

Abstract
Persistent room temperature phosphorescent materials with unique mechanical properties and robust optical properties have great potential in flexible electronics and photonics. However, developing such materials remains a formidable challenge. Here, we present highly stretchable, lightweight, and multicolored persistent luminescence elastomers, produced by incorporating ionic room temperature phosphorescent polymers and polyvinyl alcohol into a polydimethylsiloxane matrix. These prepared elastomers exhibit high optical transparency in daylight and emit bright persistent luminescence after the removal of 365 nm excitation. The homogeneous distribution of polymers within the matrix has been confirmed by confocal fluorescence microscopy, scanning electron microscopy, and atomic force microscopy. Mechanical property investigations revealed that the prepared persistent luminescence elastomers possess satisfactory stretchability. Impressively, these elastomers maintain robust optical properties even under extensive and repeated mechanical deformations, a characteristic previously unprecedented. These fantastic features make these persistent luminescence elastomers ideal candidates for potential applications in wearable devices, flexible displays, and anti-counterfeiting.
Similar content being viewed by others
Introduction
Long-afterglow luminescence, an appealing optical phenomenon that lasts for at least several seconds after the removal of the excitation source1,2, has garnered considerable attention due to its vast potential in applications such as bioimaging3,4, display5,6, sensor7,8, and anti-counterfeiting9,10,11,12. In addition to conventional inorganic materials, organic ultralong room temperature phosphorescent (RTP) materials have also been extensively investigated in recent years, due to their high biocompatibility, low cost, and easy molecular structure modification13,14,15. To date, numerous organic persistent RTP materials with high quantum yields and ultralong long emission lifetimes have been reported16,17,18,19,20. However, most of these are limited to molecular crystals21,22,23, which severely constrain their practical applications because of the stringent conditions required for cultivating the crystalline state. In this context, the achievement of organic persistent RTP polymers with good flexibility, large-area processability, and producibility were developed by incorporating small-molecule phosphors into a commercially available polymer matrix. This approach is one of the most popular methods for improving the processability of persistent RTP materials20,21,22,23. Poly(methylmethacrylate) (PMMA) and polyvinyl alcohol (PVA) are the most commonly used polymer matrices24,25,26,27, but their stretchability is poor.
Polymer composites with a combination of high-efficiency luminescence and good elastomeric properties are promising in wearable optoelectronic devices, flexible displays, and anti-counterfeiting28,29,30,31. Recently, persistent RTP elastomers have been developed by doping small-molecule phosphors into thermoplastic polymer matrices32,33. Nevertheless, upon application of drawing force, their luminescence intensity and emission lifetime in the deformation region were remarkably decreased or even quenched. The constant mechanical deformations in practical applications make those materials dissatisfy the requirements for real-world usage. This is because the compact interactions between chromophores and polymer matrix, as well as interactions among chromophores themselves, would be broken under the large-area deformation. Therefore, it is highly desirable to develop persistent RTP materials that can maintain their optical properties in the deformation region and after multiple-time bending and stretching. Alternatively, blending amorphous RTP copolymers into elastomer matrices might be an effective way to obtain stretchable films with stable persistent luminescence properties. This is because the long polymer chains of RTP copolymers can resist external stretch to some extent (Fig. 1). Addressing the instability of optical properties of elastomers in deformation area could provide great opportunities for flexible and wearable optoelectronics.

a Chemical structures of amorphous RTP copolymers P1-P6. b Schematic representation for the preparation of full-color persistent room temperature phosphorescent elastomers.
In this work, full-color persistent luminescence elastomers were prepared by blending polyacrylamide containing organic quaternary phosphonium salts and PVA into a polydimethylsiloxane (PDMS) matrix. The amino group and positively charged quaternary phosphonium salts in RTP polymers, the alcohol group in the PVA, and the ether linkage in PDMS can interact with each other through hydrogen bonding, ion–dipole, and dipole–dipole interactions, thus endowing the prepared elastomer with strong mechanical properties and intense RTP properties. The obtained elastomers exhibit satisfactory mechanical properties, high optical transparency, and bright persistent luminescence. Importantly, their afterglow RTP can be well retained in the large-area deformation region and after multiple rounds stretching. To our knowledge, this work represents an example of multi-color persistent RTP elastomers with robust and stable optical properties. Finally, we demonstrated the complex knitting and anti-counterfeiting labels with high stretchability and stable persistent RTP (Fig. 1).
Results
Preparation of persistent RTP elastomers
PDMS was selected as the matrix in this study because it is an elastomer with high flexible elasticity, optical transparency, chemical stability, and excellent resistance to high and low temperature34. Organic quaternary phosphonium derivatives were chosen as the phosphorescent monomers because they were reported to exhibit intense persistent RTP from single molecule, and their afterglow colors can be readily tuned by appropriate chemical modification35,36,37. Thus, six monomers of 4-(but-3-en-1-yl-N,N-dimethylaniline)diphenylphosphoranyl bromide (M1), but-3-en-1-yl(4-methoxynaphthalen-1-yl)diphenylphosphonium bromide (M2), but-3-en-1-yl(phenanthren-9-yl)diphenylphosphonium bromide (M3), but-3-en-1-yl(4-fluoronaphthalen-1-yl)diphenylphosphonium bromide (M4), 4-(but-3-en-1-yl-1-naphthonitrile)diphenylphosphonium bromide (M5), but-3-en-1-yldiphenyl(pyren-2-yl)phosphonium bromide (M6) were synthesized according to the previous reported methods38. Time-Dependent Density Functional Theory (TDDFT) calculations, using the B3LYP functional and def2-SVP basis set, were conducted to simulate the phosphorescence spectra of monomers M1-M6. This computational approach provided valuable insights into the energy levels of their lowest-lying triplet states (Supplementary Fig. 27). The results revealed distinct triplet energy levels for these monomers, supporting their capacity to generate multi-color RTP.
The copolymerization of M3 and acrylamide was conducted at various molar ratios, including 1:400, 1:200, 1:100, 1:50, and 1:10, resulting in a series of persistent P3 RTP polymers. As illustrated in Supplementary Fig. 28, the phosphorescence peaks for these polymers are almost identical. Upon evaluation of emission decay times, it was observed that the 1:50 molar ratio had the most substantial RTP lifetime of 1090 ms (Supplementary Fig. 28). Consequently, a series of persistent RTP polymers (P1-P6) were prepared by copolymerization of monomers with acrylamide in molar ratio of 1: 50. The detailed synthetic procedures are illustrated in the supplementary information. The obtained monomers were characterized by 1H, 13C NMR spectroscopy, and high-resolution mass spectrometry (ESI-MS). The obtained polymers were characterized by gel permeation chromatography (GPC) and 1H NMR (Supplementary Figs. 1–26 and Table S1).
Firstly, we tried to fabricate persistent RTP elastomer by simply mixing persistent RTP polymer with PDMS precursors (Sylguard-184A and Sylguard-184B). It was found that the distribution of RTP polymer in PDMS matrix was inhomogeneous, which is due to the immiscibility between hydrophilic RTP polymer and hydrophobic PDMS. To achieve homogeneous luminescence in the elastomers, the uniform dispersion of RTP polymers in the PDMS matrix is very important. Thus, to overcome this immiscibility barrier, PVA was incorporated into the elastomer because of its ability to mediate binding between the RTP polymer and PDMS through hydrogen bonding and van-der-Waals interactions39,40,41,42. The mechanism diagram for grafting of PDMS and PVA was depicted in Supplementary Fig. 31. Firstly, different RTP polymers (P1-P6) and PVA were refluxed in the aqueous solution for 6 h. Then, Sylguard-184A was added into the mixture for 2 min stirring, and Sylguard-184B was subsequently added. Next, the mixture was stirred for ten minutes under the 365 nm irradiation. Lastly, the viscous mixture was transferred into a fluoridated mold, and elastomer films (E1-E6) were obtained after cooling. The transmittance of the prepared thin films is >85% at the visible light region (410–750 nm), indicating uniform dispersion of RTP polymers in elastomers and their high transparency (Fig. 2a). Such high transparency is beneficial for the afterglow emission generated by RTP polymers to readily penetrate the elastomer matrix. It is noteworthy that increasing the contents of RTP polymers or PVA may influence the photophysical and transmittance properties of the prepared elastomers (Supplementary Figs. 32 and 33).

a Transmittance spectra of E1-E6. b Phosphorescence spectra of E1-E6 (λex = 365 nm). c CIE chromaticity diagram for E1-E6. d Lifetime decay curves of E1-E6. e Confocal fluorescence images of E3 (λex = 405 nm). f AFM images of PDMS and E3 films with different contents of RTP polymer.
Photophysical property
The photophysical properties of P1-P6 were first studied by steady-state and delayed PL spectra. The detailed photophysical data were given in the Supplementary Information (Supplementary Fig. 30 and Supplementary Tab. 2). All the polymers exhibit intense absorption peaks below 500 nm, which are attributed to intramolecular π-π, π-π* or n-π* transitions, as depicted in Supplementary Fig. 30. Upon UV excitation, all polymers showed blue or green fluorescence in the solid state under ambient condition. The delayed PL spectra exhibited that the phosphorescence peaks of these polymers are located at 450 nm, 490 nm, 505 nm, 520 nm, 555 nm, and 620 nm, respectively. Their emission decay times were measured to be 135 ms, 679 ms, 1090 ms, 413 ms, 275 ms, and 176 ms, respectively (Supplementary Fig. 30). Thus, different afterglow colors from deep blue to red were observed with naked eyes when the UV source was removed.
To confirm the long-lasting luminescence of these polymers originated from triplet state rather than singlet state, we conducted a detailed analysis of the temperature dependence of phosphorescence in a P3 sample (1/50). As depicted in Supplementary Fig. 29, over the range of 77 to 300 K, a noticeable decrease in emission intensity was observed with an increase in temperature. Further, the lifetime decay curves of P3 decreased from 1276 ms to 759 ms as the temperature increased from 77 K to 300 K. These results derived from temperature-dependent phosphorescence spectra and lifetime decay curves substantiate the conclusion that the long-lasting luminescence originates from the triplet state rather than the singlet state. The intense interactions among polymeric chains through hydrogen bonding constructed a network that can provide a rigid environment to restrict the molecular vibrations, which are responsible for reducing the nonradiative transition and promoting RTP emissions as demonstrated in the previous study43.
The elastomers E1-E6 exhibited the similar delayed PL spectra and RTP colors with P1-P6, and the Commission International de l’Eclairage (CIE) coordinates for them are (0.15, 0.12), (0.22, 0.52), (0.26, 0.53), (0.33, 0.59), (0.46, 0.52), and (0.52, 0.39), respectively (Fig. 2b, c). Particularly, it is worth noting that the CIE coordinate of delayed PL of E1 is located at deep blue region, which is very difficult to achieve for organic persistent RTP. The emission lifetimes were reduced to 82 ms, 485 ms, 792 ms, 225 ms, 144 ms, and 137 ms, because molecular interactions among polymeric chains of monomers were weakened after they were blended into PDMS matrix (Fig. 2d). Among them, E3 exhibited the brightest emission and longest RTP lifetime (Supplementary Movie 1 and 2), thus it was studied in detail as a model in the following sections.
To confirm the homogeneous afterglow RTP in the elastomers, confocal fluorescence microscopy was used to demonstrate their uniform distribution. As shown in Fig. 2e and Supplementary Fig. 18, we can see the homogeneous emission in the confocal fluorescence images, indicating that RTP polymers were uniformly dispersed in PDMS (Supplementary Fig. 34). Z-scans luminescence imaging of E3 further confirms the high uniformity as well. Furthermore, scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were employed to analyze the brittle fracture section of E3. As indicated by the EDS mapping image, phosphorous (P) elements, which are solely derived from the P3 polymer, are uniformly distributed within the PDMS matrix (Supplementary Fig. 35). This evidences the homogeneous dispersion of the RTP polymer within the PDMS matrix. In addition, atomic force microscope (AFM) was used to detect the surface morphology of E3. Figure 2f showed that pure PDMS film has a very smooth surface, while E3 exhibited a roughness surface. However, no obvious accumulation of RTP polymer aggregates can be detected. The hydrogen bonding, ion–dipole, and dipole–dipole interactions among amino group and positively charged quaternary phosphonium salts in the RTP polymers, alcohol group in the PVA, and ether linkage in PDMS made the RTP polymers distribute uniformly in the PDMS and prevented them from aggregating39.
Mechanical properties
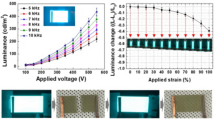
The tensile tests were carried out to study the mechanical properties of these persistent RTP elastomers. First, the tensile stress–strain curves of E3 with different thicknesses from 0.5 mm to 2.0 mm were measured, and the results show that the elastomer with 1.0 mm thickness exhibited the best performance among them, which can be stretched to 153% of its original length (Fig. 3a). Therefore, the elastomers of E1-E6 were prepared with 1.0 mm thickness for mechanical property investigation. In addition, considering that the mechanical properties are normally sensitive to the strain rates, thus different strain rates were applied for the study, and the results were recorded as shown in Fig. 3b. We can see that the engineering stress of a 1.0 mm thickness E3 decreased when the strain rate was decreased, and the strain to failure increased with a decrease in strain rate. Hence, the strain rate of 20 mm min−1 was used for the comparison of mechanical properties of E1-E6. As exhibited in Fig. 3c, these persistent RTP elastomers E1-E6 show better performance in stretchability (116–173%) than that of pure PDMS film (95%). This might be because the addition of ionic RTP polymers and PVA can provide extra hydrogen bonding, ion–dipole, and dipole–dipole interactions in the PDMS matrix, thus resulting in the increase of stretchability.

a Stress-strain curves of E3 with different thickness (0.5–2.0 mm). b Stress-strain curves for E3 (1.0 mm) samples when stretched at different rates. c Stress-strain curves of various E1-E6 elastomers (1.0 mm). d Cyclic loading and recovery of E3 (1.0 mm). e Changes of E3 (1.0 mm) in phosphorescence intensity after multiple-times stretching. f Changes of E3 (1.0 mm) in emission lifetime after multiple-times stretching.
Next, the stability of optical property was studied by stretching elastomers with large strain. The mechanical robustness of the persistent RTP elastomers were investigated by the tensile stretch-release cycles (Supplementary Movie 3). Figure 3d showed that no remarkable degradation of the tensile profiles was observed for E3 after 100 cycles of 25% strain stretch-release cycles. Simultaneously, it is exciting to see that only 15.4% and 6.5% degradation in phosphorescence intensity and lifetime of E3 were detected before and after 100 cycles of 25% strain stretch-release cycles (Fig. 3e, f). Therefore, we can conclude that these persistent RTP elastomers are mechanically robust and mechanical deformation only has little effect on their phosphorescence properties.
As shown in Fig. 4a, the strong persistent RTP can be clearly observed when the elastomer films were stretched, even in the deformation region. Thus, the detailed investigation on the photophysical properties of E3 in the deformation region was studied. As shown in Fig. 4b, the photophysical properties of E3 in the different stretched states (from 1 to 78%) were investigated. The results showed that the RTP intensity and emission lifetimes decreased to 92 and 84% of its original value, suggesting that their optical properties have good resistance against mechanical deformations (Supplementary Fig. 36). Impressively, the RTP and mechanical properties of E3 can be well retained even under water (Fig. 4c). These fantastic features make these persistent RTP elastomer films of E1-E6 perfect candidates for practical usage (Supplementary Movie 4). It is known that the generation of RTP in these materials is primarily due to the fact the polyacrylamide can effectively immobilize the phosphors due to the presence of hydrogen bond cross-linking between the polymeric chains44. When these films undergo stretching, the strain imposed may sever the hydrogen bond cross-links amongst polymeric chains in some extent. This disruption weakens the immobilization of phosphors, thereby leading to a reduction in RTP intensity and emission lifetimes.

a Schematic illustration of stretching E1-E6 and their RTP photographs. b RTP afterglow emission of E3 under strain. c Changes of E3 in phosphorescence intensity and emission lifetime under strain. d RTP afterglow photograph of stretched E3 under water.
Potential applications
The elastomers with unique persistent RTP properties and good mechanical properties can be applied in various fields, such as display, fashion, or anti-counterfeiting. The elastomers of E1, E3, and E6 were prepared as persistent RTP lines, which are almost transparent under the daylight, but showing strong RTP emissions after ceasing 365 nm UV light (Supplementary Fig. 37). As shown in Fig. 5a, the persistent RTP of these lines can be well maintained after multiple times bending and twisting, indicating that they can be directly used as functional lines to fabricate different complicated knitting. As shown in Fig. 5b, a complicated knot with blue, green, and red afterglow colors was fabricated for fashion and aesthetic purposes.

a Photographs of bending, rolling, and twisting E3 after removing 365 nm excitation. b Colorful afterglow images of different fabrics. c Schematic illustration the microelectronic printing and photographs of printed labels on PDMS substrate by using E1, E3, and E6. d Photographs of anti-counterfeiting labels under different stretched states.
In addition, the mixture of RTP polymers, PVA, and PDMS in a certain proportion can be used as security inks to print stretchable anti-counterfeiting labels through high-resolution ink-jet printing system. As shown in Fig. 5c, a multicolor label of lotus flower with a size of 5 cm × 5 cm was printed on a PDMS substrate. In Fig. 5d, bending and stretching the badge has almost no effect on its persistent RTP properties, which shows better performance to previous anti-counterfeiting labels45,46. Such robust optical properties of these persistent RTP elastomers against various mechanical deformation indicate their great potential in real-world applications.
Discussion
In summary, we present a concise and facile strategy to fabricate persistent RTP elastomers with high stretchability and robust optical property by blending ionic RTP polymers and PVA into PDMS matrix. Multicolor persistent RTP elastomers can be easily achieved by modifying the chemical structure of monomers to display different phosphorescence colors from blue to red. In addition, the prepared elastomers exhibit good mechanical properties compared to pure PDMS film, attributed to the addition of ionic RTP polymers and PVA, which provide extra hydrogen bonding, ion–dipole, and dipole–dipole interactions in the PDMS matrix. Importantly, their optical properties can be well maintained even after multiple cycles of bending, twisting, and stretching. Based on these fantastic features, we successfully prepared complex knots with different afterglow colors, demonstrating their potential in flexible display and fashion design. Moreover, the persistent RTP elastomers were used to print an anti-counterfeiting label on the PDMS substrate, which can retain intense afterglow emission upon bending and stretching. It is believed that the development of persistent luminescence elastomers with robust and stable optical properties will greatly broaden the range of organic RTP materials and promote their practical applications in wearable optoelectronic devices, flexible displays, and advanced anti-counterfeiting areas.
Methods
Measurements
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker ACF400 spectrometer at 298 K using deuterated solvents (CDCl3 or DMSO-d6). The UV-visible absorption spectra were measured by Shimadzu UV-2600 UV-vis spectrophotometer. The steady-state fluorescence and phosphorescence spectra were measured using Hitachi F-4700. The lifetimes were obtained on an Edinburgh FLS980 fluorescence spectrophotometer equipped with a Xenon arc lamp (Xe900) and a microsecond flash-lamp (uF900). The powder X-ray diffraction patterns were collected by D8 Advanced (Bruker) using Cu-Kα radiation. Aqueous gel permeation chromatography (GPC) was performed on a Series Samples used 0.05 mol/L NaNO3/H2O as mobile phase at 1.0 mL min−1 flow rate. The patterns were produced by a microelectronic printer (Shanghai Mi Fang Electronic Technology Co., Ltd). PVA-1799 was used as the calibration standard. The crystalline samples were obtained from slow evaporation of a mixture of hexane/dichloromethane (1:1, v/v).
Preparation of transparent elastomer films (E1-E6)
Firstly, The RTP polymer P1-P6 (40 mg, 30 mg, 30 mg, 40 mg, 60 mg, 60 mg, respectively.) were dissolved in 2 mL of deionized water. Subsequently, PVA (10 mg) was refluxed in the aqueous solution for 6 h. Then, Sylguard-184A (1000 mg) and Sylguard-184B (100 mg) were added into the mixture. The PDMS-PVA-P3 mixture was stirred for ten minutes under the 365 nm irradiation. Lastly, the viscous mixture was transferred into a fluoridated mold, heating slowly from 50 °C to 120 °C on the heat table for 12 h, and the elastomer films (E1-E6) were obtained after cooling.
Preparation of RTP lines
Firstly, P3 (30 mg) and PVA (10 mg) were dissolved in 2 mL of deionized water. Subsequently, the Sylguard-184A (1000 mg) and Sylguard-184B (100 mg) were poured into the mixture. The PDMS-PVA-P3 mixture was stirred for ten minutes under the 365 nm irradiation. Lastly, the viscous mixture was transferred into a line fluoridated mold, heating slowly from 50 °C to 120 °C on the heat table for 12 h, and the RTP lines were obtained after cooling.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request. Source data are provided with this paper.
References
Li, Y. et al. Long persistent phosphors-from fundamentals to applications. Chem. Soc. Rev. 45, 2090–2136 (2016).
Xu, J. et al. Persistent luminescence instead of phosphorescence: History, mechanism, and perspective. J. Lumin. 205, 581–620 (2019).
Zhang, K. Y. et al. Long-lived emissive probes for time-resolved photoluminescence bioimaging and biosensing. Chem. Rev. 118, 1770–1839 (2018).
Zhi, J. et al. Organic room temperature phosphorescence materials for biomedical applications. Chem.-Asian J. 15, 947–957 (2020).
Zhou, B. et al. Boosting wide-range tunable long-afterglow in 1D metal-organic halide micro/nanocrystals for space/time-resolved information photonics. Adv. Mater. 33, 2007571 (2021).
Xu, S. et al. Excited state modulation for organic afterglow: Materials and applications. Adv. Mater. 28, 9920–9940 (2016).
Li, F. et al. Halide-containing organic persistent luminescent materials for environmental sensing applications. Chem. Sci. 13, 2184–2201 (2022).
Qin, W. et al. Simultaneous promotion of efficiency and lifetime of organic phosphorescence for self-referenced temperature sensing. Chem. Eng. J. 400, 125934 (2020).
Li, D. et al. Completely aqueous processable stimulus responsive organic room temperature phosphorescence materials with tunable afterglow color. Nat. Commun. 13, 347 (2022).
She, P. F. et al. Controlling organic room temperature phosphorescence through external heavy-atom effect for white light emission and luminescence printing. Adv. Opt. Mater. 8, 1901437 (2019).
Peng, H. et al. On-demand modulating afterglow color of water-soluble polymers through phosphorescence FRET for multicolor security printing. Sci. Adv. 8, eabk2925 (2022).
Zhang, J. et al. Stimuli-responsive deep-blue organic ultralong phosphorescence with lifetime over 5 s for reversible water-jet anti-counterfeiting printing. Angew. Chem. Int. Ed. 60, 17094–17101 (2021).
Kabe, R. et al. Organic long persistent luminescence. Nature 550, 384–387 (2017).
Liang, X. et al. Organic room-temperature phosphorescence with strong circularly polarized luminescence based on paracyclophanes. Angew. Chem. Int. Ed. 58, 17220 (2019).
Hamzehpoor, E. D. et al. Crystal engineering of room temperature phosphorescence in organic solids. Angew. Chem. Int. Ed. 132, 10282 (2020).
Ma, L. et al. Highly efficient room‐temperature phosphorescence based on single‐benzene structure molecules and photoactivated luminescence with afterglow. Adv. Funct. Mater. 31, 2010659 (2021).
Ma, X. et al. A color-tunable single molecule white light emitter with high luminescence efficiency and ultra-long room temperature phosphorescence. J. Mater. Chem. C. 9, 727–735 (2021).
Zhang, Z. et al. Highly efficient and stable deep-blue room temperature phosphorescence via through-space conjugation. Chem. Eng. J. 442, 136179 (2022).
Zhang, X. P. et al. Highly efficient and persistent room temperature phosphorescence from cluster exciton enables ultrasensitive off-on VOC sensing. Matter 10, 3499–3512 (2022).
Qian, C. More than carbazole derivatives activate room temperature ultralong organic phosphorescenceof benzoindole derivatives. Adv. Mater. 34, 2200544 (2022).
Li, T. T. et al. Crosslink-enhanced strategy to achieve multicolor long-lived room temperature phosphorescent films with excellent photostability. Chin. Chem. Lett. 33, 4238–4242 (2022).
Wang, Z. H. et al. Regulation of irradiation-dependent long-lived room temperature phosphorescence by controlling molecular structures of chromophores and matrix. Adv. Opt. Mater. 10, 2200481 (2022).
Wu, H. Z. et al. Tailoring noncovalent interactions to activate persistent room-temperature phosphorescence from doped polyacrylonitrile films. Adv. Funct. Mater. 31, 2101656 (2021).
Yang, Y. F. et al. Tunable photoresponsive behaviors based on triphenylamine derivatives: the pivotal role of pi-conjugated structure and corresponding application. Adv. Mater. 33, 2104002 (2021).
Wang, C. et al. Photo‐induced dynamic room temperature phosphorescence based on triphenyl phosphonium containing polymers. Adv. Funct. Mater. 32, 2111941 (2022).
Wang, Z. et al. Color-tunable polymeric long-persistent luminescence based on polyphosphazenes. Adv. Mater. 32, 1907355 (2020).
Wang, D. et al. Unveiling the crucial contributions of electrostatic and dispersion interactions to the ultralong room-temperature phosphorescence of H-bond crosslinked poly(vinyl alcohol) films. Mater. Horiz. 9, 1081–1088 (2022).
Gan, N. et al. Recent advances in polymer-based metal-free room-temperature phosphorescent materials. Adv. Funct. Mater. 28, 1802657 (2018).
Nam, S. et al. Pronounced side chain effects in triple bond-conjugated polymers containing naphthalene diimides for n‑channel organic field-effect transistors. ACS Appl. Mater. Interfaces. 10, 12921 (2018).
Sakurai, T. et al. Electron-transporting foldable alternating copolymers of perylenediimide and flexible macromolecular chains. Mater. Chem. Front. 2, 718 (2018).
Yao, X. et al. Room-temperature phosphorescence enabled through nacre-mimetic nanocomposite design. Adv. Mater. 33, 2005973 (2021).
Zhang, Y. F. et al. Room temperature phosphorescent (RTP) hermoplastic elastomers with dual and variable RTP emission, photo-patterning memory effect, and dynamic deformation RTP response. Adv. Sci. 9, 2103402 (2022).
Tao, W. J. et al. Strain-responsive persistent room-temperature phosphorescence from halogen-free polymers for early damage reporting through phosphorescence lifetime and image analysis. Adv. Opt. Mater. 10, 2102449 (2022).
Li, C. H. et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 8, 618–624 (2016).
Alam, P. et al. Two are better than one: a design principle for ultralong-persistent luminescence of pure organics. Adv. Mater. 22, 2001026 (2020).
Alam, P. et al. Organic long-persistent luminescence from a single-component aggregate. J. Am. Chem. Soc. 144, 3050–3062 (2022).
Chen, G. et al. Photophysical tuning of organic ionic crystals from ultralong afterglow to highly efficient phosphorescence by variation of halides. J. Phys. Chem. Lett. 9, 6305–6311 (2018).
She, P. et al. Controllable photoactivated persistent organic room temperature phosphorescence for information encryption and visual temperature detection. Cell Rep. Phys. Sci. 2, 100505 (2021).
Li, J. Y. et al. Chemical modification on top of nanotopography to enhance surface properties of PDMS. Surf. Coat. Technol. 206, 2161 (2012).
Perween, S. et al. PVA-PDMS-stearic acid composite nanofibrous mats with improved mechanical behavior for selective filtering applications. Sci. Rep. 8, 16038 (2018).
Shin, M. et al. Swollen Behavior of crosslinked network hydrogels based on poly(vinyl alcohol) and polydimethylsiloxane. J. Appl. Polym. Sci. 85, 957 (2002).
Subramanyam, U. et al. PVA networks grafted with PDMS branches. J. Appl. Polym. Sci. A: Polym. Chem. 47, 5272 (2009).
Wei, J. et al. Conformation-dependent dynamic organic phosphorescence through thermal energy driven molecular rotations. Nat. Commun. 14, 627 (2023).
Chen, H. et al. Amorphous, efficient, room-temperature phosphorescent metal-free polymers and their applications as encryption ink. Adv. Opt. Mater. 4, 1397–1401 (2016).
Zhang, Y. et al. Large-area, flexible, transparent, and long-lived polymer-based phosphorescence films. J. Am. Chem. Soc. 143, 13675–13685 (2021).
Yang, Y. et al. Efficient and color-tunable dual-mode afterglow from large-area and flexible polymer-based transparent films for anti-counterfeiting and information encryption. Angew. Chem. Int. Ed. 61, e202201820 (2022).
Acknowledgements
We gratefully acknowledge the financial support from National Funds for Distinguished Young Scientists (61825503), National Natural Science Foundation of China (62288102, 62322508, and 62075101), National Key R&D Program of China (2022YFA1204404), and Natural Science Foundation of Jiangsu Province of China (BK20200095).
Author information
Authors and Affiliations
Contributions
Y.M., W.H., and Q.Z. conceived the idea for this work and designed the experiments. J.W. contributed to the synthesis work, photophysical property study, measurements, and application study. M.Z., J.L., T.D., P.D., C.L., J.D., X.Z., S.Z., L.G., and H.W. contributed to implementation of the experiments. S.J.L. revised the manuscript and provided suggestions. All authors discussed the results and commented on the manuscript at all stages.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, J., Zhu, M., Du, T. et al. Full-color persistent room temperature phosphorescent elastomers with robust optical properties. Nat Commun 14, 4839 (2023). https://doi.org/10.1038/s41467-023-40193-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40193-1
- Springer Nature Limited
This article is cited by
-
Afterglow bio-applications by utilizing triplet excited states of organic materials
Science China Chemistry (2023)