

Abstract
Endothelin system comprises three endogenous 21-amino-acid peptide ligands endothelin-1, -2, and -3 (ET-1/2/3), and two G protein-coupled receptor (GPCR) subtypes—endothelin receptor A (ETAR) and B (ETBR). Since ET-1, the first endothelin, was identified in 1988 as one of the most potent endothelial cell-derived vasoconstrictor peptides with long-lasting actions, the endothelin system has attracted extensive attention due to its critical role in vasoregulation and close relevance in cardiovascular-related diseases. Here we present three cryo-electron microscopy structures of ETAR and ETBR bound to ET-1 and ETBR bound to the selective peptide IRL1620. These structures reveal a highly conserved recognition mode of ET-1 and characterize the ligand selectivity by ETRs. They also present several conformation features of the active ETRs, thus revealing a specific activation mechanism. Together, these findings deepen our understanding of endothelin system regulation and offer an opportunity to design selective drugs targeting specific ETR subtypes.
Similar content being viewed by others

Introduction
Endothelin receptors (ETRs) are comprised of two subtypes (ETAR1 and ETBR2) with ~60% sequence similarity, both of which belong to the class A G protein-coupled receptors (GPCRs)3. ETRs are widely expressed in the human body, with ETAR primarily expressed in the cardiovascular system4 and ETBR in the brain5. They are activated by three types of endothelin (ET-1, ET-2, and ET-3), 21-amino-acid peptide hormones6,7. Besides, naturally occurring sarafotoxins (s6a–d), isolated from snake venom, show a high degree of sequence similarity and biological effects with ET-1 (Fig. 1a)8. Both ETAR and ETBR show different responses to these peptide agonists. ETAR exhibits similar subnanomolar affinities to ET-1 and ET-2 but a 100-fold weaker affinity to ET-3, while three ETs are equally effective for ETBR9,10. Sarafotoxin s6c shows a 100- to 10,000-fold reduced affinity for ETAR compared with s6b, thus producing a substantial ETBR selectivity11. Upon binding by agonists, ETAR primarily couples to the Gq protein, while ETBR couples to multiple G proteins, including Gs, Gi, and Gq proteins, to transduce signals (Fig. 1b, data from GPCRdb.org12). Intriguingly, two ETR subtypes exert opposing actions on vasoregulation: ET-1-mediated ETAR activation promotes long-lasting vasoconstriction, while ETBR mediates vasodilation and can inhibit the ET-1-mediated effects upon activated by ET-3, thus being considered a physiological antagonist of ETAR13,14.

a Sequence comparison of peptides, including ET-1, ET-3, IRL1620, sarafotoxin S6b, and S6c. The N-terminal, α-helical, and C-terminal regions of peptides are colored blue, green, and yellow, respectively. Residues different from that of ET-1 are colored black. b Schematic illustration of G protein-coupling of ETRs activated by endothelin and IRL1620. c–e Cryo-EM density maps (left panel) and cartoon representation (right panel) of ET-1-ETAR-Gq-scFv16 (c), ET-1-ETBR-Gq-scFv16 (d), and IRL1620-ETBR-Gi-scFv16 complexes (e). Components of ETRs complexes are colored as indicated.
The endothelin system has ignited extensive interest in its utmost importance in cardiovascular system regulation. Dysregulation of the endothelin system, especially the ETAR receptor, is closely associated with arterial hypertension, atherosclerosis, heart failure, renal disease, diabetes, etc., making ETRs promising drug targets for the treatment of these diseases9,10,14. Several non-selective ETRs antagonists (bosentan and macitentan) and ETAR-selective antagonists (ambrisentan and clazosentan) have been approved for the treatment of pulmonary arterial hypertension (PAH). ETBR-selective agonist Succinyl-[Glu9, Ala11,15]-ET-1(8-21), also named IRL162015, is also in clinical trials for adjuvant cancer therapy to enhance the delivery of anti-tumor drugs by improving blood flow16. Thus, developing selective ligands targeting specific endothelin receptor subtypes is of great pharmacology interest.
Extensive efforts have been made to clarify the recognition and activation mechanism of ETRs. Structural and relationship analysis had clarified residues on ET-1 crucial for binding17,18,19. Crystal structures of ETBR bound to endogenous peptides, including ET-1, ET-3, s6b, and synthesized agonists or antagonists were subsequently released, thus offering molecular evidence on ETBR recognition by ligands and receptor activity regulation11,20,21,22,23. In addition, different architectures of ECL2 of ETAR and ETBR were suggested to account for the selectivity of ET isopeptides22. A structure-based molecular simulation revealed that E9 in sarafotoxin s6c may rearrange its interaction with extracellular regions, subsequently destabilize its interaction with ETAR, thus leading to reduced ETAR affinity and ETBR specificity11. However, the inaccessible structural information on the ETAR subtype hampers our understanding of mechanisms of ligand recognition by ETAR, ligand selectivity, and ETAR activation. In this study, we present cryo-electron microscopy (cryo-EM) structures of engineered G protein-coupled ETAR and ETBR bound to ET-1 and ETBR bound to the selective peptide IRL1620. These structures reveal the molecular mechanism of peptide agonists recognition by ETAR and characterize the ligand selectivity and activation features for ETRs.
Results
Structure determination of ETAR and ETBR complexes
The NanoBiT tethering strategy, which has been used to stabilize the GPCR-G protein complex24, was introduced to facilitate the assembly of three endothelin receptor complexes. The large subunit (LgBiT, 18 kDa) and a complimentary peptide (HiBiT, VSGWRLFKKIS) with high affinity for LgBiT were fused to C-termini of the receptor and the Gβ subunit, respectively (Supplementary Fig. 1). The complementation of LgBiT and HiBiT facilitates the stabilization of ETR and G protein. The chimeric mini-Gαq used for the assembly of ETR complexes is the GTPase domain of the Gαq subunit with its N-terminal 35 amino acids replaced by the corresponding sequence in Gαi2 to facilitate binding of a single-chain variable fragment (scFv16). It has been successfully applied in the structural determination of the 5-HT2A-Gαq complex25. The Gαi subunit was engineered by introducing two dominant-negative mutations, G203A and A326S, to increase the stability of the ETBR-Gi protein complex26. Unless otherwise specified, Gq and Gi refer to respective engineered G proteins, which are used in the structural studies of ETR complexes. The structure of ET-1 bound ETAR and ETBR in complex with engineered Gq heterotrimer were determined by single-particle cryo-EM at global resolutions of 3.0 and 3.5 Å. The cryo-EM structure of the engineered Gi-coupled ETBR bound to the selective peptide IRL1620 was solved at a resolution of 3.0 Å (Fig. 1c–e, Supplementary Fig. 2, and Supplementary Table 1). The high-quality map allowed accurate model building for receptor residues N66NTD to N3788.52, I85NTD to F3978.54, and T84NTD to L4018.58 (superscripts refer to Ballesteros–Weinstein numbering27) for the ET-1-ETAR-Gq, ET-1-ETBR-Gq, and IRL1620-ETBR-Gi complexes, respectively, which can provide detailed atomic information of the ligand-binding pocket and the receptor-G protein-coupling interface (Fig. 1c–e and Supplementary Fig. 3a–c).
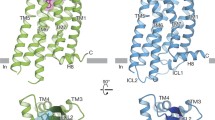
Globally, ETAR and ETBR adopt the canonical seven-transmembrane fold and G protein recruitment manner. All three complexes show a similar overall arrangement, with a root mean square deviation (RMSD) value for Cα atoms of 0.7–1.0 Å (Fig. 2a). At the receptor extracellular portion, both ETRs exhibit a large-scale outward kink at their extracellular end of TM5, which is distinguished by other class A peptide GPCRs. The kink is stabilized by a polar interaction network comprised of K3.33, E4.60, K5.38, Y5.34, and W21 of ET-1 and creates a wide-open pocket to accommodate peptide agonists (Fig. 2b, c). In addition, besides the conserved TM3-ECL2 disulfide bond, ETRs have an additional disulfide bond between C69/C90NTD and C341/C3587.25 for ETAR/ETBR. The featured disulfide bond links the receptor N-terminus and the extracellular end of TM7, thus capping the peptide-binding pocket. The compact extracellular interaction may prevent the agonist dissociation and explain the long-lasting activation of ET-1 (Fig. 2d).

a Structure superposition of ET1-bound and IRL1620-bound ETRs. b Structure superposition of ET-1-bound ETRs and other active class A peptide GPCRs, GHSR (PDB: 7F9Y), and NMUR2 (PDB: 7W55). The notable outward kink of the extracellular part of TM5 upon ETRs activation is highlighted. c Polar interaction network that stabilizes the kink conformation of TM5 of ETAR. d The disulfide bond between C69/C90NTD and C341/C3587.25 for ETAR/ETBR.
Recognition mechanism of ET-1 by ETAR
The ET-1 comprises the N-terminal (residues C1-M7), α-helical (D8-L17), and C-terminal regions (D18-W21) (Fig. 3a). It adopts a bicyclic architecture, formed by two pairs of intramolecular disulfide bonds (C1–C15 and C3–C11) (Fig. 3b), as observed in crystal and nuclear magnetic resonance (NMR) structures18,28. A previous NMR study identified a stable conformation of the N-terminal and α-helical regions in solution, whereas the C-terminal region is flexible28. The C-terminal region of ETs fixes its conformation by inserting it into the receptor core. The extreme C-terminal amino acid W21 makes an H-bond with K1663.33 and faces hydrophobic residues V1693.36, L2595.42, W3196.48, L3226.51, and I3557.39. Its backbone CO also forms an H-bond with K1663.33. I19 and I20 are buried in a hydrophobic cavity comprising I1362.60, W146ECL1, P1623.29, F2244.64, Y3527.36, and I3557.39 (Fig. 3c and Supplementary Fig. 3d). In addition, D18 forms a salt bridge with R3266.55, which makes a polar contact with D3517.35 and further S3256.54, thus constituting an extensive polar interaction network with TM6 and TM7 of ETAR (Fig. 3d and Supplementary Fig. 3d). These structural observations are supported by the dramatically decreased ET-1 activity on alanine mutants of K1663.33, R3266.55, and D3517.35 (Supplementary Fig. 4a and Table 2). These findings are consistent with a previous report that the C-terminal region is essential for the potency of ET-129, of which W21 shows critical roles in receptor binding and activation30. Small molecular antagonists of ETBR bosentan and K8794 and reverse agonist IRL2500 mimic the hexapeptide of the C-terminal region21,23, also indicating the importance of this segment in regulating receptor activity. Intriguingly, ET-3 and IRL1620 show an identical C-terminus with ET-1, while s6b and s6c bear a similar hydrophobic replacement from isoleucine to valine at position 19 relative to ET-1 (Fig. 1a). Correspondingly, C-terminus-interacting residues in both ETRs are conserved (Fig. 3e and Supplementary Fig. 5), proposing a conserved receptor-binding mode across these peptides.

a Sequence of ET-1. The N-terminal, α-helical, and C-terminal regions of ET-1 are colored blue, green, and yellow, respectively. b Cross-section of the ET-1-binding pocket in ETAR. Two pairs of intramolecular disulfide bonds of ET-1 are displayed as sticks. ET-1 is shown as a ribbon model with its three regions colored as in a. c, d Detailed interactions between ET-1 and ETAR at the C-terminal region. H-bonds are shown as blue dotted lines. e The profile of the interaction residues between the C-terminal region of ligand and endothelin receptors. f Detailed interactions between ET-1 and ETAR at the α-helical region.
The N-terminal region of ET-1 is anchored to the α-helical region by two pairs of intramolecular disulfide bonds. Residues D8-L17 in ET-1 form a helical fold, constituting the architecture of the α-helical region (Fig. 3b). The side chain of D8 forms an H-bond with Y3336.62. The phenyl ring of F14 makes hydrophobic interactions with L3447.28, L3477.31, and L3487.32 (Fig. 3f). Residues on ECL2 of ETAR, including F229, Y231, R232, T238, and M240, are also critical for ET-1 activity through association with the α-helical region of the peptide (Supplementary Fig. 4a and Table 2). The N-terminal region of ET-1 points to the kinked N-terminus of receptor TM5 (Fig. 3b). Compared with the α-helical and C-terminal regions, the N-terminus of ET-1 exposes to the solvent with seldom ETAR residues making a minimal contribution to ET-1 activity. In contrast to the C-terminus region, the α-helical region, especially the N-terminus of peptide agonists, show lower sequence identity (Fig. 1a), indicating non-conserved binding modes of these two regions and their potential roles in regulating peptide selectivity against ETAR and ETBR.
Ligand recognition selectivity for ETAR and ETBR
The structures of ETAR and ETBR provide templates to explore the mechanism of ligand-binding selectivity. Sequence comparison of ET-1 with two ETBR selective agonists, ET-3 and IRL1620, reveals that the most noticeable difference resides in the N-terminal region of peptides, in which all replaced amino acids in ET-3 from ET-1 are bulkier (Fig. 1a). This sequence difference elicits a hypothesis that ETBR has a larger pocket space to accommodate the bulkier N-terminal region of ET-3, which is supported by a smaller pocket size (1976 Å3, calculated by POVME31) for ETAR relative to ETBR (2059 Å3). This conclusion is in agreement with a previous finding that increasing intramolecular ring size decreased peptide activity at ETAR and shifted the peptide toward ETBR selectivity32.
Different from ETs, the N-terminal region is absent in agonist IRL1620 (Fig. 1a), which shows over 120,000-fold ETBR selectivity (Ki = 16 pM) over ETAR (Ki = 1900 nM)15. Substituting the K9 of ET-1 with the glutamic acid (E9) in IRL1620 increases the electronegativity, which may facilitate the electrostatic interaction with the positively charged extracellular part of ETBR (Fig. 4a, b)22. S6c bears an identical substitution from K9 to E9 compared with s6b (Fig. 1a). The charge of the ninth residue has been thought to determine the ETR selectivity of s6c over s6b33, supporting the importance of electrostatic interaction on peptide selectivity. This theory is also consistent with a previous finding that the net negative electrostatic potential of D8-K9-E10 is essential for ETs binding to ETBR15. In addition, the Nα-succinylation of IRL1620 and its analogs increased the binding affinity for ETBR, but not ETAR, thus producing ETBR preference15. The negative charge of succinylation is more compatible with the positively charged extracellular region of ETBR relative to its cognate receptor, probably contributing to the increased ETBR binding affinity and ETBR selectivity (Fig. 4b).

The electrostatic surface presentation of the extracellular regions of ET-1 bound ETAR (a) and IRL1620-bound ETBR (b) are shown. ET-1 in the ET-1-ETAR complex structure is omitted for a clear presentation. The electrostatic potential was calculated and presented using PyMOL. Deep blue represents positively charged and red represents negatively charged areas.
Activation of endothelin receptors
A structural comparison of ETR-G protein complexes with the crystal structures of ETBR in apo and agonist-, antagonist-, and reverse agonist-bound states reveals that these ETRs are indeed in the active state. TM6 of the ET-1 bound ETBR-G protein complex exhibits a notable outward displacement compared with that of crystal structures of apo (PDB: 5GLI, 4.4 Å, measured at Cα of R3186.30), antagonists/inverse agonist bound (PDB: 5XPR and 5 × 93, 5.4, and 4.6 Å), and the agonist ET-1 bound ETBR (PDB: 5GLH, 6.8 Å) due to the engagement of G proteins, the hallmark of class A GPCR activation (Fig. 5a). The almost identical active conformation of the ETAR relative to ETBR reveals a conserved activation mechanism in ETRs (Fig. 2a). Thus, structural comparison of the ET-1-ETBR-Gq complex with the apo ETBR was performed to analyze the activation mechanism of ETRs.

a Structure superposition of ETR-G protein complexes with the crystal structures of ETBR in apo (PDB: 5GLI) and agonist- (PDB: 5GLH), antagonist- (PDB: 5XPR), and reverse agonist-bound states (PDB: 5X93). b Overall conformational changes of the extracellular side of ETBR upon ET-1 binding. c The potential steric clash between L17 of ET-1 and K971.28 of ETBR disturbs the receptor TM1 α-helix from Q911.22 to K971.28. The potential clash is highlighted by the red dashed oval. d Detailed Interaction between W21, I19 of ET-1 and W3366.48, I3727.39 of ETBR. The movement orientations of W3666.48, I3727.39, and TM7 upon ETBR activation are indicated as black arrows. e Conformation comparison of the extracellular part of TM1 between apo and ET-1 bound ETBR. The TM1 segment from Q911.22 to K971.28, highlighted in the blue dashed oval, undergoes a helix-to-loop conformational change upon ETBR activation. The clash between N1041.35 and Y3697.36 are indicated by a red dashed oval. f Differences between the conformational changes of N7.49P7.50xxY7.53 motif in ETBR and CCKAR. Y7.53 in the NPxxY motif is replaced by L7.53 in ETRs. The movement of L3867.53 in ETBR and Y3707.53 in CCKAR are indicated by black and red arrows, respectively. g Structure superposition of ET-1-bound ETBR, CCK-8-bound CCKAR (PDB: 7EZM), the crystal structures of ETBR in apo (PDB: 5GLI), and devazepide-bound CCKAR states (PDB: 7F8Y) in cytoplasmic view. Black arrows show the movement directions of the cytoplasmic ends of ETBR helices upon receptor activation, while the red arrow indicates the shift of TM7 upon CCKAR activation. h The polar interaction linking N357 of the Gαq subunit and N1372.40 and S3908.47 of ETBR.
On the extracellular side, TM6 and TM7 of ETBR undergo inward movement upon ET-1 binding (3.7 and 4.8 Å, measured at Cα of Y3506.62 and R3577.24, respectively), compressing the ligand-binding pocket to form compact interactions with the peptide. Meanwhile, the extracellular end of TM2 occurs a lateral shift (2.9 Å, measured at Cα of A1642.67), while TM1 shows a smaller extent of inward bend (2.4 Å, measured at Cα of Q911.22) and undergoes a helix-to-loop conformational change (Fig. 5b, c). The potential steric clash between L17 of ET1 and K971.28 of ETBR disturbs the receptor TM1 α-helix from Q911.22 to K971.28 (Fig. 5c). The resulting loop approaches the center of the ligand-binding pocket and forms hydrophobic and van der Waal interactions with the α-helical region of ET-1. In addition, the α-helical region of ET-1 clashes with the N-terminus of ETBR in the apo state, disrupting the latter’s interaction with ECL2. Instead, the N-terminus of ETBR covers the ligand-binding pocket and form weak interactions with the N-terminus of the α-helical region of ET-1 (Fig. 5c). These movements of the receptor extracellular ends may propagate to the corresponding cytoplasmic end of the receptor to correspond with the seesaw model of class A GPCR activation, leading to the opening of the cytoplasmic cavity of the helical bundle to accommodate the G protein34.
Insertion of ET-1 deep into the ligand-binding pocket leads to direct contact of W21 with the toggle switch residue W3366.48 in ETBR and triggers their downward movement. Intriguingly, the side chain of I3727.39 of ETBR undergoes a downward rotamer shift and a hydrophobic rearrangement with I19 of ET-1, leading to a half-helical downward movement of TM7 (Fig. 5d). In addition, the side chain of Y3697.36 occurs a remarkable rotation, which probably clashes with N1041.35 and pushes TM1 apart from the helical core (Fig. 5e).
The agonist binding propagates agonism signaling downward and triggers the rearrangement of conserved motifs, such as toggle switch, P5.50V3.40F6.44, and D3.49R3.50Y3.51 (Supplementary Fig. 6a–c). Intriguingly, Y7.53 in the conserved N7.49P7.50xxY7.53 motif is replaced by leucine in ETRs (Supplementary Fig. 6d). In the classic activation mechanism, the side chain of Y7.53 occurs a large amplitude of rotation towards TM3, causing the inward movement of TM7 and strengthening its packing with TM335. Differently, upon ETBR activation, L3867.53 undergoes a lateral movement towards TM1 and TM2 (Fig. 5f), concomitant with the slight outward movement of these helices together with ICL1 (Fig. 5g), reflective of a unique activation mechanism of ETRs. This conformation may be further stabilized by the insertion of the α5 helix of the Gα subunit, in which N359 makes polar interactions with S3908.47 in the TM7-helix 8 hinge region and N1372.40 (Fig. 5h). Besides the abovementioned movements of helices, the downward propagated agonism signal eventually leads to the pronounced outward displacement of the cytoplasmic end of TM6, the hallmark of class A GPCRs activation, thus opening the cytoplasmic cavity of the receptor helix bundle to recruit G proteins (Fig. 5g).
Discussion
Even though extensive efforts have been made, we are still far from fully understanding important issues in the regulation of the endothelin system, although several crystal structures of ETBR in the inactive state have been released. By determining cryo-EM structures of ETAR and ETBR in complex with peptide agonists and G proteins, here we provide molecular evidence to deepen our understanding of the rationale for ligand recognition, subtype selectivity, and activation of endothelin receptors. Our structures present several conformation features upon ETR activation, including the helix-to-loop change of the extracellular portion of TM5, upward half-helical, and shift towards TM1-ICL2-TM2 of TM7 due to unique inter-helical interactions (Fig. 2). Our structures also reveal a conserved recognition mechanism of endothelin isopeptides (ET-1/-2/-3) between both endothelin receptor subtypes (Fig. 3c–e). The identical C-terminus of ETs inserts deep into the ligand-binding pocket of ETRs and is critical for the ET-1-induced ETR activation. The extreme C-terminal residue W21 of ETs directly contacts with toggle switch residue W6.48 and triggers familial ETR activation (Fig. 3c). We further provide the structural explanations on peptide selectivity for ETRs, including differences between two ETRs in the pocket sizes and electrostatic potential of the receptor extracellular surface (Fig. 4), and also demonstrate the activation features of ETRs (Fig. 5). Thus, our findings offer an opportunity for designing drugs targeting specific ETR subtypes for the treatment of diseases caused by an endothelin system malfunction.
Methods
Cell culture
Spodoptera frugiperda 9 (Sf9) insect cells (Expression Systems, #94-001 F) were grown in ESF 921 serum-free medium (Expression Systems) at 27 °C. AD293 cells (Agilent) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher), and maintained in a humidified chamber with 5% CO2 at 37 °C.
Constructs
The human wild-type (WT) ETAR and ETBR gene was codon-optimized for Sf9 expression and synthesized by Synbio Technologies. To facilitate the expression and purification, human ETAR (residues 21–406) and human ETBR (residues 27–424) DNA was cloned into a modified pFastBac vector (Thermo Fisher), which contains an N-terminal FLAG tag (DYKDDDD) followed by an 8 × His-tag before the receptor using homologous recombination (CloneExpress One Step Cloning Kit, Vazyme). The native signal peptide of the receptors was replaced with the prolactin precursor sequence to increase protein expression.
NanoLuc Binary Technology (NanoBiT, Promega) is originally a structurally complementary reporter system to evaluate protein-protein interaction in cells36. NanoBiT is composed of a large subunit (LgBiT, 18 kDa) and an 11 amino acid complementary peptide (SmBiT, peptide, VTGYRLFEEIL) with low affinity for LgBiT, both of which are connected to two interacting target proteins of interest, respectively. When the two proteins interact, the two subunits assemble into an active enzyme and generate a bright luminescent signal in the presence of a substrate. To improve the homogeneity and stability of ETR-G protein complexes, we applied the NanoBiT tethering strategy24 by fusing a LgBiT subunit at the receptor C-terminus. Rat Gβ1 fuses a HiBiT subunit (peptide 86, VSGWRLFKKIS), a homolog peptide of SmBiT with high affinity for LgBiT36, with a 15-amino acid (15AA) polypeptide linker (GSSGGGGSGGGGSSG) at its C-terminus (Supplementary Fig. 1).
The Gαq was designed based on a mini-Gαs skeleton with an N-terminus replaced by Gαi for the binding of scFv16. Bovine Gαi were incorporated with two dominant-negative mutations (G203A and A326S) by site-directed mutagenesis to increase the stability of the Gαβγ complex and decrease the affinity of nucleotide-binding. The engineered Gαq, Gαi, Gβ1-15AA-HiBiT, and bovine Gγ2 were cloned into the pFastBac vector, respectively. scFv16 was cloned into a modified pFastBac vector which contains a GP67 secretion signal peptide at its N-terminus.
Expression and purification of complexes
Baculoviruses were prepared using the Bac-to-Bac Baculovirus Expression System (Thermo Fisher). Sf9 insect cells were cultured to a density of 3 × 106 cells per mL and co-infected with FLAG-His 8-ETAR(21–406)/ETBR(27–424)-LgBiT, engineered Gαq/Gαi, Gβ1-15AA-HiBiT, Gγ2, and scFv16 baculoviruses at a 1:1:1:1:1 ratio. The cells were then harvested by centrifugation 48 h post-infection and stored at −80 °C for future use.
The frozen cells were thawed on ice and resuspended in lysis buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 10 mM MgCl2, 5 mM CaCl2, and supplemented with EDTA-free protease inhibitor cocktail (TargetMol). For ET-1-ETAR-Gq/ IRL1620-ETBR-Gi complex, cells were lysed by dounce homogenization and complex formation was initiated with the addition of 25 mU/mL Apyrase (Sigma-Aldrich) and 10 μM ET-1/ IRL1620 (GenScript) for 1.5 h at room temperature (RT). The membrane was then solubilized by adding 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) and 0.1% (w/v) cholesterol hemisuccinate (CHS, Anatrace) for 2 h at 4 °C. The sample was clarified by centrifugation at 80,000×g for 30 min and the supernatant was then incubated with anti-DYKDDDDK Affinity Beads (GenScript) for 3 h at 4 °C. After incubation, the resin was collected by centrifugation (600×g, 10 min) and loaded into a gravity flow column, followed by a wash with 20-column volumes of 20 mM HEPES, pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 1 μM ET-1/IRL1620, 0.01% (w/v) LMNG, and 0.002% (w/v) CHS. The protein was finally eluted with ten-column volumes of 20 mM HEPES, pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 1 μM ET-1/IRL1620, 0.01% (w/v) LMNG, and 0.2 mg/mL FLAG peptide. The purified complexes were concentrated using an Amicon Ultra centrifugal filter (molecular weight cut-off of 100 kDa, Millipore) and then subjected to a Superose 6 Increase 10/300 GL column (GE Healthcare) that was pre-equilibrated with buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1 μM ET-1/IRL1620, 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN, and 0.0002% (w/v) CHS. The purification procedure of ET-1-ETBR-Gq complex was similar to ETAR, expect the addition of 0.05% (w/v) digitonin, the final size-column equilibrated with 0.05% (w/v) digitonin instead of 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN, and 0.0002% (w/v) CHS. The monomeric fractions of the complex were collected and concentrated for cryo-EM examination.
Cryo-EM grid preparation and data collection
Three microliters of the purified ET-1-ETAR-Gq-scFv16, ET-1-ETBR-Gq-scFv16, and IRL1620-ETBR-Gi-scFv16 complexes at the concentration of about 18, 10, and 20 mg/mL, respectively, were applied onto glow-discharged holey carbon grids (Quantifoil R1.2/1.3). Excess samples were blotted for 3 s with a blot force of 1 and were vitrified by plunging into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Frozen grids were transferred to liquid nitrogen and stored for data acquisition. Cryo-EM imaging was performed on a Titan Krios G4 at 300 kV in the Advanced Center for Electron Microscopy at Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China).
A total of 11,657 movies were collected for the ET-1-ETAR-Gq-scFv16 complex using a Gatan K3 detector in super-resolution mode with a pixel size of 0.824 Å using the EPU. Movies were obtained at a dose rate of about 15 electrons per Å2 per second with a defocus ranging from −0.8 to −1.8 μm. The total exposure time was 2.35 s, and 36 frames were recorded per micrograph.
For ET-1-ETBR-Gq-scFv16 and IRL1620-ETBR-Gi-scFv16 complexes, A total of 6750 and 4496 movies were collected by a Gatan K3 detector at a pixel size of 1.04 Å using the EPU, respectively. The micrographs were recorded in super-resolution mode at a dose rate of about 15 electrons per Å2 per second with a defocus ranging from −0.8 to −2 μm. The total exposure time was 3.13 s, and 36 frames were recorded per micrograph.
Cryo-EM data processing
Cryo-EM image stacks were aligned using RELION-4.0-beta37. Contrast transfer function (CTF) parameters for each micrograph were estimated by CTFFIND 4.038. The following data processing was performed using RELION-4.0-beta37.
For the ET-1-ETAR-Gq-scFv16 complex, automated particle selection yielded 13,462,594 particle projections. The projections were subjected to reference-free 2D classification to discard poorly defined particles, producing 665,480 particle projections for further processing. This subset of particle projections was subjected to a round of maximum-likelihood-based three-dimensional classification with a pixel size of 0.824 Å. A selected subset containing 510,362 projections was used to obtain the final map using a pixel size of 0.824 Å. Further 3D classification without image alignment using a mask including only the receptor produced one good subset accounting for 510,197 particles, which were subsequently subjected to 3D refinement and Bayesian polishing. The final refinement generated a map with an indicated global resolution of 3.0 Å and was subsequently post-processed by DeepEMhancer.
For the ET-1-ETBR-Gq-scFv16 complex, automated particle selection yielded 7,414,897 particle projections. The projections were subjected to reference-free 2D classification to discard poorly defined particles, producing 1,375,681 particle projections for further processing. This subset of particle projections was subjected to a round of maximum-likelihood-based three-dimensional classification with a pixel size of 1.04 Å. A selected subset containing 415,508 projections was used to obtain the final map using a pixel size of 1.04 Å. Further 3D classification focusing on the receptor without alignment using a mask to mask out the detergent and G protein produced one good subset accounting for 373,655 particles, which were subsequently subjected to 3D refinement, CTF refinement, and Bayesian polishing. The final refinement generated a map with an indicated global resolution of 3.5 Å, and was subsequently post-processed by DeepEMhancer.
For the IRL1620-ETBR-Gi-scFv16 complex, automated particle selection yielded 4,078,200 particle projections. The projections were subjected to reference-free 2D classification to discard poorly defined particles, producing 967,642 particle projections for further processing. This subset of particle projections was subjected to a round of maximum-likelihood-based three-dimensional classification with a pixel size of 1.04 Å. A selected subset containing 410,751 projections was used to obtain the final map using a pixel size of 1.04 Å. Further 3D classification focusing the alignment on the receptor produced one good subset accounting for 191,493 particles, which were subsequently subjected to 3D refinement, CTF refinement, and Bayesian polishing. The final refinement generated a map with an indicated global resolution of 3.0 Å.
Model building and refinement
The initial templates of ETAR and ETBR were derived from Alphafold239. Models were docked into the EM density map using UCSF Chimera40. The initial models were then subjected to iterative rounds of manual adjustment based on the side-chain densities of bulky aromatic amino acids in Coot41 and automated refinement in Rosetta and PHENIX42. The final refinement statistics were validated using the module “comprehensive validation (cryo-EM)” in PHENIX42. The final refinement statistics are provided in Supplementary Table 1. All structural figures were prepared using Chimera40, Chimera X43, and PyMOL (https://pymol.org/2/).
NanoBiT assay for β-arrestin recruitment
The NanoBiT assay was used to assess the interaction between ETRs and β-arrestin, which reflects the activity of β-arrestin recruitment by ETRs. Briefly, the full-length ETAR/ETBR and mutants were cloned into the pBiT1.1 vector (Thermo Fisher) with a FLAG tag at its N-terminus and the LgBiT at its C-terminus. Human β-arrestin 2 (residues 1-393, with I386A, V387A, and F388A mutations) was cloned into a pBiT2.1 vector (Thermo Fisher) with the SmBiT at its N-terminus. AD293 cells were cultured in DMEM/high Glucose (GE healthcare) supplemented with 10% (w/v) FBS (Gemini). Cells were maintained at 37 °C in a 5% CO2 incubator with 300,000 cells per well in a six-well plate. Cells were grown overnight and then transfected with 1.5 μg ETAR/ETBR and 1.5 μg β-arrestin constructs by FuGENE® HD transfection reagent in each well for 24 h. Cells were harvested and resuspended in Hanks’ balanced salt solution buffer (HBSS) at a density of 5 × 105 cells/ml. The cell suspension was seeded in a 384-well plate at a volume of 10 μl per well, followed by 10 μl HBSS, 10 μl HBSS containing different concentrations of ET-1, and another 10 μl the NanoLuc substrate (furimazine, 1:25 dilution, Promega) diluted in the detection buffer. The luminescence signal was measured with an EnVision plate reader at room temperature.
Receptor surface expression
Cell-surface expression levels of WT or mutants ETAR/ETBR were quantified by flow cytometry. AD293 cells were seeded at a density of 1.5 × 105 per well into 12-well culture plates. Cells were grown overnight and then transfected with 1.0 μg ETAR/ETBR construct by FuGENE® HD transfection reagent in each well for 24 h. After 24 h of transfection, cells were washed once with PBS and then detached with 0.2% (w/v) EDTA in PBS. Cells were blocked with PBS containing 5% (w/v) BSA for 15 min at room temperature before incubating with primary anti-Flag antibody (diluted with PBS containing 5% BSA at a ratio of 1:300, Sigma-Aldrich, F3165) for 1 h at room temperature. Cells were then washed three times with PBS containing 1% (w/v) BSA and then incubated with anti-mouse Alexa-488-conjugated secondary antibody (diluted at a ratio of 1:1000, Thermo Fisher, A-11029) at 4 °C in the dark for 1 h. After another three times of washing, cells were collected, and fluorescence intensity was quantified in a BD Accuri C6 flow cytometer system (BD Biosciences) through a BD Accuri C6 software1.0.264.21 at excitation 488 nm and emission 519 nm. Approximately 10,000 cellular events per sample were collected, and data were normalized to the wild-type ETAR/ETBR. Experiments were performed at least three times, and data were presented as means ± SEM.
Statistical analysis
All functional study data were analyzed using Prism 9 (GraphPad) and presented as means ± SEM from at least three independent experiments. Concentration-response curves were evaluated with a three-parameter logistic equation. EC50 is calculated with the Sigmoid three-parameter equation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon reasonable request. Density maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-34663 (ET-1-ETAR-Gq-scFv16 complex); EMD-34667 (ET-1-ETBR-Gq-scFv16 complex); EMD-34619 (IRL1620-ETBR-Gi-scFv16 complex). Structure coordinates have been deposited in the Protein Data Bank (PDB) with accession codes 8HCQ (ET-1-ETAR-Gq-scFv16 complex); 8HCX (ET-1-ETBR-Gq-scFv16 complex); 8HBD (IRL1620-ETBR-Gi-scFv16 complex). Previously published structures can be accessed via accession codes 7EZM; 7F8Y; 7F9Y; 7W55; 5GLI; 5XPR; 5GLH; 5X93. Source data are provided with this paper.
References
Arai, H., Hori, S., Aramori, I., Ohkubo, H. & Nakanishi, S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 348, 730–732 (1990).
Sakurai, T. et al. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348, 732–735 (1990).
Davenport, A. P. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharm. Rev. 54, 219–226 (2002).
Regard, J. B., Sato, I. T. & Coughlin, S. R. Anatomical profiling of G protein-coupled receptor expression. Cell 135, 561–571 (2008).
Schinelli, S. Pharmacology and physiopathology of the brain endothelin system: an overview. Curr. Med. Chem. 13, 627–638 (2006).
Yanagisawa, M. et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415 (1988).
Inoue, A. et al. The human preproendothelin-1 gene. Complete nucleotide sequence and regulation of expression. J. Biol. Chem. 264, 14954–14959 (1989).
Kloog, Y. et al. Sarafotoxin, a novel vasoconstrictor peptide: phosphoinositide hydrolysis in rat heart and brain. Science 242, 268–270 (1988).
Davenport, A. P. et al. Endothelin. Pharm. Rev. 68, 357–418 (2016).
Kedzierski, R. M. & Yanagisawa, M. Endothelin system: the double-edged sword in health and disease. Annu. Rev. Pharm. Toxicol. 41, 851–876 (2001).
Izume, T., Miyauchi, H., Shihoya, W. & Nureki, O. Crystal structure of human endothelin ETB receptor in complex with sarafotoxin S6b. Biochem. Biophys. Res. Commun. 528, 383–388 (2020).
Pandy-Szekeres, G. et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–D446 (2018).
Barton, M. & Yanagisawa, M. Endothelin: 30 years from discovery to therapy. Hypertension 74, 1232–1265 (2019).
Rubanyi, G. M. & Polokoff, M. A. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharm. Rev. 46, 325–415 (1994).
Takai, M. et al. A potent and specific agonist, Suc-[Glu9,Ala11,15]-endothelin-1(8-21), IRL 1620, for the ETB receptor. Biochem. Biophys. Res. Commun. 184, 953–959 (1992).
Maguire, J. J. & Davenport, A. P. Endothelin@25 - new agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR review 12. Br. J. Pharm. 171, 5555–5572 (2014).
Huggins, J. P., Pelton, J. T. & Miller, R. C. The structure and specificity of endothelin receptors: their importance in physiology and medicine. Pharm. Ther. 59, 55–123 (1993).
Janes, R. W., Peapus, D. H. & Wallace, B. A. The crystal structure of human endothelin. Nat. Struct. Biol. 1, 311–319 (1994).
Orry, A. J. & Wallace, B. A. Modeling and docking the endothelin G-protein-coupled receptor. Biophys. J. 79, 3083–3094 (2000).
Shihoya, W. et al. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 537, 363–368 (2016).
Shihoya, W. et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 24, 758–764 (2017).
Shihoya, W. et al. Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation. Nat. Commun. 9, 4711 (2018).
Nagiri, C. et al. Crystal structure of human endothelin ETB receptor in complex with peptide inverse agonist IRL2500. Commun. Biol. 2, 236 (2019).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Kim, K. et al. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 182, e1519 (2020).
Liu, P. et al. The structural basis of the dominant negative phenotype of the Galphai1beta1gamma2 G203A/A326S heterotrimer. Acta Pharm. Sin. 37, 1259–1272 (2016).
Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 63 (1995).
Takashima, H. et al. Distributed computing and NMR constraint-based high-resolution structure determination: applied for bioactive Peptide endothelin-1 to determine C-terminal folding. J. Am. Chem. Soc. 126, 4504–4505 (2004).
Galantino, M. et al. D-amino acid scan of endothelin: importance of amino acids adjacent to cysteinyl residues in isomeric selectivity. Pept. Res. 8, 154–159 (1995).
Tam, J. P. et al. Alanine scan of endothelin: importance of aromatic residues. Peptides 15, 703–708 (1994).
Wagner, J. R. et al. POVME 3.0: software for mapping binding pocket flexibility. J. Chem. Theory Comput. 13, 4584–4592 (2017).
Wolf, P. & Beck-Sickinger, A. G. The ring size of monocyclic ET-1 controls selectivity and signaling efficiency at both endothelin receptor subtypes. J. Pept. Sci. 27, e3325 (2021).
Takayanagi, R. et al. Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett. 282, 103–106 (1991).
Yin, W. et al. Crystal structure of the human 5-HT1B serotonin receptor bound to an inverse agonist. Cell Disco. 4, 12 (2018).
Zhou, Q. et al. Common activation mechanism of class A GPCRs. Elife https://doi.org/10.7554/eLife.50279 (2019).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. https://doi.org/10.1002/pro.3943 (2020).
Acknowledgements
The Cryo-EM data of the ET-1-ETAR-Gq-scFv16, ET-1-ETBR-Gq-scFv16, and IRL1620-ETBR-Gi-scFv16 complexes were collected at the Advanced Center for Electron Microscopy, Shanghai Institute of Materia Medica (SIMM). We thank all the staff at the cryo-EM facilities for their technical support. This work was partially supported by the National Natural Science Foundation (32171187 to Y.J., 82121005 to Y.J. and H.E.X., and 32130022 to H.E.X.); the Ministry of Science and Technology (China) grants (2018YFA0507002 to H.E.X.); the Shanghai Municipal Science and Technology Major Project (2019SHZDZX02 to H.E.X.); Shanghai Municipal Science and Technology Major Project (H.E.X.); the CAS Strategic Priority Research Program (XDB37030103 to H.E.X.). H.E.X. is the founder and stakeholder of Cascade Pharmaceutics.
Author information
Authors and Affiliations
Contributions
Y.-J.J. designed the expression constructs, purified the ETR proteins, prepared the final samples for negative stain and cryo-EM data collection, conducted functional studies, and participated in figure and manuscript preparation with the help of J.D.; Q.Y. performed cryo-EM data calculations, model building, and participated in supplementary figure preparation; G.Y., S.Z., and T.G. participated in protein expression and functional studies; X.H. participated in figure preparation; H.J. and X.C. supervised X.H. in figure preparation; K.W. and W.H. participated in cryo-EM grid preparation and data collection; Y.J. and H.E.X. conceived and supervised the project, analyzed the structures, and wrote the manuscript with inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jennifer Cash and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, Y., Duan, J., Yuan, Q. et al. Structural basis of peptide recognition and activation of endothelin receptors. Nat Commun 14, 1268 (2023). https://doi.org/10.1038/s41467-023-36998-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36998-9
- Springer Nature Limited
This article is cited by
-
Cryo-electron microscopy for GPCR research and drug discovery in endocrinology and metabolism
Nature Reviews Endocrinology (2024)