

Abstract
The Alder–ene type reaction between alkenes and alkynes provides an efficient and atom-economic method for the construction of C-C bond, which has been widely employed in the synthesis of natural products and other functional molecules. The intramolecular enantioselective Alder-ene cycloisomerization reactions of 1,n-enynes have been extensively investigated. However, the intermolecular asymmetric version has not been reported, and remains a challenging task. Herein, we describe a rhodium-catalyzed intermolecular enantioselective Alder-ene type reaction of cyclopentenes with silylacetylenes. A variety of chiral (E)-vinylsilane tethered cyclopentenes bearing one quaternary carbon and one tertiary carbon stereocenters are achieved in high yields and enantioselectivities. The reaction undergoes carbonyl-directed migratory insertion, β-H elimination and desymmetrization of prochiral cyclopentenes processes.
Similar content being viewed by others
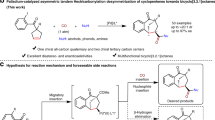

Introduction
The production of synthetically valuable products from readily accessible substrates in a redox-neutral, atom- and step-economical approach is a long-standing goal in organic synthesis. The Alder-ene type reaction comprises a particularly efficient subset of this target1,2,3,4,5,6, which provides an appealing tool to construct C–C bond. Over the past decades, transition metal-catalyzed intramolecular enantioselective Alder-ene type cycloisomerization reactions of 1,6-enynes or 1,7-enynes have been extensively investigated for the rapid assembly of chiral five- or six membered carbo- and hetero-cyclic frameworks (Fig. 1a)7,8,9,10,11,12,13,14,15,16,17. However, the intermolecular Alder-ene reactions were mainly confined to less-hindered terminal alkenes18,19,20,21,22,23,24,25, and the studies on internal alkenes have rarely been reported26,27. Very recently, Trost and coworkers successfully implemented ruthenium-catalyzed intermolecular alkene–alkyne coupling reactions with well-designed vinyl boronate compounds to synthesize boron-functionalized 1,4-dienes (Fig. 1b)28,29, in which the boron substituent played a vital role to facilitate the transformations. However, the intermolecular enantioselective Alder-ene type reaction remains an unexplored territory.

a Intramolecular enantioselective Alder-ene type cycloisomerization reaction (well-developed). b Intermolecular Alder-ene type reaction of internal alkenes (Trost’s work). c Intermolecular enantioselective Alder-ene type reaction (this work).
All-carbon chiral quaternary stereocenters are fundamental structural motifs present in natural products and pharmaceuticals, which could improve the metabolic stability and target selectivity of biologically active compounds30. However, the synthesis of chiral quaternary carbon centers is a challenging task, especially those that are not formed at the direct reaction site31,32,33,34,35,36,37,38,39,40,41. Asymmetric desymmetrization of prochiral compounds or meso-compounds offers a commendable synthetic tool for achieving this objective42,43,44,45,46,47,48,49,50,51,52,53,54.
Herein, we describe an asymmetric desymmetrization of prochiral cyclopentenes with silylacetylenes enabled by rhodium-catalyzed intermolecular enantioselective Alder-ene type reaction. This protocol allows access to chiral (E)-vinylsilane tethered cyclopentenes bearing one quaternary carbon and one tertiary carbon stereocenters in high yields and enantioselectivities (Fig. 1c).
Results
Reaction optimization
We commenced our studies with the employment of N,1-diphenylcyclopent-3-ene-1-carboxamide 1a and triisopropylsilylyne 2a as the model substrates. After considerable screening of the reaction parameters (see the Supplementary Table 1 for details), the desired product 3a was obtained in 96% yield and 95% ee with [Rh(COD)OMe]2 as the catalyst, phosphoramidite L6 as the ligand, PhMe2CCO2H and NaBARF as the additives in DCM at 80 °C (Table 1, entry 1). The P,P-ligands L1, L2 and N,P-ligand L3 inhibited the transformation (entry 2). Phosphoramidite ligands L4 and L5 performed this reaction in less efficiency (entries 3 and 4). Rh(COD)2OTf and [Rh(COD)Cl]2 gave inferior results compared with [Rh(COD)OMe]2, [Cp*RhCl2]2 and Pd(dba)2 delivered trace amount yield of product 3a (entries 5–8). AcOH and PhCO2H offered 3a in diminished enantioselectivities (entries 9 and 10), and no product was detected in the presence of TsOH (entry 11). Using other additives, such as AgSbF6 and AgPF6 could not perform this transformation as well as NaBARF (entries 12 and 13). Conducting the reaction in CHCl3 and toluene led to lower yields and enantioselectivities (entries 14 and 15), and the reaction was completely suppressed in THF (entry 16).
Substrate scope
With the optimized reaction conditions in hand, we then explored the generality of this rhodium-catalyzed intermolecular enantioselective Alder-ene type reaction (Fig. 2). Various aryl and heteroaryl substituted cyclopentenes (see the Supplementary Methods for details) performed the reactions well, affording the products 3b-3k in 90–94% ee. Replacing the aryl groups with a benzyl group or an alkyl group delivered the products 3l and 3m in 96 and 95% ee. Notably, the substrate 1n containing two alkene moieties, performed the reaction selectively with the endocyclic double bond to give the product 3n in 84% yield with 92% ee. The amino substituted cyclopentene 1o was also suitable substrate for this transfomation, furnishing chiral α-amino acid derivative 3o in 93% ee. N-benzylcyclopent-3-ene-1-carboxamide (R1 = H) offered the desired product 3p in 90% yield and 94% ee.

Reaction conditions: 1a (0.10 mmol), 2a (0.30 mmol), [Rh(COD)OMe]2 (2.5 mol%), L6 (6.0 mol%), PhMe2CCO2H (60 mol%), NaBARF (10 mol%), DCM (1.0 mL), 80 oC, 36 h, under argon. Isolated yields, d.r. > 20:1, E/Z > 20:1, determined by 1H NMR analysis. The ee values were determined by chiral HPLC analysis. a[Rh(COD)OMe]2 (5.0 mol%), 72 h. b72 h. c[Rh(COD)OMe]2 (5.0 mol%). d[Rh(COD)Cl]2 (5.0 mol%), L5 (12 mol%), Ph3CCO2H (60 mol%), NaBARF (20 mol%), d.r. > 20:1, Z/E > 20:1.
Subsequently, the compatibilities of amide moiety were investigated, and the products 3q-3w with aryl and alkyl groups were achieved in 86–94% ee. The substrate 1x with tertiary amide group gave 3x in 63% yield with 96% ee under slightly modified conditions. Besides, the enantioenriched spirolactam 3y could also be synthesized in 78% yield and 90% ee. Ester or ketone groups substituted cyclopentenes proceeded this reaction efficiently, affording the products 3z-3ad in 72–93% yields with 90–93% ee. When substrate 1ae was subjected to the standard reaction conditions, no desired product 3ae was detected.
After checking the character of cyclopentenes, we then turned our attention to the scope of alkynes. The reactions proceeded smoothly with diverse silylacetylenes, delivering products 3af-3ai in 93–94% ee. In addition, sterically hindered alkyl alkyne was also proved to be qualified for this reaction, as demonstrated by the formation of product 3aj with 90% ee. Unfortunately, simple alkyl and aryl substituted acetylenes were not yet compatible with the reaction system to give the desired products 3ak and 3al. The 1-heptyne caused the reaction to become confusing, 4-ethynylanisole offered a branched byproduct in a very low yield (see Supplementary Note 1 for details). The internal alkyne biphenylacetylene could participate in this transformation to afford product 3am in 66% yield with 47% ee. The absolute configuration of the product was confirmed by the X-ray analysis of the compound from iododesilylation of 3f (see Supplementary Note 2 for details).
Gram-scale experiment and further transformations of the products
To demonstrate the synthetic utility of this reaction, a gram-scale experiment (Fig. 3a) and further transformations of the products 3p and 3ai were carried out (Fig. 3b). Product 3p (1.72 g) could be synthesized in 93% yield and 95% ee on a 5.0 mmol scale under the standard conditions. A palladium-catalyzed hydrogenation reaction converted 3p to compound 4 in 95% yield. The selective reduction of the amide group of 3p using LiAlH4 afforded compound 5 in 93% yield. Moreover, compound 3p could be decorated with NBS to give bromination compounds 6 in 40% yield and 7 in 45% yield, respectively. The desilylation of 3ai with TFA offered compound 8 in 94% yield. Arylated product 9 could be constructed through palladium-catalyzed Hiyama cross-coupling reaction, maintaining the E/Z-stereochemistry and enantioselectivity.

a Gram-scale experiment. b Transformations of the products. aPd/C (10 wt %), B2(OH)4, H2O, DCM, rt. bLiAlH4, 1,4-dioxane, 0 oC-80 oC, 15 h. cNBS, Ag2CO3, HFIP, rt, 2 h. dTFA, DCM, 60 oC, 12 h. ePd(dba)2, TBAF·3H2O, Iodobenzene, THF, rt, 24 h. c Mechanistic studies. d Proposed mechanism.
Mechanistic investigations
To gain insight into the mechanism of this reaction, several control experiments and deuterium-labeling experiments were carried out (Fig. 3c). When [D]-2a was employed, the reaction resulted in [D]1-3aa with 63% D atom at β-position to the TIPS group and 35% D atom at α-position. This result implied that a metal vinylidene intermediate could be involved55,56,57. The reaction was drastically prohibited in the absence of the acid. Conducting the reaction with PhCO2D provided [D]2-3aa with 10% D atom at β-position to the TIPS group and 6% D atom at α-position, which indicated that the acid may work with the rhodium(I) to catalyze this reaction by the formation of RhH species. The generation of product [D]3-3aa from [D]-2a with PhCO2D further supported the hypothesis.
Based on the above experiment results and previous works45,46,47,55,56,57,58,59,60, a possible reaction pathway is depicted in Fig. 3d with 1a and 2a as the model substrates. Oxidative addition of rhodium(I) into PhMe2CCO2H generates RhH species A, and the presence of NaBARF plays a pivotal role for the generation and stabilization of this cationic rhodium complex59,60. Regioselective alkyne insertion into the species A forms vinylrhodium intermediate B1 directly (path a). On the other hand, η2-coordination of the alkyne to the species A followed by rearrangement generates an η1-vinylidenerhodium species C55,56,57, which could produce intermediate B2 via migratory insertion (path b). Coordination of B1 or B2 to the double bond of 1a directed by carbonyl group forms the rhodium(III) intermediate D45,46,47,58, which undergoes syn-migratory insertion to afford the intermediate E. Subsequent β-hydride elimination of E offers the desired product 3a.
Discussion
In conclusion, we have described a rhodium-catalyzed intermolecular enantioselective Alder-ene/desymmetrization reaction of prochiral cyclopentenes with alkynes directed by carbonyl group. This method provides a practical route for the synthesis of chiral (E)-vinylsilane tethered cyclopentenes bearing one quaternary carbon and one tertiary carbon stereocenters in high efficiency and atom-economy. Further studies on the reaction mechanism and expansion of the asymmetric intermolecular Alder-ene type reactions are underway in our laboratory.
Methods
General procedure for the rhodium-catalyzed intermolecular enantioselective Alder-ene type reaction of cyclopentenes with silylacetylenes
To an oven-dried 10-mL Schlenk tube equipped with a teflon-coated magnetic stir bar was added 1a (26.3 mg, 0.100 mmol, 1.00 equiv), [Rh(COD)OMe]2 (1.2 mg, 2.5 mol%), L6 (2.7 mg, 6.0 mol%), NaBARF (8.9 mg, 10 mol%) and PhMe2CCO2H (10 mg, 60 mol%). The vial was thoroughly flushed with argon, and 2a (54.7 mg, 0.600 mmol, 3.00 equiv), as well as DCM (1.0 mL) was added under argon atmosphere. Then the reaction mixture was stirred at 80 °C for 36 h. After the reaction vessel was cooled to room temperature, the solution was concentrated in vacuum and purified by careful chromatography on silica gel (PE/EA = 50/1) to afford the desired product 3a.
Data availability
All data generated and analyzed in this study are provided in this article and its Supplementary Information, and also available from the corresponding authors upon request. The X-ray crystallographic coordinate for structure 10 reported in this study has been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2064563. These data could be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Source data are provided with this paper.
References
Trost, B. M. Palladium-catalyzed cycloisomerizations of enynes and related reactions. Acc. Chem. Res. 23, 34–42 (1990).
Fairlamb, I. J. S. Asymmetric cycloisomerization of 1,6- and 1,7-enynes by transition-metal catalysts. Angew. Chem. Int. Ed. 43, 1048–1052 (2004).
Marinetti, A., Jullien, H. & Voituriez, A. Enantioselective, transition metal catalyzed cycloisomerizations. Chem. Soc. Rev. 41, 4884–4908 (2012).
Watsona, I. D. G. & Toste, F. D. Catalytic enantioselective carbon-carbon bond formation using cycloisomerization reactions. Chem. Sci. 3, 2899–2919 (2012).
Stathakis, C. I., Gkizis, P. L. & Zografos, A. L. Metal-catalyzed cycloisomerization as a powerful tool in the synthesis of complex sesquiterpenoids. Nat. Prod. Rep. 33, 1093–1117 (2016).
Trost, B. M., Frederiksen, M. U. & Rudd, M. T. Ruthenium-catalyzed reactions—a treasure trove of atom-economic transformations. Angew. Chem. Int. Ed. 44, 6630–6666 (2005).
Trost, B. M., Lee, D. C. & Rise, F. A new palladium catalyst for intramolecular carbametalations of enynes. Tetrahedron Lett. 30, 651–654 (1989).
Goeke, A., Sawamura, M., Kuwano, R. & Ito, Y. Enantioselective cycloisomerization of 1,6-enynes catalyzed by chiral diphosphane-palladium complexes. Angew. Chem. Int. Ed. Engl. 35, 662–663 (1996).
Hatano, M., Terada, M. & Mikami, K. Highly enantioselective palladium-catalyzed ene-type cyclization of a 1,6-enyne. Angew. Chem. Int. Ed. 40, 249–253 (2001).
Hatano, M. & Mikami, K. Highly enantioselective quinoline synthesis via ene-type cyclization of 1,7-enynes catalyzed by a cationic BINAP-palladium(ii) complex. J. Am. Chem. Soc. 125, 4704–4705 (2003).
Hatano, M. & Mikami, K. Highly enantioselective alkaloid synthesis via ene-type cyclizations catalyzed by cationic chiral palladium(II) complexes of PN-ligands with an achiral oxazoline unit. Org. Biomol. Chem. 1, 3871–3873 (2003).
Hatano, M., Yamanaka, M. & Mikami, K. A new N,P-ligand with achiral gem-dimethyloxazoline for Palladium(II)-catalyzed cyclization of 1,6-enynes: transition state probe for the N/C trans mode in Mizoroki-Heck-type C-C bond formation. Eur. J. Org. Chem. 2003, 2552–2555 (2003).
Cao, P. & Zhang, X. The first highly enantioselective Rh-catalyzed enyne cycloisomerization. Angew. Chem. Int. Ed. 39, 4104–4106 (2000).
Deng, X. et al. Enantioselective rhodium-catalyzed cycloisomerization of (E)-1,6-Enynes. Angew. Chem. Int. Ed. 55, 6295–6299 (2016).
Wu, C. & Yoshikai, N. Cobalt-catalyzed intramolecular reactions between a vinylcyclopropane and an alkyne: switchable [5+2] cycloaddition and homo-ene pathways. Angew. Chem. Int. Ed. 57, 6558–6562 (2012).
Martínez, A., García-García, P., Fernández-Rodríguez, M. A., Rodríguez, F. & Sanz, R. Gold(I)-catalyzed enantioselective synthesis of functionalized indenes. Angew. Chem. Int. Ed. 49, 4633–4637 (2010).
Liang, R.-X. et al. Palladium-catalyzed enantioselective heteroarenyne cycloisomerization reaction. Angew. Chem. Int. Ed. 60, 7412–7417 (2021).
Trost, B. M. & Indolese, A. Ruthenium-catalyzed addition of alkenes to acetylenes. J. Am. Chem. Soc. 115, 4361–4362 (1993).
Trost, B. M., Pinkerton, A. B., Toste, F. D. & Sperrle, M. Synthesis of 1,1-disubstituted alkenes via a Ru-catalyzed addition. J. Am. Chem. Soc. 123, 12504–12509 (2001).
Trost, B. M. & Surivet, J.-P. An atom-economic three-carbon chain extension to give enamides. Angew. Chem. Int. Ed. 40, 1468–1471 (2001).
Hansen, E. C. & Lee, D. Synthesis of β, β-disubstituted vinyl boronates via the ruthenium-catalyzed alder ene reaction of borylated alkynes and alkenes. J. Am. Chem. Soc. 127, 3252–3253 (2005).
Hilt, G. & Treutwein, J. Cobalt-catalyzed Alder–ene Reaction. Angew. Chem. Int. Ed. 46, 8500–8502 (2007).
Hilt, G., Erver, F. & Harms, K. Regioselective cobalt-catalyzed Alder-ene reaction toward silicon- and boron-functionalized building blocks. Org. Lett. 13, 304–307 (2011).
Trost, B. M., Koester, D. C. & Herron, A. N. Stereocontrolled synthesis of vinyl boronates and vinyl silanes via atom-economical ruthenium-catalyzed alkene–alkyne coupling. Angew. Chem. Int. Ed. 54, 15863–15866 (2015).
Trost, B. M. & Cregg, J. J. Ruthenium-catalyzed alkene-alkyne coupling of disubstituted olefins: application to the stereoselective synthesis of trisubstituted enecarbamates. J. Am. Chem. Soc. 137, 620–623 (2015).
Hilt, G., Paul, A. & Treutwein, J. Cobalt catalysis at the crossroads: cobalt-catalyzed Alder-ene reaction versus [2+2] cycloaddition. Org. Lett. 12, 1536–1539 (2010).
Rummelt, S. M., Cheng, G.-J., Gupta, P., Thiel, W. & Fürstner, A. Hydroxy-directed ruthenium-catalyzed alkene/alkyne coupling: increased scope, stereochemical implications, and mechanistic rationale. Angew. Chem. Int. Ed. 56, 5637–5652 (2017).
Trost, B. M. et al. Ruthenium-catalysed multicomponent synthesis of the 1,3-dienyl-6-oxy polyketide motif. Nat. Chem. 12, 629–637 (2020).
Trost, B. M. & Zhang, G. Ruthenium-catalyzed intermolecular coupling of vinylic 1,2-bisboronates with alkynes: stereoselective access to borylsubstituted homoallylic alcohols. J. Am. Chem. Soc. 142, 7312–7316 (2020).
Talele, T. T. Opportunities for tapping into three-dimensional chemical space through a quaternary carbon. J. Med. Chem. 63, 13291–13315 (2020).
Quasdorf, K. W. & Overman, L. E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 516, 181–191 (2014).
Feng, J., Holmes, M. & Krische, M. J. Acyclic quaternary carbon stereocenters via enantioselective transition metal catalysis. Chem. Rev. 117, 12564–12580 (2017).
Li, C., Ragab, S. S., Liu, G. & Tang, W. Enantioselective formation of quaternary carbon stereocenters in natural product synthesis: a recent update. Nat. Prod. Rep. 37, 276–292 (2020).
Süsse, L. & Stoltz, B. M. Enantioselective formation of quaternary centers by allylic alkylation with first-row transition-metal catalysts. Chem. Rev. 121, 4084–4099 (2021).
Shi, B.-F., Zhang, Y.-H., Lam, J. K., Wang, D.-H. & Yu, J.-Q. Pd(II)-catalyzed enantioselective C-H olefination of diphenylacetic acids. J. Am. Chem. Soc. 132, 460–461 (2010).
Krautwald, S., Sarlah, D., Schafroth, M. A. & Carreira, E. M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 340, 1065–1068 (2013).
Qiu, H. et al. Highly enantioselective trapping of zwitterionic intermediates by imines. Nat. Chem. 4, 733–738 (2012).
Mei, T.-S., Patel, H. H. & Sigman, M. S. Enantioselective construction of remote quaternary stereocentres. Nature 508, 340–344 (2014).
Masarwa, A. et al. Merging allylic carbon–hydrogen and selective carbon–carbon bond activation. Nature 505, 199–203 (2014).
Ohmatsu, K., Imagawa, N. & Ooi, T. Ligand-enabled multiple absolute stereocontrol in metal-catalysed cycloaddition for construction of contiguous all-carbon quaternary stereocentres. Nat. Chem. 6, 47–51 (2014).
Zhu, S., Niljianskul, N. & Buchwald, S. L. A direct approach to amines with remote stereocentres by enantioselective CuH-catalysed reductive relay hydroamination. Nat. Chem. 8, 144–150 (2016).
Zeng, X.-P., Cao, Z.-Y., Wang, Y.-H., Zhou, F. & Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev. 116, 7330–7396 (2016).
Petersen, K. S. Nonenzymatic enantioselective synthesis of all-carbon quaternary centers through desymmetrization. Tetrahedron Lett. 56, 6523–6535 (2015).
Zhou, F. et al. Asymmetric copper(i)-catalyzed azide−alkyne cycloaddition to quaternary oxindoles. J. Am. Chem. Soc. 135, 10994–10997 (2013).
Wu, H., Wang, Q. & Zhu, J. Copper-catalyzed enantioselective arylative desymmetrization of prochiral cyclopentenes with diaryliodonium salts. Angew. Chem. Int. Ed. 57, 2721–2725 (2018).
Chen, G., Cao, J., Wang, Q. & Zhu, J. Desymmetrization of prochiral cyclopentenes enabled by enantioselective palladium-catalyzed oxidative heck reaction. Org. Lett. 22, 322–325 (2020).
He, Y.-P., Cao, J., Wu, H., Wang, Q. & Zhu, J. Catalytic enantioselective aminopalladation-heck cascade. Angew. Chem. Int. Ed. 60, 7093–7097 (2021).
Yuan, Z. et al. Palladium-catalyzed asymmetric intramolecular reductive heck desymmetrization of cyclopentenes: access to chiral bicyclo-[3.2.1]octanes. Angew. Chem. Int. Ed. 58, 2884–2888 (2019).
You, C., Li, X., Gong, Q., Wen, J. & Zhang, X. Nickel-catalyzed desymmetric hydrogenation of cyclohexadienones: an efficient approach to all-carbon quaternary stereocenters. J. Am. Chem. Soc. 141, 14560–14564 (2019).
Zhu, C., Wang, D., Zhao, Y., Sun, W. Y. & Shi, Z. Enantioselective palladium-catalyzed intramolecular α-arylative desymmetrization of 1,3-diketones. J. Am. Chem. Soc. 139, 16486–16489 (2017).
Yuan, Z. et al. Constructing chiral bicyclo[3.2.1]octanes via palladium-catalyzed asymmetric tandem Heck/carbonylation desymmetrization of cyclopentenes. Nat. Commun. 11, 2544 (2020).
Yang, B. et al. Desymmetrization of 1,3-diones by catalytic enantioselective condensation with hydrazine. J. Am. Chem. Soc. 143, 4179–4186 (2021).
Cheng, Y.-F. et al. Catalytic enantioselective desymmetrizing functionalization of alkyl radicals via Cu(I)/CPA cooperative catalysis. Nat. Catal. 3, 401–410 (2020).
Hu, X. D. et al. Enantioselective synthesis of α-all-carbon quaternary center containing carbazolones via amino-palladation/desymmetrizing nitrile addition cascade. J. Am. Chem. Soc. 143, 3734–3740 (2021).
Bruneau, C. & Dixneuf, P. H. Metal vinylidenes and allenylidenes in catalysis: applications in anti-Markovnikov additions to terminal alkynes and alkene metathesis. Angew. Chem. Int. Ed. 45, 2176–2203 (2006).
Sun, X., Zhao, W. & Li, B.-J. Iridium-catalyzed, ligand-controlled directed alkynylation and alkenylation of arenes with terminal alkynes. Chem. Commun. 56, 1298–1301 (2020).
Lumbroso, A., Vautravers, N. R. & Breit, B. Rhodium-catalyzed selective anti-Markovnikov addition of carboxylic acids to alkynes. Org. Lett. 12, 5498–5510 (2010).
Yang, Z.-D., Pal, R., Hoang, G., Zeng, X. & Takacs, J. Mechanistic insights into carbonyl-directed rhodium-catalyzed hydroboration: ab initio study of a cyclic γ,δ-unsaturated amide. ACS Catal. 4, 763–773 (2014).
Brookhart, M., Grant, B. & Volpe, A. F. [(3,5-(CF3)2C6H3)4B]-[H(OEt2)2]+: a convenient reagent for generation and stabilization of cationic, highly electrophilic organometallic complexes. Organometallics 11, 3920–3922 (1992).
Krossing, I. & Raabe, I. Noncoordinating anions—fact or fiction? a survey of likely candidates. Angew. Chem. Int. Ed. 43, 2066–2090 (2004).
Acknowledgements
The authors thank the National Natural Science Foundation of China (NSFC21572272), the Innovation Team of “the Double-First Class” Disciplines (CPU2018GY04 and CPU2018GY35), and the Foundation of the Open Project of State Key Laboratory of Natural Medicines (SKLNMZZCX201818) for the financial support.
Author information
Authors and Affiliations
Contributions
D.Z. planned and conducted most of the experiments; D.Z., M.L. and J.L. prepared substrates for the reaction scope evaluation; A.L. and H.Y. directed the projects and cowrote the manuscript. All authors contributed to the discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, D., Li, M., Li, J. et al. Rhodium-catalyzed intermolecular enantioselective Alder–ene type reaction of cyclopentenes with silylacetylenes. Nat Commun 12, 6627 (2021). https://doi.org/10.1038/s41467-021-26955-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26955-9
- Springer Nature Limited
This article is cited by
-
Catalytic desymmetrization reactions to synthesize all-carbon quaternary stereocentres
Nature Synthesis (2023)
-
Cobalt-catalyzed branched selective hydroallylation of terminal alkynes
Nature Communications (2022)