

Abstract
The detailed information on the surface structure and binding sites of oxide nanomaterials is crucial to understand the adsorption and catalytic processes and thus the key to develop better materials for related applications. However, experimental methods to reveal this information remain scarce. Here we show that 17O solid-state nuclear magnetic resonance (NMR) spectroscopy can be used to identify specific surface sites active for CO2 adsorption on MgO nanosheets. Two 3-coordinated bare surface oxygen sites, resonating at 39 and 42 ppm, are observed, but only the latter is involved in CO2 adsorption. Double resonance NMR and density functional theory (DFT) calculations results prove that the difference between the two species is the close proximity to H, and CO2 does not bind to the oxygen ions with a shorter O···H distance of approx. 3.0 Å. Extensions of this approach to explore adsorption processes on other oxide materials can be readily envisaged.
Similar content being viewed by others


Introduction
Metal oxides play an essential role in many fields of chemistry, physics, material science, biology, and environmental science1,2,3. Their corresponding nanomaterials, which have a high surface area, often exhibit unique or improved properties, especially for applications in gas sensing, gas storage, and catalysis4,5,6. Identifying gas adsorption sites in oxide nanomaterials and gas-surface interactions are crucial in designing and tailoring materials with improved performances. A variety of characterization methods, including electron microscopy and spectroscopic techniques, have been used to explore this issue, which is challenging due to the complex nature of the surface7. Despite the very high-resolution microscopy can possibly achieve these days, the images may not be representative of the whole structure and therefore quantitative analysis cannot be performed8. Spectroscopic investigations, such as IR, often require probe molecules, e.g., CO, making such methods indirect9 and unsuitable for in situ studies, in which probe molecules may interfere. Furthermore, many of these techniques require ultrahigh or high vacuum conditions10,11, thus the information obtained may not correspond to the real application. Therefore, it is required to develop alternative approaches to understand the gas binding sites in oxide-based materials.
Recently, we showed that 17O solid-state NMR spectroscopy provides very high resolution and oxygen species at different facets or different layers in oxide nanostructures can be clearly distinguished based on 17O NMR shifts12,13,14,15. This approach does not introduce any external species that may potentially alter the structure or properties of oxide surface, such as probe molecules12. Therefore, it provides unprecedented opportunities to explore the specific gas adsorption sites and the interactions on the surface for metal oxides.
MgO, a common oxide with a rock-salt structure, is of fundamental importance in physics16,17, chemistry18,19,20, nanotechnology21, as well as earth and space science22,23,24. In particular, chemisorption using MgO represents a promising strategy for reducing the levels of atmospheric CO2, the major greenhouse gas causing rapid and adverse climate change25. This strategy has attracted a lot of research attention owing to its wide operation temperature, low cost, and insusceptibility to humidity25,26,27. For example, MgO was recently spread over land for direct CO2 capture and demonstrated its scalable potential to remove more than 2 Gt CO2 per year at a low cost28. Here, we demonstrate that 17O solid-state NMR spectroscopy can be used as a primary method to study details of CO2 adsorption on MgO nanosheets. The approach, which is potentially extendable to other oxides, can distinguish two surface oxygen species only slightly different on the third coordination shell, identify the sorption sites of CO2, and reveal the nature of these sites as well as the reasons behind the differences in adsorption properties.
Results
Previous studies have shown that the surface structure and CO2 adsorption capacity of MgO vary significantly after calcining at a temperature higher than 800 K29,30. Thus, two different temperatures of 773 and 1073 K were used to prepare MgO nanomaterials. The powder X-ray diffraction (XRD) patterns of the MgO materials calcined at both temperatures are indexed and periclase MgO structure is confirmed (PDF No.71-1176) (Supplementary Fig. 1). High-resolution transmission electron microscopy (HRTEM) images show the nanosheet morphology of the MgO materials and the nanosheets are denoted as NS-773 and NS-1073 (NS for nanosheets), according to the corresponding thermal treatment temperatures (Supplementary Fig. 2). The primary exposed facets are determined as (111), and the thicknesses of both nanosheet samples are approx. 10 nm, which agrees with the average crystal sizes of MgO nanosheets calculated based on the XRD data using the Debye–Scherrer equation.
MgO nanosheets were then mixed with 17O2 and heated at 623 K for 17O enrichment. First, 17O single-pulse MAS NMR was used to study NS-773 and NS-1073 (Fig. 1). For the data obtained at an external field of 19.6 T, the well-resolved sharp peak at 47 ppm found in both spectra can be ascribed to 6-coordinated oxygen ions (O6C) in the bulk part of the MgO nanosheets31,32. Lower frequency resonances are also observed, including a relatively sharp and intense signal at 42 ppm, a broader component centered at around 39 ppm along with weaker peaks at approx. 34 and 23 ppm. Since 17O is a quadrupolar nucleus, peaks or discontinuities may not necessarily mean different sites and they can just arise from non-zero second-order quadrupolar interactions. Therefore, NMR spectra were also obtained for both samples at 9.4 T (Supplementary Fig. 3b). The linewidths of the peaks owing to large quadrupolar couplings become much wider at a lower external magnetic field. The fact that the data at 9.4 T do not show significant line broadening, indicates that these peaks are due to oxygen species in different chemical environments, which are associated with small quadrupolar interactions. An 17O NMR resonance centered at 41 ppm was observed in 17O-enriched nanophase MgO by Chadwick et al., which was assigned to oxygen sites perturbed by nearby H or CH3 at the third-nearest-neighbor on the surface33. Therefore, the peaks at 42 and 39 ppm in our data are tentatively assigned to surface oxygen species. 17O enrichment of oxides with 17O2 gas usually requires high temperature and most 17O isotopes diffuse into the bulk part of the sample, leading to poor sensitivity of 17O NMR observations of surface species. In our 17O NMR data, however, the intensities of the resonances for O6C in the bulk are only a small fraction of the total spectral intensity (~10%), indicating that a large fraction of the signal comes from the surface of the nanosheets, which is probably owing to the relative low enrichment temperature and/or the high surface area of the MgO nanosheets (Supplementary Fig. 3). The small fraction of the ‘bulk’ signal is also related to the relatively short recycle delay used, which is long enough for quantitative measurements of surface oxygen species yet not enough for bulk, because the longitudinal relaxation time (T1) is shorter for the surface than the bulk (Supplementary Fig. 4)14. Comparing the NMR data of NS-1073 and NS-773, the peak centered at 39 ppm has the strongest intensity for NS-773, while this resonance is much weaker and the peak at 42 ppm dominates in the spectrum of NS-1073, indicating that the concentrations of the two corresponding surface species vary when the MgO nanosheets are thermally treated at different temperatures.

External field: 19.6 T; MAS rate: 16 kHz; recycle delay: 5 s. The spectra are plotted such that the highest peak in each spectrum is at the same height and the relative intensities of different peaks can be conveniently compared.
The 17O-enriched MgO nanosheets were activated under vacuum and then dosed with 13CO2 to explore the adsorption capacity and the surface sites involved in CO2 adsorption. The 13C MAS NMR spectrum of NS-773 shows a single peak at 167 ppm, which can be ascribed to unidentate carbonate species34,35, suggesting that CO2 was chemisorbed on the surface of MgO nanosheets (Fig. 2a and Supplementary Fig. 5). Since the experiments were performed at a relatively low temperature of 313 K, the carbonate is formed mostly on the surface36,37. The 13C NMR spectrum of NS-1073 is similar and the peak maximum is observed at a slightly lower frequency of 166 ppm. CO2 adsorption data (Supplementary Fig. 6) and quantitative analysis on the 13C NMR spectral intensities show that the CO2 adsorption capacity (per mass) is around 55% higher for NS-1073 than NS-773, despite the fact that the former showed a much smaller surface area (142 m2 ∙ g−1) than the latter (227 m2 ∙ g−1) (Supplementary Fig. 7), which is similar to previous observations30. Such differences probably arise from the different relative concentrations of different surface species in the two samples.

a Single-pulse 13C MAS NMR spectra of NS-773 and NS-1073 after 13CO2 chemisorption at 9.4 T. MAS rate: 16 kHz; recycle delay: 40 s. 17O NMR spectra of NS-773 (b) and NS-1073 (c) before and after 13CO2 chemisorption along with the difference spectrum obtained at 9.4 T. MAS rate: 14 kHz; recycle delay: 5 s.
To identify the surface oxygen species involved in CO2 adsorption, 17O MAS NMR spectroscopy was applied to study the changes on the surface of MgO nanosheets. For NS-773, although both the peak at 42 ppm and the shoulder at 39 ppm have a very strong intensity in the spectrum before adsorption, only the intensity of the peak at 39 ppm remains after adsorption while the resonance at 42 ppm becomes much weaker (Fig. 2b). The difference spectrum clearly shows that the surface species giving rise to the peak at 42 ppm are likely involved exclusively in binding CO2, converting to surface carbonate species upon adsorption, as suggested by 13C NMR results. Carbonates oxygens have a 17O chemical shift of 223–274 ppm and a large quadrupolar coupling constant (CQ) of approx. 7 MHz, according to previous 17O NMR calculations on MgCO3 clusters38 and experimental observations of CaCO339. However, such a signal is not observed in our data, presumably due to the large linewidth arising from the large second-order quadrupolar interaction. Similar results were obtained for NS-1073 (Fig. 2c). Although the difference spectrum also shows weak signals at 52, 34, and 23 ppm, which may arise from a small amount of different surface sites, the majority of the intensity comes from the peak at 42 ppm, implying that the corresponding species are the key to CO2 adsorption for MgO nanosheets. Therefore, only the peaks at 39 and 42 ppm are focused on in the following study.
Since the peaks at around 41 ppm may be related to the surface H or CH3 according to previous studies33, 1H NMR spectroscopy was used to study the H species in MgO nanosheets (Supplementary Fig. 8). The overlapping resonances observed in the spectra of NS-773 and NS-1073 centered at around −1 to −0.5 ppm can be ascribed to the hydrogen-bond acceptor and/or isolated hydroxyl groups40,41,42. Quantitative measurements show that the total 1H NMR intensity of NS-1073 is only around 20% of NS-773, indicating significant H loss during thermal treatment. While the CH3 proton is not resolved in the 1H NMR spectrum, neither 13C single pulse (Supplementary Fig. 9) nor 1H→13C cross-polarization (CP) MAS NMR (Supplementary Fig. 10) spectrum of NS-773 exhibits the signal for CH3, implying the proposed CH3 is not present in our MgO nanosheets.
Despite strong evidence for the presence of hydroxyl sites in MgO nanosheets, no peak attributed to these species appear in the single-pulse 17O NMR data (Fig. 1 and Supplementary Fig. 11). The hydroxyl groups are expected to have a similar chemical environment as oxygen ions in Mg(OH)2, and thus an isotropic chemical shift of around 20 ppm and a very large CQ of approx. 6.8 MHz (obtained on Mg(OH)2)43. The absence of such signals is presumably due to the relatively low concentration of H in MgO nanosheets as well as the wide linewidth. On the other hand, broad resonances at low frequencies (~20 to −250 ppm) appear in the spectra of the MgO nanosheets exposed to water vapor, indicating hydroxyl species can be observed at a higher concentration due to large quadrupolar interaction (see Supplementary Figs. 12, 13 and related discussion).
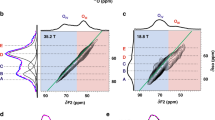
17O-1H REDOR NMR spectroscopy, which probes the internuclear proximity between oxygen and proton, was further applied to explore the nature of the resonances at 42 and 39 ppm (Fig. 3)44. Similar intensities in both the control and double resonance spectra are observed for the sharp resonance at 47 ppm, even at a very long recoupling time of 2.75 ms, because oxygen ions in the bulk part are far away from any proton, which is on the surface (Fig. 3a). In contrast, both the peaks at 42 and 39 ppm show lower intensity at a long recoupling time in the double resonance spectrum compared to the control spectrum, indicating that these oxygen species are near protons and thus are indeed on the surface. Nonetheless, the relatively small REDOR fractions (ΔS/S0, where ΔS and S0 are the intensity of the difference spectrum and the control spectrum, respectively) with short recoupling time suggest these oxygen sites are not directly bound to proton (Fig. 3b and Supplementary Fig. 14)14,15,45,46. The difference spectrum clearly shows that the signal at 39 ppm is associated with a greater REDOR fraction than the peak at 42 ppm, suggesting the oxygen ions in the former environment are closer to the proton.

a REDOR NMR spectra with a recoupling time of 2.75 ms. b REDOR fraction (ΔS/S0) as a function of recoupling time with the schematic surface structure of MgO. The yellow and orange triangles represent 39 and 42 ppm, respectively. External field: 18.8 T; MAS rate: 16 kHz; recycle delay: 5 s.
The distances between O and H were extracted by measuring the REDOR fractions as a function of recoupling time and performing numerical simulations47. Based on a model containing a single O–H spin pair, O–H distances of ~3.7 Å and 3.0 Å are determined for the surface oxygen ions giving rise to the peak at 42 and 39 ppm, respectively, the latter consistent with the distance between an oxygen ion and a proton in its third coordination shell (3.2 Å, see Fig. 3b). A longer O–H distance for the peak at 42 ppm suggests that this type of oxygen ion is less affected by the closest proton, which is probably in the fifth coordination shell or further away. Therefore, considering the observation that the 1H NMR intensity of NS-773 is much stronger than NS-1073, and the fact that the peak at 39 ppm dominates the 17O NMR spectrum of NS-773 while the resonance at 42 ppm is the strongest signal for NS-1073, the 17O NMR peaks at 39 and 42 ppm are assigned to surface 3-coordinated oxygen ions (O3C) with and without proton in the third coordination shell on the MgO (111) facets (Fig. 3b). Since CO2 only binds to the species giving rise to the peak at 42 ppm, the absence of proton nearby is crucial for successful CO2 adsorption. It can also explain the stronger spectral intensity for the peak at 39 ppm compared to the hydroxyl species (not observed in 17O NMR) because one proton can generate up to six bare surface oxygen ions in the surrounding giving rise to the resonance at 39 ppm (Supplementary Fig. 15).
The above results are also in good agreement with UV-Vis diffuse reflectance spectra of MgO nanosheets (Supplementary Fig. 16). Before CO2 adsorption, only one intense adsorption band at ~280 nm (4.4 eV) is observed for both samples, which can be attributed to the excitation of O3C48,49,50. The intensity of this band decreases significantly after CO2 adsorption, while the intensity of the band corresponding to O4C sites (230 nm or 5.4 eV) increases, indicating O3C species are converted to O4C by forming unidentate carbonate species during the process (Fig. 2).
Finally, density functional theory (DFT) calculations were performed to calculate the free energies of CO2 adsorption (Gads) on pristine MgO(111) surface and hydrogen pre-adsorbed MgO(111) surface (denoted as MgO(111)-4H), which correspond to the adsorption on the surface oxygen ion without and with a proton at the third coordination shell, respectively (Fig. 4). The calculated Gads for unidentate adsorption of CO2 on pristine MgO(111) is −0.81 eV, indicating strong CO2 adsorption on this surface. In contrast, the calculated Gads is much closer to zero (−0.27 eV) for MgO(111)-4H, suggesting that CO2 molecules are much less inclined to be adsorbed at these sites. This result, in combination with the NMR data, confirms that protons can poison nearby bare oxygen species at the MgO(111) facets and inhibit CO2 adsorption on these sites. Thus, a decrease in the proton concentration can enhance the CO2 adsorption capacity significantly.

CO2 unidentate adsorption on pristine MgO(111) (a) and MgO(111)-4H (b). Hydrogen, oxygen, carbon, and magnesium atoms are in white, red, gray, and green, respectively. Oxygen and magnesium atoms uncoordinated to CO2 are displayed with lines.
Discussion
With isotopic labeling 17O in MgO nanomaterials, the surface structure and CO2-surface interaction can be conveniently probed with 17O solid-state NMR spectroscopy. We show that bare surface oxygen sites on MgO nanosheets different only on the third coordination shell are distinguished according to 17O NMR shifts (42 and 39 ppm), and the oxygen species responsible for CO2 adsorption (42 ppm) are clearly identified based on the spectral intensity change before and after adsorption. Double resonance NMR techniques and DFT calculations show that CO2 adsorption on the surface oxygen atom is strongly inhibited by H species at its third coordination shell and the results shed light on the variations in the CO2 adsorption capacity upon thermal treatment.
Unlike the previous probe-molecule-assisted NMR method51,52,53, in which the surface structural information is observed indirectly through adsorbed molecules, our 17O NMR-based approach focuses on the solid surface itself and can follow its evolution upon sorption directly at high resolution. This 17O NMR method may also be combined with signal enhancement techniques, such as dynamic nuclear polarization54,55,56,57,58, to study materials with larger sizes and smaller surface area, and thus potentially be extended to a variety of other metal oxides to investigate gas adsorption processes and help to design related materials with improved properties for adsorption and catalysis. Despite these possible promising applications, it is challenging to detect species with a large quadrupolar interaction at a very low concentration by using this approach. Although oxygen ions in most metal oxides do have a small to medium CQ due to the small electronegativity values of common metals59, large quadrupolar interactions may be found in oxygen atoms bound to elements with a relatively large electronegativity, such as hydroxyl groups in this case, or in surface sites with lower coordination numbers and/or dangling bonds14. Obtaining data at a higher external magnetic field, which decreases the line broadening due to quadrupolar interactions, may alleviate the problem.
Methods
Synthesis of MgO nanosheets
MgO nanosheets were prepared by a modified aero-gel method60. In a typical synthesis, 1.0 g Mg belt was polished by using sandpaper and cleaned with acetone before it was dissolved in methanol (~44 mL), forming a 10 wt % Mg(OCH3)2 solution in methanol. Then 4-methoxybenzyl alcohol (BZ) was added to the above solution (with a ratio of Mg:BZ = 2:1). After the mixture was stirred for 5 h, 30 mL water-methanol solution (molar ratio of H2O to Mg is 2:1) was added dropwise. Then the mixture was further stirred for 12 h before it was transferred to an autoclave. After purging the autoclave with Ar for 10 min, the pressure was raised to 10 bar with Ar. Then the autoclave was heated to 538 K and kept at the temperature for 15 h before the vapor in the autoclave was released in 1 min. The resulting white power was heated to 773 K with a ramping step of 3 K/min, and it was kept at the temperature for 6 h to obtain the white powder of MgO nanosheets exposing mostly {111} facets used in this work.
17O-Enrichment procedures and CO2 chemisorption
MgO nanosheets were first heated at elevated temperature (773 or 1073 K) under vacuum for 2 h before they were cooled to room temperature. After that, 90% 17O-enriched O2 gas was introduced to the samples through a vacuum line. In order to selectively label mostly the surface oxygen species, the mixture was heated to 623 K and kept at the temperature for 14 h. For CO2 chemisorption, 17O-labeled MgO nanosheets were first exposed to vacuum and then 170 mbar 13CO2 at 313 K for 6 h. After that, the samples were exposed to vacuum again to remove physisorbed CO2.
Characterization
The X-ray powder diffraction (XRD) measurements were carried out using a SHIMADZU XRD-6000 diffractometer with a Cu Kα source (λ = 1.54178 Å) operating at 40 kV and 40 mA, in the 2θ range of 20−80°. High-resolution transmission electron microscopy (HRTEM) images were acquired on a JEOL JEM-2100 instrument with an acceleration voltage of 200 kV. The Brunauer−Emmett−Teller (BET) specific surface area was obtained by nitrogen adsorption-desorption at 77 K with a Micromeritics Tristar 2020 apparatus. The UV-Vis measurements were operated using a Shimadzu UV-3600 at room temperature (200–800 nm). 17O MAS NMR spectra obtained at 19.6 T were recorded with a Bruker Avance NEO spectrometer with 3.2 mm MAS probes. Excitation pulses with a width of 2 μs, corresponding to a solution π/6 flip angle, were used. 17O-1H REDOR NMR spectra acquired at 18.8 T were recorded with a Bruker Avance III spectrometer with a double-tuned 3.2 mm MAS probe. A direct excitation pulse sequence was used 17O pulses of 5 and 10 μs (solution π/2 and π pulses) and 1H π pulses of 6.7 μs for recoupling. O–H distances were extracted assuming there is only a single O–H spin pair47. 17O, 1H, and 13C MAS NMR data obtained at 9.4 T were recorded with a Bruker Avance III spectrometer with 3.2 and 4.0 mm MAS probes. Excitation pulses with a width of 1.2 μs, corresponding to a solution π/6 flip angle, were used for single-pulse 17O NMR experiments. π/2 pulses with a length of 2.1 and 2.9 μs were applied, for single-pulse 1H and 13C MAS NMR, respectively. 17O, 1H, and 13C chemical shifts were referenced to H2O at 0.0 ppm, adamantane at 1.92, and 38.5 (CH2) ppm, respectively. All the samples were packed in an N2 glove box.
Data availability
All data supporting the findings presented here is included in the manuscript, its supporting information, or from the authors upon request.
References
Wells, A. F. Structural Inorganic Chemistry (Oxford Univ. Press, 2012).
Harrison, W. A. Electronic Structure and the Properties of Solids (Dover, 1989).
Noguera, C. Physics and Chemistry at Oxide Surfaces (Cambridge Univ. Press, 1996).
Comini, E. Metal oxide nano-crystals for gas sensing. Analytica Chim. Acta 568, 28–40 (2006).
Kim, T. K. et al. Nanoporous metal oxides with tunable and nanocrystalline frameworks via conversion of metal−organic frameworks. J. Am. Chem. Soc. 135, 8940–8946 (2013).
Rodriguez, J. A. et al. Activity of CeOX and TiOX nanoparticles grown on Au(111) in the water-gas shift reaction. Science 318, 1757–1760 (2007).
Mudunkotuwa, I. A. & Grassian, V. H. The devil is in the details (or the surface): impact of surface structure and surface energetics on understanding the behavior of nanomaterials in the environment. J. Environ. Monit. 13, 1135–1144 (2011).
Vogt, T., Dahmen, W. & Binev, P. Modeling Nanoscale Imaging in Electron Microscopy (Springer, 2012).
Lamberti, C., Zecchina, A., Groppo, E. & Bordiga, S. Probing the surfaces of heterogeneous catalysts by in situ IR spectroscopy. Chem. Soc. Rev. 39, 4951–5001 (2010).
Guo, J., Bian, K., Lin, Z. & Jiang, Y. Perspective: Structure and dynamics of water at surfaces probed by scanning tunneling microscopy and spectroscopy. J. Chem. Phys. 145, 160901 (2016).
Wang, Y. & Wöll, C. IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: bridging the materials gap. Chem. Soc. Rev. 46, 1875–1932 (2017).
Du, J.-H. & Peng, L. Recent progress in investigations of surface structure and properties of solid oxide materials with nuclear magnetic resonance spectroscopy. Chin. Chem. Lett. 29, 747–751 (2018).
Shen, L. & Peng, L. 17O solid-state NMR studies of oxygen-containing catalysts. Chin. J. Catal. 36, 1494–1504 (2015).
Wang, M. et al. Identification of different oxygen species in oxide nanostructures with 17O solid-state NMR spectroscopy. Sci. Adv. 1, e1400133 (2015).
Li, Y. et al. Distinguishing faceted oxide nanocrystals with 17O solid-state NMR spectroscopy. Nat. Commun. 8, 581 (2017).
You, Y. S., Reis, D. A. & Ghimire, S. Anisotropic high-harmonic generation in bulk crystals. Nat. Phys. 13, 345–349 (2017).
Uzan, A. J. et al. Attosecond spectral singularities in solid-state high-harmonic generation. Nat. Photonics 14, 183–187 (2020).
Wang, H. et al. Strong metal–support interactions on gold nanoparticle catalysts achieved through Le Chatelier’s principle. Nat. Catal. 4, 418–424 (2021).
Song, Y. et al. Dry reforming of methane by stable Ni–Mo nanocatalysts on single-crystalline MgO. Science 367, 777–781 (2020).
Millet, M.-M. et al. Ni single atom catalysts for CO2 activation. J. Am. Chem. Soc. 141, 2451–2461 (2019).
Oh, Y.-W. et al. Field-free switching of perpendicular magnetization through spin-orbit torque in antiferromagnet/ferromagnet/oxide structures. Nat. Nanotechnol. 11, 878–884 (2016).
McWilliams, R. S. et al. Phase transformations and metallization of magnesium oxide at high pressure and temperature. Science 338, 1330–1333 (2012).
Cordier, P., Amodeo, J. & Carrez, P. Modelling the rheology of MgO under Earth’s mantle pressure, temperature and strain rates. Nature 481, 177–180 (2012).
Coppari, F. et al. Implications of the iron oxide phase transition on the interiors of rocky exoplanets. Nat. Geosci. 14, 121–126 (2021).
Bhagiyalakshmi, M., Lee, J. Y. & Jang, H. T. Synthesis of mesoporous magnesium oxide: its application to CO2 chemisorption. Int. J. Greenh. Gas. Control 4, 51–56 (2010).
Lee, S. C. et al. CO2 absorption and regeneration of alkali metal-based solid sorbents. Catal. Today 111, 385–390 (2006).
Zhao, X. et al. Mesoporous MgO promoted with NaNO3/NaNO2 for rapid and high-capacity CO2 capture at moderate temperatures. Chem. Eng. J. 332, 216–226 (2018).
McQueen, N., Kelemen, P., Dipple, G., Renforth, P. & Wilcox, J. Ambient weathering of magnesium oxide for CO2 removal from air. Nat. Commun. 11, 3299 (2020).
King, B. V. & Freund, F. Surface charges and subsurface space-charge distribution in magnesium oxides containing dissolved traces of water. Phys. Rev. B 29, 5814–5824 (1984).
Mutch, G. A. et al. Carbon capture by metal oxides: unleashing the potential of the (111) facet. J. Am. Chem. Soc. 140, 4736–4742 (2018).
Bastow, T. J. & Stuart, S. N. 17O NMR in simple oxides. Chem. Phys. 143, 459–467 (1990).
Turner, G. L., Chung, S. E. & Oldfield, E. Solid-state oxygen-17 nuclear magnetic resonance spectroscopic study of the Group II oxides. J. Mag. Reson. 64, 316–324 (1985).
Chadwick, A. V., Poplett, I. J. F., Maitland, D. T. S. & Smith, M. E. Oxygen speciation in nanophase MgO from solid-state 17O NMR. Chem. Mater. 10, 864–870 (1998).
Ito, T., Kobayashi, H. & Tashiro, T. Roles of low-coordinated surface ions in adsorption of gases on MgO. Il Nuovo Cim. D. 19, 1695–1705 (1997).
Fu, Y., Zhang, L., Yue, B., Chen, X. & He, H. Simultaneous characterization of solid acidity and basicity of metal oxide catalysts via the solid-state NMR technique. J. Phys. Chem. C. 122, 24094–24102 (2018).
Gregg, S. J. & Ramsay, J. D. Adsorption of carbon dioxide by magnesia studied by use of infrared and isotherm measurements. J. Chem. Soc. A 2784–2787 (1970).
Ward, S. M., Braslaw, J. & Gealer, R. L. Carbon dioxide sorption studies on magnesium oxide. Thermochim. Acta 64, 107–114 (1983).
Chen, M., Jackson, V. E., Felmy, A. R. & Dixon, D. A. Structures and Energetics of (MgCO3)nClusters (n≤ 16). J. Phys. Chem. A 119, 3419–3428 (2015).
Smith, M. E., Steuernagel, S. & Whitfield, H. J. 17O magic-angle spinning nuclear magnetic resonance of CaCO3. Solid State Nucl. Magn. Reson. 4, 313–316 (1995).
Chizallet, C., Costentin, G., Lauron-Pernot, H., Maquet, J. & Che, M. 1H MAS NMR study of the coordination of hydroxyl groups generated upon adsorption of H2O and CD3OH on clean MgO surfaces. Appl. Catal. A Gen. 307, 239–244 (2006).
Mastikhin, V. M. Characterization of surface active sites of catalysts with high-resolution solid-state NMR. Colloids Surf. A 78, 143–166 (1993).
Chizallet, C. et al. Study of the structure of OH groups on MgO by 1D and 2D 1H MAS NMR combined with DFT cluster calculations. J. Phys. Chem. C. 111, 18279–18287 (2007).
van Eck, E. R. H. & Smith, M. E. Orientation of the quadrupole and dipole tensors of hydroxyl groups by 17O quadrupole separated local field NMR. J. Chem. Phys. 108, 5904–5912 (1998).
Gullion, T. & Schaefer, J. Rotational-echo double-resonance NMR. J. Magn. Reson. 81, 196–200 (1989).
Peng, L., Liu, Y., Kim, N., Readman, J. E. & Grey, C. P. Detection of brønsted acid sites in zeolite HY with high-field 17O-MAS-NMR techniques. Nat. Mater. 4, 216–219 (2005).
Chen, J. et al. Polar surface structure of oxide nanocrystals revealed with solid-state NMR spectroscopy. Nat. Commun. 10, 5420 (2019).
Gullion, T. Introduction to rotational-echo, double-resonance NMR. Concepts Magn. Reson. 10, 277–289 (1998).
Stankic, S. et al. Size-dependent optical properties of MgO nanocubes. Angew. Chem. Int. Ed. 44, 4917–4920 (2005).
Coluccia, S., Deane, A. M. & Tench, A. J. Photoluminescent spectra of surface states in alkaline earth oxides. J. Chem. Soc. Faraday Trans. 1 74, 2913–2922 (1978).
Shluger, A. L., Sushko, P. V. & Kantorovich, L. N. Spectroscopy of low-coordinated surface sites: Theoretical study of MgO. Phys. Rev. B 59, 2417–2430 (1999).
Hu, Y. et al. Facet-dependent acidic and catalytic properties of sulfated titania solid superacids. Chem. Commun. 51, 14219–14222 (2015).
Peng, Y.-K. et al. Trimethylphosphine-assisted surface fingerprinting of metal oxide nanoparticle by 31P solid-state NMR: A zinc oxide case study. J. Am. Chem. Soc. 138, 2225–2234 (2016).
Peng, Y.-K. et al. Mapping surface-modified titania nanoparticles with implications for activity and facet control. Nat. Commun. 8, 675 (2017).
Zagdoun, A. et al. Large molecular weight nitroxide biradicals providing efficient dynamic nuclear polarization at temperatures up to 200 K. J. Am. Chem. Soc. 135, 12790–12797 (2013).
Zagdoun, A. et al. A slowly relaxing rigid biradical for efficient dynamic nuclear polarization surface-enhanced NMR spectroscopy: Expeditious characterization of functional group manipulation in hybrid materials. J. Am. Chem. Soc. 134, 2284–2291 (2012).
Perras, F. A., Wang, Z., Naik, P., Slowing, I. I. & Pruski, M. Natural abundance 17O DNP NMR provides precise O−H distances and insights into the brønsted acidity of heterogeneous catalysts. Angew. Chem. Int. Ed. 56, 9165–9169 (2017).
Blanc, F. et al. Dynamic nuclear polarization enhanced natural abundance 17O spectroscopy. J. Am. Chem. Soc. 135, 2975–2978 (2013).
Reif, B., Ashbrook, S. E., Emsley, L. & Hong, M. Solid-state NMR spectroscopy. Nat. Rev. Methods Prim. 1, 2 (2021).
Schramm, S. & Oldfield, E. High-resolution oxygen-17 NMR of solids. J. Am. Chem. Soc. 106, 2502–2506 (1984).
Zhu, K., Hu, J., Kübel, C. & Richards, R. Efficient preparation and catalytic activity of MgO(111) nanosheets. Angew. Chem. Int. Ed. 118, 7435–7439 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (91745202 and 21972066), NSFC—Royal Society Joint Program (21661130149). L.P. thanks the Royal Society and Newton Fund for a Royal Society—Newton Advanced Fellowship. The ECUST group thanks to the Programme of Introducing Talents of Discipline to Universities (B16017). A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by the National Science Foundation Cooperative Agreement No. DMR-1644779 and the state of Florida.
Author information
Authors and Affiliations
Contributions
J.-H.D. and L.P. supervised the project. J.-H.D., B.Z., K.C., M.W., Y.W., I.H., and Z.G. designed and carried out the experiments. L.C., X.-P.W., and X.-Q.G. carried out the DFT calculations. J.-H.D. and L.P wrote the paper; and all authors participated in the analysis of the data and discussions of the results, as well as preparation of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, JH., Chen, L., Zhang, B. et al. Identification of CO2 adsorption sites on MgO nanosheets by solid-state nuclear magnetic resonance spectroscopy. Nat Commun 13, 707 (2022). https://doi.org/10.1038/s41467-022-28405-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28405-6
- Springer Nature Limited