgenic seeds were surface sterilized with 50% bleach followed by 70% ethanol for 2 min, washed with sterile deionized water and sowed on agar plates (0.5X MS medium, 1% sucrose, pH 5.7) supplemented with cefotaxime (250 mg/L, Duchefa) and hygromycin (25 mg/L, Duchefa). Plates were placed in a growth chamber at 23 °C, 70% humidity, 120 μE m–2 light with a 16-h light/8-h dark (long days) photoperiod for two weeks, and seedlings were then transferred to soil and grown in greenhouse long-day conditions to complete the life cycle (16 h light and 8 h dark).
RT-qPCR analysis
Pollen was purified by collecting open flowers into a 50 mL falcon tube, vortexing for 3 min in 20 mL of 100 mM phosphate buffer, and filtering through a 50μm mesh. Pollen was then concentrated by centrifugation (3 min, 5000 RPM), and disrupted by shacking in the presence of glass beads (Sigma) with a Retsch homogenizer. Total RNA was then extracted using the Direct-zol RNA Microprep kit (Zymo) following the manufacturer’s instructions. Developing siliques were collected 7 days after pollination, and total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) following the manufacturer’s recommendations for seed tissues (RLC buffer). 1 µg of total RNA was then subjected to DNase (Invitrogen) treatment and converted into cDNA using Superscript II RT and random primers (Invitrogen). RT-qPCR was performed on a CFX Connect Real-Time PCR machine (BioRad) using SsoAdvanced Universal SYBR Green Supermix (BioRad). Primers used for RT-qPCR reaction are listed in Supplementary Table 1. MGH3 and ACT2 were used as internal controls.
Fluorescence-activated cell sorting
The FTL1230 line contains a pollen-expressed transgene genetically linked to NRPD1a (Supplementary Fig. 6), and is composed of a DsRed marker gene driven by the LAT52 promoter26. This line was crossed with the homozygous mutant nrpd1a-3, and the resulting heterozygous nrpd1a-3/ + F1 plants allowed purification of wild-type (DsRed positive) and mutant nrpd1a-3 (DsRed negative) pollen by Fluorescence-Activated Cell Sorting (FACS) (Supplementary Fig. 6). Pollen was purified by collecting open flowers into Eppendorf tubes, vortexing in 2 mL of 100-mM sodium phosphate buffer (pH 7) for 3 min, and filtering through a 50 µm nylon mesh (CellTrics, Sysmex). Pollen was purified using a MoFlo Astrios EQ cell sorter (Beckman-Coulter) equipped with a 561 nm laser (200 mW) for DsRed excitation. Pollen populations are characterized by an elevated high angle scatter (SSC) and autofluorescence, which combined with the differences in DsRed signal intensity allowed to sort WTFTL (DsRed +) and nrpd1a-3FTL (DsRed -) pollen simultaneously (Supplementary Fig. 4c). Approximately 500,000 pollen grains of each population were used for downstream analysis.
Small RNA sequencing and analysis
Library construction and sequencing was performed at BGI Genomics (Hong Kong). Single-end 50-nt reads were pre-processed by filtering collapsed reads according to length and quality. Filtered reads were mapped to the Arabidopsis TAIR10 genome with bowtie44, reporting all multi-mappers. Only perfect-match reads were used for all downstream analysis. Reads were normalized by dividing non-redundant read counts by the number of genomic hits, and subsequently calculating the number of reads per million of filtered (18–30 nt) and perfectly mapped reads. Differential expression analysis was performed with the R package DESeq245 using an FDR adjusted p-value of 0.05. Additional downstream analyses and plots were done with custom R scripts. Graphical outputs were produced using the R package “ggplot2”. A summary of all small-RNA sequencing data generated in this study is presented in Supplementary Data 4.
Whole-genome bisulfite sequencing and DNA methylation analysis
Genomic DNA was purified from total pollen and two-week-old seedlings, and library preparation and sequencing were performed by BGI Genomics (Hong Kong) as paired-end 100 bp reads using DNBSEQ technology. Pre-processed and high-quality reads were mapped to the TAIR10 genome using bismark with default settings for paired-end libraries46, and all figures and downstream analysis were performed using custom R scripts. Graphical outputs were produced using the R package “ggplot2” and “ComplexHeatmap”. Methylome data from nrpd1a-3 seedlings was already available47. DMRs were defined as 100-bp bins containing at least 4, 5, or 6 differentially methylated CGs, CHGs, or CHH and with an absolute methylation difference of at least 0.4, 0.2, or 0.1, respectively. Bins localizing within 200 bp of each other were merged and considered as DMRs. The statistical significance of the observed overlaps between DMRs was calculated using the R package “SuperExactTest”48, and the analysis is presented in Supplementary Data 5. A summary of all WGBS sequencing data generated in this study is presented in Supplementary Data 4.
RNA sequencing and analysis
Sequencing of messenger RNA was performed by BGI Genomics (Hong Kong) as paired-end 100 bp reads using DNBSEQ technology. High-quality raw reads were aligned to the TAIR10 genome using STAR49. Normalization and analysis of differential gene expression was performed using the R package DESeq245, with an FDR adjusted p-value of 0.05. Graphical outputs were produced using the R packages “ggplot2”, “pheatmap”, and “ComplexHeatmap”. A summary of all RNA sequencing data generated in this study is presented in Supplementary Data 4.
Triploid block quantification and statistical analysis
The triploid block response in jas-3 lines was quantified by imaging dry seeds from six siliques under a stereoscopic microscope (Nikon), and counting the number of normal and collapsed seeds. Statistically significant differences in the percentage of collapsed seeds were calculated by one-way analysis of variance (ANOVA) with a post hoc Dunnett test, using the R packages “ggpubr” and “multcomp”. These results are presented in Supplementary Data 5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing datasets generated in this study are publicly available in the NCBI’s Gene Expression Omnibus under the accession number GSE231670. Expression profiling of Col-0 pollen nuclei is available in GSE155369. Source data are provided with this paper.
References
Vaucheret, H. & Voinnet, O. The plant siRNA landscape. Plant Cell https://doi.org/10.1093/plcell/koad253. (2023).
Chow, H. T. & Mosher, R. A. Small RNA-mediated DNA methylation during plant reproduction. Plant Cell https://doi.org/10.1093/plcell/koad010. (2023).
Pachamuthu, K. & Borges, F. Epigenetic control of transposons during plant reproduction: From meiosis to hybrid seeds. Curr. Opin. Plant Biol. 75, 102419 (2023).
D’Ario, M., Griffiths-Jones, S. & Kim, M. Small RNAs: big impact on plant development. Trends Plant Sci. 22, 1056–1068 (2017).
Borges, F. & Martienssen, R. A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 16, 727–741 (2015).
Bologna, N. G. & Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 65, 473–503 (2014).
Matzke, M. A. & Mosher, R. A. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408 (2014).
Wang, L. et al. Reinforcement of CHH methylation through RNA-directed DNA methylation ensures sexual reproduction in rice. Plant Physiol. 188, 1189–1209 (2021).
Wang, Z. et al. Polymerase IV plays a crucial role in pollen development in Capsella. Plant Cell 32, 950–966 (2020).
Dorweiler, J. E. et al. mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell 12, 2101–2118 (2000).
Borg, M. & Twell, D. Life after meiosis: patterning the angiosperm male gametophyte. Biochem. Soc. Trans. 38, 577–582 (2010).
Calarco, J. P. et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151, 194–205 (2012).
Ibarra, C. A. et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337, 6 (2012).
Slotkin, R. K. et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136, 461–472 (2009).
Borges, F. et al. Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat. Genet. 50, 186–192 (2018).
Oliver, C. et al. The miRNome function transitions from regulating developmental genes to transposable elements during pollen maturation. Plant Cell 34, 784–801 (2022).
Borges, F. et al. Loss of small-RNA-directed DNA methylation in the plant cell cycle promotes germline reprogramming and somaclonal variation. Curr. Biol. 31, 591–600 (2021).
Martínez, G., Panda, K., Köhler, C. & Slotkin, R. K. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat. Plants 2, 16030 (2016).
Wu, W. et al. Heterochromatic silencing is reinforced by ARID1‐mediated small RNA movement in Arabidopsis pollen. New Phytol. https://doi.org/10.1111/nph.16871 (2020).
Long, J. et al. Nurse cell–derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 373, eabh0556 (2021).
Shamandi, N. et al. Plants encode a general siRNA suppressor that is induced and suppressed by viruses. PLoS Biol. 13, e1002326 (2015).
Brownfield, L. et al. A plant germline-specific integrator of sperm specification and cell cycle progression. PLoS Genet. 5, e1000430 (2009).
Grant-Downton, R. et al. Artificial microRNAs reveal cell-specific differences in small RNA activity in pollen. Curr. Biol. 23, R599–R601 (2013).
Borg, M. et al. Epigenetic reprogramming rewires transcription during the alternation of generations in Arabidopsis. eLife 10, e61894 (2021).
Hsieh, P.-H. et al. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl Acad. Sci. USA 113, 15132–15137 (2016).
Francis, K. E. et al. Pollen tetrad-based visual assay for meiotic recombination in Arabidopsis. Proc. Natl Acad. Sci. USA 104, 3913–3918 (2007).
Martinez, G. et al. Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat. Genet. 50, 193–198 (2018).
Panda, K., McCue, A. D. & Slotkin, R. K. Arabidopsis RNA polymerase IV generates 21–22 nucleotide small RNAs that can participate in RNA-directed DNA methylation and may regulate genes. Phil. Trans. R. Soc. B 375, 20190417 (2020).
Erdmann, R. M., Satyaki, P. R. V., Klosinska, M. & Gehring, M. A small RNA pathway mediates allelic dosage in endosperm. Cell Rep. 21, 3364–3372 (2017).
Satyaki, P. R. V. & Gehring, M. Paternally acting canonical RNA-directed dna methylation pathway genes sensitize arabidopsis endosperm to paternal genome dosage. Plant Cell 31, 1563–1578 (2019).
Storme, N. D. & Geelen, D. The Arabidopsis Mutant jason produces unreduced first division restitution male gametes through a parallel/fused spindle mechanism in meiosis II. Plant Physiol. 155, 1403–1415 (2011).
Kradolfer, D., Wolff, P., Jiang, H., Siretskiy, A. & Köhler, C. An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev. Cell 26, 525–535 (2013).
Batista, R. A. et al. The MADS-box transcription factor PHERES1 controls imprinting in the endosperm by binding to domesticated transposons. eLife 8, e50541 (2019).
Schatlowski, N. et al. Hypomethylated pollen bypasses the interploidy hybridization barrier in Arabidopsis. Plant Cell 26, 3556–3568 (2014).
Huc, J. et al. Bypassing reproductive barriers in hybrid seeds using chemically induced epimutagenesis. Plant Cell 34, 989–1001 (2022).
Schoft, V. K. et al. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 10, 1015–1021 (2009).
Borg, M. et al. Targeted reprogramming of H3K27me3 resets epigenetic memory in plant paternal chromatin. Nat. Cell Biol. https://doi.org/10.1038/s41556-020-0515-y. (2020).
Buttress, T. et al. Histone H2B.8 compacts flowering plant sperm through chromatin phase separation. Nature https://doi.org/10.1038/s41586-022-05386-6. (2022).
Schröder, J. A., Bonnet, D. M. V. & Jullien, P. E. Non-cell-autonomous small RNA silencing in Arabidopsis female gametes. Curr. Biol. 33, 183–188.e3 (2023).
Jullien, P. E., Schröder, J. A., Bonnet, D. M. V., Pumplin, N. & Voinnet, O. Asymmetric expression of Argonautes in reproductive tissues. Plant Physiol. 188, 38–43 (2022).
Herridge, R. P. et al. Pseudouridine guides germline small RNA transport and epigenetic inheritance. Preprint at https://doi.org/10.1101/2023.05.27.542553 (2023).
Satyaki, P. R. V. & Gehring, M. RNA Pol IV induces antagonistic parent-of-origin effects on Arabidopsis endosperm. PLoS Biol. 20, e3001602 (2022).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium -mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Wang, Z. et al. Transgenerational effect of mutants in the RNA-directed DNA methylation pathway on the triploid block in Arabidopsis. Genome Biol. 22, 141 (2021).
Wang, M., Zhao, Y. & Zhang, B. Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Acknowledgements
This work was supported by the grant EpiHYBRIDS from the French National Agency of Research (ANR-19-CE120008 to F.B.). We thank all members of the EPIREP team for daily discussions, Hervé Vaucheret for critical reading of the manuscript and sharing the p35S::RTL1-Myc plasmid and seeds, and Gregory Copenhaver for the FTL1230 line. The authors acknowledge technical support from Mickael Bourge and Nicolas Valentin from the cytometry facility of the I2BC. This work has benefited from the support of IJPB’s Plant Observatory technological platforms. The IJPB benefits from the support of Saclay Plant Sciences-SPS (ANR-17-EUR-0007).
Author information
Authors and Affiliations
Contributions
F.B. designed the study; K.P. and M.S. performed the experiments; F.B. analyzed the data and wrote the manuscript with contributions from K.P. and M.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pachamuthu, K., Simon, M. & Borges, F. Targeted suppression of siRNA biogenesis in Arabidopsis pollen promotes triploid seed viability. Nat Commun 15, 4612 (2024). https://doi.org/10.1038/s41467-024-48950-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48950-6
- Springer Nature Limited