

Abstract
Despite paramount applications of chiral trifluoromethylated compounds in medicinal chemistry and materials science, limited strategies have been developed for catalytic asymmetric synthesis of such valuable fluorinated structures. Here, we report a nickel catalyzed enantioselective dicarbofunctionalization of inexpensive industrial chemical 3,3,3-trifluoropropene (TFP) with readily available tertiary alkyl and aryl iodides. The reaction overcomes the β-F elimination side reaction of TFP, and proceeds efficiently under mild reaction conditions. The protocol possesses advantages, such as synthetic convenience, high enantioselectivity, and excellent functional group tolerance, providing rapid and straightforward access to chiral trifluoromethylated compounds of medicinal interest.
Similar content being viewed by others
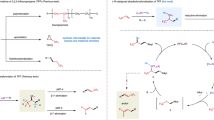

Introduction
Trifluoromethy group (CF3) represents one of the prominent functional groups in developing pharmaceuticals, agrochemicals, and advanced functional materials1,2,3,4. Particularly, the tactical introduction of CF3 into organic molecules can significantly improve their biological properties5,6,7. Consequently, numerous therapeutic drugs contain CF3 moiety. Over the past decades, impressive achievements on the site-selective trifluoromethylations have been made. However, many of developed methods mainly focus on the synthesis of trifluoromethylated arenes8,9,10, the construction of Csp3–CF3 bond at a stereogenic center remains a challenging topic (Fig. 1a). To date, only limited strategies for catalytic asymmetric trifluoromethylation have been reported11,12,13. Using Lewis acids14,15 or organocatalysis11,13,16,17,18,19 can enantioselectively construct the Csp3–CF3 bond at a stereogenic center, but shows efficiency with carbonyl compounds and their derivatives. Transition-metal catalyzed asymmetric trifluoromethylation via cross-coupling reactions is an alternative route to access chiral trifluoromethylated compounds20,21. However, the lack of an efficient catalytic system and limited substrate scope restrict its widespread synthetic applications. To circumvent these limitations, the use of low-cost and widely available prochiral CF3-containing chemical feedstocks as starting materials to asymmetrically synthesize chiral trifluoromethylated compounds shows great advantages, because they are ready for cost-efficiently diversified transformations.

a Biologically active molecules bearing an α-CF3 stereogenic center. b Pathways for α-CF3 metal species. c Nickel-catalyzed enantioselective dicarbofunctionalization of TFP (this work).
As an inexpensive and bulk industrial chemical, 3,3,3-trifluoropropene (TFP) has important applications in the production of fluorinated materials22 and refrigerants23, and should be a cost-effective and versatile linchpin to access chiral trifluoromethylated compounds. However, only limited efficient strategies for transformations of TFP have been developed24. To date, the asymmetric synthesis of trifluoromethylated compounds from TFP remains severely underdeveloped. Only rare examples that rely on the asymmetric dihydroxylation25 and hydroformylation26 of TFP have been reported. The asymmetric formation of a C–C bond at α-CF3 stereogenic center from TFP remains challenging. Usually, the installation of a functional group, such as an electron-withdrawing group and π-system, onto TFP is needed to facilitate the asymmetric process12,27,28. Despite effectiveness of these approaches, they require additional steps to prepare the trifluoromethylated alkenes. As such, to overcome these limitations, it is highly desirable to develop an efficient method that can provide a straightforward and cost-efficient access to chiral trifluoromethylated compounds through stereoselective transformations of TFP.
In 2016, we developed a chelation-assisted strategy for nickel-catalyzed dicarbofunctionalization of alkenes, in which the newly formed chiral center could be stereoselectively controlled by a chiral nickel catalyst to provide an 18% ee29. Inspired by this work, we hypothesized that the nickel-catalyzed asymmetric dicarbofunctionalization of TFP would be a promising strategy to construct the valuable chiral trifluomethylated compounds30,31,32,33,34,35,36,37. Because such a strategy can simultaneously form two new carbon-carbon bonds and generate one new α-CF3 stereogenic center without tedious synthetic procedure, thereby providing an attractive opportunity to diversely construct complex chiral trifluoromethylated structures. However, this process is plagued by the high tendency of β-H38,39,40 and β-F39,40,41,42,43 eliminations from the resulting α-CF3 alkyl-metal intermediate (Fig. 1b). We envisioned that with the aid of a suitable chiral ligand, the α-CF3 stereogenic center would be enantioselectively controlled by reaction of a chiral nickel(II) species with the α-CF3 alkyl radical that was generated by fast radical addition of TFP catalyzed by nickel complex; subsequently, the fast reductive elimination of the resulting highly active α-CF3 alkylnickel(III) intermediate would suppress the β-H and β-F eliminations (Fig. 1c).
Herein, we disclose an enantioselective nickel-catalyzed dicarbofunctionalization of TFP with aryl and tertiary iodides. The approach features excellent functional group tolerance and synthetic convenience without the preparation of organometallic reagents, providing efficient and straightforward access to a variety of chiral trifluoromethylated compounds.
Results
Optimization of nickel-catalyzed enantioselective alkyl-arylation of TFP
To test our hypothesis, we chose two electrophiles to couple with TFP, as this strategy could omit the preparation of organometallic reagents, and would provide rapid and straightforward access to chiral trifluoromethylated compounds. Initially, tert-butyl iodide 2a and ethyl 4-iodobenzoate 3a were chosen as the model substrates for asymmetric dicarbofunctionalization of TFP (Table 1). In a reaction that was carried out with TFP (1.6 equiv), 2a (1.5 equiv), and 1a (1.0 equiv) in the presence of NiBr2·DME (10 mol%) and reductant Zn in dioxane at room temperature, a high yield (84%) of 4a was obtained using an achiral ligand 4,4’-dit-Bu-bpy (L1) (entry 1). Encouraged by this result, a survey of chiral ligands was conducted (entries 2-8). We found that bis(oxazoline) (Biox) ligands showed a beneficial effect on the enantioselectivity (entries 2-6), and isobutyl-substituted chiral Biox (L6) was identified as the best ligand, providing 4a in 69% yield and 90% ee (entry 6). The same enantioselectivity with a slightly lower yield (66%) could be obtained by using Cy-Biox (L3) (entry 3), but other Biox ligands led to lower yields and enantioselectivities (entries 2, 4, 5). Pyox ligand was proved to be less effective (entry 7), and even no reaction occurred using Box or diamine ligands (entry 8). Further optimization of the reaction conditions by using DME instead of 1,4-dioxane as the solvent could improve the yield of 4a to 70% with 91% ee (entry 9). Other nickel catalysts were also examined. Slightly lower yields and comparable enantioselectivities were obtained by using NiCl2·DME or NiBr2·diglyme, but no reaction occurred with NiCl2 or NiBr2 (Supplementary Table 3). The absence of nickel catalyst or Biox ligand failed to provide desired product 4a (entries 10, 11), demonstrating the essential role of [Ni/L] in promoting the reaction.
Scope of the nickel-catalyzed enantioselective alkyl-arylation of TFP
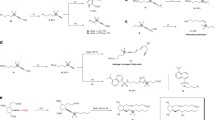
With the viable reaction conditions in hand, the substrate scope of aryl iodides 3 toward dicarbofunctionalization of TFP with tert-butyl iodide 2a was examined. As shown in Fig. 2, a variety of aryl iodides bearing substituents with different electronic nature were applicable to the reaction, providing the corresponding products with good yields and high enantioselectivities, ranging from 88 to 92% ee. Generally, electron-deficient aryl iodides (4a–4h, 4n) provided higher yields than electron-rich substrate (4m). Aryl bromide was also applicable to the reaction, with a lower yield and comparable ee obtained (4a). However, aryl chlorides were not suitable substrates. The reaction exhibited excellent functional group tolerance. Synthetically versatile handles, such as base and nucleophile sensitive ester, enolizable ketone, aldehyde, and cyanide, were compatible with the reaction (4a–4h). The successful formation of 4i with intact boronate further demonstrated the synthetic advance of the current nickel-catalyzed process. Notably, substrates bearing acidic proton, such as sulfonamide (4j) and alcohol (4k), are competent coupling partners, with good yields and high enantioselectivities obtained. Pyridine- and thiophene-containing aryl iodides underwent the coupling smoothly without loss of enanotioselectivity (4o, 4p). The substrate scope of aryl iodides can also be extended to complex molecules. D-Serine derived substrate with unprotected hydroxyl group furnished corresponding product 4q in synthetically useful yield and high enantioselectivity. Sulbactam-derived aryl iodide bearing two additional chiral centers did not influence the stereoselectivity, and afforded 4r in good yield and high diastereoselectivity (90% de), thus providing a potential opportunity for applications in medicinal chemistry. However, the reaction was sensitive to the sterically hindered substrates. For instance, ortho-substituted aryl iodide led to poor yield (4c). The reaction can also be extended to vinyl bromide, providing corresponding product 4s with a synthetically useful yield and moderate ee. In some cases (4k, 4m, 4p, 4q), the addition of NaI to the reaction could provide slightly higher yields, possibly because NaI can facilitate the reduction of the nickel catalyst or form a nickelate species to facilitate the catalytic cycle44,45.

aConditions A (unless otherwise specified): NiBr2·DME (10 mol%), L6 (10 mol%), 1 (1.2 M in DMA, 1.6 equiv), 2 (1.5 equiv), 3 (0.4 mmol, 1.0 equiv), DME (3.2 mL). All reported yields are isolated yields. bAryl bromide or vinyl bromide was used. c0.5 equiv of NaI was used. d10 mol% DMAP, 2 (0.4 mmol, 1.0 equiv) and 3 (0.6 mmol, 1.5 equiv) were used. eNiBr2·DME (13 mol%) and L6 (13 mol%) were used. fConditions B (unless otherwise specified): NiBr2·DME (12 mol%), L6 (10 mol%), 1 (1.2 M in DMA, 1.6 equiv), 2 (1.5 equiv), 3 (0.2 mmol, 1.0 equiv), FeCl3 (0.25 equiv), DME (1.6 mL). All reported yields are isolated yields. gFeBr2 (0.25 equiv) instead of FeCl3 (0.25 equiv) was used. h0.5 equiv of NaI instead of FeCl3 (0.25 equiv) was used. iNo additive was used. DMAP, N,N-dimethylpyridin-4-amine.
In addition to tert-butyl iodide 2a, tertiary alkyl iodides 2 bearing different side chains were also examined (Fig. 2). We found that low yields were obtained under standard reactions due to the relatively low conversion of aryl iodides. Increasing the loading amount of NiBr2·DME from 10 to 12 mol% with FeCl3 or FeBr246,47,48 as the additive benefited the conversion of aryl iodides and provided the chiral trifluoromethylated compounds in good yields and high ee (Supplementary Tables 4–10). The possible role of the ion salts is likely to facilitate the reduction of nickel(II) species, thus benefiting the catalytic cycle. However, the exact role of these iron salts in the reaction remains elusive at this stage. Tertiary alkyl iodide substituted with a phthalimide underwent the reaction efficiently, allowing the dicarbofunctionalization of TFP with a variety of aryl iodides with high ee and excellent functional group tolerance (5a–5e). Remarkably, 1-bromo-3-iodobenzene was also amenable to the reaction with selective formation of C–C bond at aryl iodide position (5f). Given that aryl bromides have widespread synthetic applications in organic synthesis, the survival of aryl bromide in the current process further demonstrated the synthetic utility of this protocol. The side chains in tertiary alkyl iodides with different chain lengths did not affect the reaction efficiency and enantioselectivities. For instance, good yields and high ee were provided with substrate bearing a long aliphatic side chain of eight carbons (5g), as well as that bearing a shorter carbon chain of three carbons (5h). In these cases, NaI instead of iron salts, was needed to facilitate the reaction. In addition, tertiary alkyl iodides 3 substituted with different important functional groups, such as ester, alkyl bromide, cyanide, ketone, and indole moieties, furnished the corresponding products efficiently (5i–5n). Last, the cyclic substrate was also applicable to the reaction, showing high ee (89–90%) and 1.2:1 dr (5o). The absolute configuration of the chiral trifluoromethylated products 4 and 5 was determined to be R by X-ray crystal structure analysis of compounds 4i and 5b. However, the primary and secondary alkyl halides as well as the substituted TFPs were not applicable to the reaction.
To demonstrate the synthetic practicability of this protocol, gram-scale synthesis of 4b was conducted (Fig. 3a), with even a higher yield (76%) and comparable ee obtained (90%). The resulting products can serve as a useful building block for various transformations. As illustrated in Fig. 3b, deprotection of the phthalimide group on compound 5b with N2H4·H2O provided amine 6 in almost quantity yield. Compound 6 can be easily converted into alkyl azide 7a49, a useful functional group for click chemistry. Condensation of 6 with carboxylic acid 8 led to amide 7b efficiently. The reaction of 2-chloropyrimidine with 6 also proceeded smoothly, providing pyrimidine containing compound 7c in good yield.

a Gram-scale synthesis of 4b. b Transformations of compound 5b. EDCI, (1-ethyl-3(3-dimethylpropylamine) carbodiimide.
Discussion
To gain mechanistic insight into the reaction, we conducted several radical trapping experiments (Fig. 4). The addition of an electron transfer inhibitor 1,4-dinitrobenzene or a radical scavenger TEMPO (2,2,6,6-tetramethylpiperidinooxy) to the reaction mixture of 1, 2a, and 3a under standard reaction conditions totally shut down the reaction (Fig. 4a). Radical clock experiment by using α-cyclopropylstyrene50 10 as a probe provided a mixture of ring-opening products 11a and 11b (Fig. 4b). These results clearly suggest that the alkyl radicals are involved in the reaction. This deduction was further supported by electron paramagnetic resonance (EPR) studies (Fig. 4c and Supplementary Figs. 5–7) with phenyl tert-butyl nitrone (PBN) as the spin trapping agent, in which the formation of spin adducts of the trapped tert-butyl radical and/or α-CF3 alkyl radical were detected. On the basis of these results and previous reports51,52,53,54,55,56, two plausible mechanisms were proposed (Fig. 5). Path I (Fig. 5a): The reaction begins with the formation of a nickel(I) species [NiILnX] (A)54,55. A subsequently reacts with tertiary alkyl iodide via a single electron transfer (SET) pathway to produce a tertiary alkyl radical and nickel(II) species [NiIILnX2] (B). The resulting B is reduced by Zn to provide nickel(0) [Ni0(Ln)], which undergoes oxidative addition with aryl iodide to generate arylnickel(II) complex [Ar-NiII(Ln)-X] (C). Meanwhile, the tertiary alkyl radical is trapped by TFP to form a new α-CF3 alkyl radical D. The combination of D with C produces the key intermediate nickel(III) complex E, in which the chiral ligand controls the stereoselectivity to form a highly reactive chiral E. Finally, E undergoes reductive elimination to deliver the chiral trifluoromethylated product and regenerate Ni(I) (A). Path II (Fig. 5b): A bimolecular oxidative addition of [NiILnX] (A) with aryl iodide initiates the reaction to provide arylnickel(II) complex C56, wherein complex A is generated by the reduction of nickel(II) complex B with zinc56,57. Subsequently, reduction of C with zinc provides arylnickel(I) complex [Ar-NiI(Ln)] (F), which reacts with tertiary alkyl iodide via a SET pathway to produce C and the tertiary alkyl radical. Radical addition of TFP delivers a new alkyl radical D. Combination of D with C provides the key intermediate E. Finally, reductive elimination of E affords the final product and regenerates A.

a Radical inhibition experiments. b Radical clock experiment. c EPR experiment.

a Possible mechanism I (path I). b Possible mechanism II (path II).
In conclusion, we have developed a highly enantioselective nickel-catalyzed alkyl-arylation of TFP with aryl and tertiary alkyl iodides. The combination of nickel catalyst with chiral Biox ligand renders the approach efficient to construct a variety of chiral trifluoromethylated compounds with high enantioselectivity. The protocol features synthetic convenience without laborious process to prepare organometallic reagents, as well as excellent functional group tolerance, even towards aryl bromide, boronate, and free alcohol, providing rapid and straightforward access to chiral trifluoromethylated compounds. In particular, the use of inexpensive industrial chemical TFP and successful suppression of its β-F elimination side reaction pave a new way to access chiral trifluoromethylated compounds of great interest in life and materials sciences. Preliminary mechanistic studies reveal that a radical tandem process is involved in the reaction.
Methods
General procedure for the enantioselective nickel-catalyzed alkyl-arylation of TFP
To a 25 mL of Schlenk tube were added Zn dust (1.5 equiv), tertiary alkyl iodide 2 (1.5 equiv), aryl iodide 3 (0.4 mmol, 1.0 equiv), L6 (10 mol%), and NiBr2·DME (10 mol%) in a glovebox. The tube was then taken out of the glovebox and evacuated and backfilled with Ar (three times). Anhydrous DME (3.2 mL) and TFP solution (1.2 M in DMA, 0.54 mL, 1.6 equiv) were added under Ar. The Schlenk tube was screw capped and stirred (800 rpm) for 12 h at room temperature. The reaction mixture was then diluted with EtOAc and filtered through a pad of Celite. The filtrate was washed with Na2S2O3, water, and brine, the combined organic layers were dried over Na2SO4, filtered. The filtrate was concentrated. The residue was purified with silica gel chromatography to provide corresponding product 4.
Data availability
The data that support the findings of this study are available within the paper and its supplementary information files. 1H, 13C, 19F NMR spectra, and mass spectrometry data are available in the Supplementary Information. The crystallographic data generated in this study have been deposited in the Cambridge Crystallographic Data Center database under accession code CCDC 2191969 (4i) and 2191970 (5b) (www.ccdc.cam.ac.uk/data_request/cif). Source data are provided with this paper.
References
Berger, R., Resnati, G., Metrangolo, P., Weber, E. & Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev. 40, 3496–3508 (2011).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 114, 2432–2506 (2014).
Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 23, 101467 (2020).
Inoue, M., Sumii, Y. & Shibata, N. Contribution of organofluorine compounds to pharmaceuticals. ACS omega 5, 10633–10640 (2020).
Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 51, 4359–4369 (2008).
Gillis, E. P., Eastman, K. J., Hill, M. D., Donnelly, D. J. & Meanwell, N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 58, 8315–8359 (2015).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Furuya, T., Kamlet, A. S. & Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature 473, 470–477 (2011).
Tomashenko, O. A. & Grushin, V. V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 111, 4475–4521 (2011).
Alonso, C., Marigorta, E. M., Rubiales, G. & Palacios, F. Carbon trifluoromethylation reactions of hydrocarbon derivatives and heteroarenes. Chem. Rev. 115, 1847–1935 (2015).
Shibata, N., Mizuta, S. & Kawai, H. Recent advances in enantioselective trifluoromethylation reactions. Tetrahedron.: Asymmetry 19, 2633–2644 (2008).
Nie, J., Guo, H.-C., Cahard, D. & Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 111, 455–529 (2011).
Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 115, 826–870 (2015).
Deng, Q. H., Wadepohl, H. & Gade, L. H. Highly enantioselective copper-catalyzed electrophilic trifluoromethylation of β-ketoesters. J. Am. Chem. Soc. 134, 10769–10772 (2012).
Liu, J. et al. Enantioselective di-/perfluoroalkylation of β-ketoesters enabled by cooperative photoredox/nickel catalysis. Org. Lett. 20, 461–464 (2018).
Iseki, K., Nagai, T. & Kobayashi, Y. Asymmetric trifluoromethylation of aldehydes and ketones with trifluoromethyltrimethylsilane catalyzed by chiral quaternary ammonium fluorides. Tetrahedron Lett. 35, 3137–3138 (1994).
Nagib, D. A., Scott, M. E. & MacMillan, D. W. C. Enantioselective trifluoromethylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 131, 10875–10877 (2009).
Kawai, H., Kusuda, A., Nakamura, S., Shiro, M. & Shibata, N. Catalytic enantioselective trifluoromethylation of azomethine imines with trimethyl(trifluoromethyl)silane. Angew. Chem. Int. Ed. 48, 6324–6327 (2009).
Furukawa, T. et al. Organocatalyzed regio- and enantioselective allylic trifluoromethylation of Morita-Baylis-hillman adducts using Ruppert-Prakash reagent. Org. Lett. 13, 3972–3975 (2011).
Gao, X., Xiao, Y.-L., Wan, X. & Zhang, X. Copper-catalyzed highly stereoselective trifluoromethylation and difluoroalkylation of secondary propargyl sulfonates. Angew. Chem. Int. Ed. 57, 3187–3191 (2018).
Trost, B. M., Gholami, H. & Zell, D. Palladium-catalyzed asymmetric allylic fluoroalkyla-tion/trifluoromethylation. J. Am. Chem. Soc. 141, 11446–11451 (2019).
Kostov, G. & Améduri, B. Telomerization reaction of 3,3,3-trifluoropropene. In Modern Synthesis Processes and Reactivity of Fluorinated Compounds (eds Groult, H. et al.) 533–559 (Elsevier, 2017).
Lai, N. A. Thermodynamic properties of HFO-1243zf and their application in study on a refrigeration cycle. Appl. Therm. Eng. 70, 1–6 (2014).
Vargaa, B. et al. Application of industrially relevant hydrofluoroolefin (HFO) gases in organic syntheses. Synthesis 53, 4313–4326 (2021).
Becker, H. & Sharpless, K. B. A new ligand class for the asymmetric dihydroxylation of olefins. Angew. Chem. Int. Ed. 35, 448–451 (1996).
Nozaki, K. et al. Highly enantioselective hydroformylation of olefins catalyzed by rhodium(I) complexes of new chiral phosphine-phosphite ligands. J. Am. Chem. Soc. 119, 4413–4423 (1997).
Dong, K., Li, Y., Wang, Z. & Ding, K. Catalytic asymmetric hydrogenation of α-CF3- or β-CF3-substituted acrylic acids using rhodium(I) complexes with a combination of chiral and achiral ligands. Angew. Chem. Int. Ed. 52, 14191–14195 (2013).
Kojima, Y., Miura, M. & Hirano, K. Copper-catalyzed regio- and enantioselective hydroallylation of 1‑trifluoromethylalkenes: effect of crown ether. ACS Catal. 11, 11663–11670 (2021).
Gu, J.-W., Min, Q.-Q., Yu, L.-C. & Zhang, X. Tandem difluoroalkylation-arylation of enamides catalyzed by nickel. Angew. Chem. Int. Ed. 55, 12270–12274 (2016).
Luo, Y.-C., Xu, C. & Zhang, X. Nickel-catalyzed dicarbofunctionalization of alkenes. Chin. J. Chem. 38, 1371–1394 (2020).
Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287–4296 (2020).
Poremba, K. E., Dibrell, S. E. & Reisman, S. E. Nickel-catalyzed enantioselective reductive cross-coupling reactions. ACS Catal. 10, 8237–8246 (2020).
Wei, X., Shu, W., Garcia-Dominguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515–13522 (2020).
Tu, H.-Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604–9611 (2020).
Qiao, J.-B. et al. Enantioselective reductive divinylation of unactivated alkenes by nickel-catalyzed cyclization coupling reaction. J. Am. Chem. Soc. 143, 12961–12967 (2021).
Wang, K., Ding, Z., Zhou, Z. & Kong, W. Ni-catalyzed enantioselective reductive diarylation of activated alkenes by domino cyclization/cross-coupling. J. Am. Chem. Soc. 140, 12364–12368 (2018).
Pan, Q., Ping, Y., Wang, Y., Guo, Y. & Kong, W. Ni-catalyzed ligand-controlled regiodivergent reductive dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 143, 10282–10291 (2021).
Li, Y. et al. Oxidative Heck reaction of fluorinated olefins with arylboronic acids by palladium catalysis. Eur. J. Org. Chem. 2015, 4340–4343 (2015).
Casnati, A., Gemoets, H. P. L., Motti, E., Della Ca, N. & Noel, T. Homogeneous and gas-liquid catellani-type reaction enabled by continuous-flow chemistry. Chem. Eur. J. 24, 14079–14083 (2018).
Cheng, R., Xu, C. & Zhang, X. Nickel-catalyzed regioselective coupling reaction of 3,3,3-trifluoropropene with arylzinc reagents. Chin. J. Org. Chem. 40, 3307–3313 (2020).
Ichitsuka, T., Fujita, T. & Ichikawa, J. Nickel-catalyzed allylic C(sp3)–F bond activation of trifluoromethyl groups via β-fluorine elimination: synthesis of difluoro-1,4-dienes. ACS Catal. 5, 5947–5950 (2015).
Paioti, P. H. S. et al. Catalytic enantioselective boryl and silyl substitution with trifluoromethyl alkenes: scope, utility, and mechanistic nuances of Cu-F β-elimination. J. Am. Chem. Soc. 141, 19917–19934 (2019).
Zhang, C., Lin, Z., Zhu, Y. & Wang, C. Chromium-catalyzed allylic defluorinative ketyl olefin coupling. J. Am. Chem. Soc. 143, 11602–11610 (2021).
Colon, I. & Kelsey, D. R. Coupling of aryl chlorides by nickel and reducing metals. J. Org. Chem. 51, 2627–2637 (1986).
Prinsell, M. R., Everson, D. A. & Weix, D. J. Nickel-catalyzed, sodium iodide-promoted reductive dimerization of alkyl halides, alkyl pseudohalides, and allylic acetates. Chem. Commun. 46, 5743–5745 (2010).
Gomes, P., Gosmini, C. & Perichon, J. New chemical cross-coupling between aryl halides and allylic acetates using a cobalt catalyst. Org. Lett. 5, 1043–1045 (2003).
Durandetti, M. & Périchon, J. Iron-catalysed reformatsky-type reactions. Synthesis 2006, 1542–1548 (2006).
Li, X., Feng, Z., Jiang, Z.-X. & Zhang, X. Nickel-catalyzed reductive cross-coupling of (hetero)aryl iodides with fluorinated secondary alkyl bromides. Org. Lett. 17, 5570–5573 (2015).
Meng, G. et al. Modular click chemistry libraries for functional screens using adiazotizing reagent. Nature 574, 86–89 (2019).
Baldwin, J. E. Thermal rearrangements of vinylcyclopropanes to cyclopentenes. Chem. Rev. 103, 1197–1212 (2003).
Weix, D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015).
Gu, J., Wang, X., Xue, W. & Gong, H. Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front. 2, 1411–1421 (2015).
Xu, C. et al. Difluoromethylation of (hetero)aryl chlorides with chlorodifluoromethane catalyzed by nickel. Nat. Commun. 9, 1170 (2018).
Somerville, R. J., Hale, L. V. A., Gómez-Bengoa, E., Burés, J. & Martin, R. Intermediacy of Ni–Ni species in sp2 C–O bond cleavage of aryl esters: relevance in catalytic C–Si bond formation. J. Am. Chem. Soc. 140, 8771–8780 (2018).
García-Domínguez, A., Li, Z. & Nevado, C. Nickel-catalyzed reductive dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 139, 6835–6838 (2017).
Lin, Q. & Diao, T. Mechanism of Ni-catalyzed reductive 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 141, 17937–17948 (2019).
He, R.-D. et al. Reductive alkylation of alkenyl acetates with alkyl bromides by nickel catalysis. Angew. Chem. Int. Ed. 61, e202114556 (2022).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21931013 (X.Z.), 22193072 (X.Z.), 21991122 (X.Z.)), the National Key R&D Program of China 2021YFF0701700 (X.Z.), and the Science and Technology Committee of Shanghai Municipality (21XD1404400 (X.Z.), 22JC1403500 (X.Z.)).
Author information
Authors and Affiliations
Contributions
Y.-Z.L. performed the experiments. N.R. and L.A. prepared some starting materials and purified some products. Y.Z. conducted EPR experiments. X.-L.W. conducted HPLC analysis. X.Z. conceived the research concept, directed the project, and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, YZ., Rao, N., An, L. et al. Enantioselective nickel-catalyzed dicarbofunctionalization of 3,3,3-trifluoropropene. Nat Commun 13, 5539 (2022). https://doi.org/10.1038/s41467-022-33159-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33159-2
- Springer Nature Limited