

Abstract
The catalytic generation of homoenolates and their higher homologues has been a long-standing challenge. Like the generation of transition metal enolates, which have been used to great affect in synthesis and medicinal chemistries, homoenolates and their higher homologues have much potential, albeit largely unrealized. Herein, a nickel-catalyzed generation of homoenolates, and their higher homologues, via decarbonylation of readily available cyclic anhydrides has been developed. The utility of nickel-bound homoenolates and their higher homologues is demonstrated by cross-coupling with unactivated alkyl bromides, generating a diverse array of aliphatic acids. A broad range of functional groups is tolerated. Preliminary mechanistic studies demonstrate that: (1) oxidative addition of anhydrides by the catalyst is faster than oxidative addition of alkyl bromides; (2) nickel bound metallocycles are involved in this transformation and (3) the catalyst undergoes a single electron transfer (SET) process with the alkyl bromide.
Similar content being viewed by others

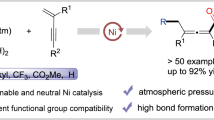
Introduction
Homoenolates and their higher homologs represent important synthetic intermediates that have found broad applications in natural product synthesis and pharmaceutical sciences1,2,3. In comparison to ketones, which exhibit nucleophilic character upon deprotonation of the relatively acidic α-C–H’s, the β- and γ-C–Hs are usually unreactive or, in the case of α,β-unsaturated derivatives, the β-carbon is electrophilic. Thus, with respect to α,β-unsaturated carbonyl compounds, the β-position of homoenolates exhibits umpolung reactivity4,5,6. Despite great progress in the generation and catalytic reactions of homoenolates7,8, the demand for more straightforward approaches to these reactive species and their higher homologs remains high.
Early routes for stoichiometric generation of homoenolates include (a) β-deprotonation of camphenilone with KOtBu by Nickon and Lambert9; (b) TiCl4-mediated ring opening of silyloxycyclopropanes by Nakamura and Kuwajima10; (c) Brønsted base-mediated metalation of allyl carbamates11; and (d) directed β-metalation of N,N-diisopropyl amides by Beak and colleagues12,13, among others14,15,16,17,18,19,20. The generation and reactions of γ-metallocarbonyls, however, remains relatively unexplored21.
Kuwajima, Nakamura, and their colleagues22 pioneered catalytic reactions of palladium-bound homoenolates derived from the ring opening of cyclopropanols. The Pd-homoenolates coupled with aryl halides to furnish β-arylcarbonyl derivatives (Fig. 1a)22. However, this ring opening strategy cannot afford γ-metallocarbonyl analogs. In addition to arylations23,24, other transformations such as acylation25, benzylation26, and allenylation27 of palladium-homoenolates were also reported via ring opening of cyclopropanols. In the case of β-alkylation of homoenolates, only activated alkyl halides have been successfully employed28,29,30,31. For example, this year the Fu group developed a remarkable Ni-catalyzed coupling of racemic β-zincated amides with racemic propargylic bromides in a doubly stereoconvergent process to provide amides with high ee and dr (Fig. 1b)32. In addition to ring opening of cyclopropanols, directing group-assisted palladium-catalyzed β-C–H activations are an alternative strategy to generate and functionalize palladium-homoenolates of carboxylic acid derivatives33,34,35,36. More recently, a palladium-catalyzed functionalization of γ-C–H’s of carboxylic acid derivatives was realized. This method employed 3,3-disubstituted butyric acid derivatives to circumvent the β-C–H functionalization pathway (Fig. 1c)21,37. To the best of our knowledge, the use of inexpensive nickel catalysts for the generation and functionalization of β-, or γ-bound carboxylic acid derivatives remains underdeveloped, especially related to coupling of these intermediates with alkyl groups38,39. To circumvent the use of unactivated alkyl halides, Rousseaux and colleagues40 developed an elegant alkylation of homoenolates derived from cyclopropanols that involves use of redox active esters (Fig. 1d). In this work, redox active esters undergo in situ decarboxylation to provide alkyl radicals, leading to alkylated homoenolates40. To the best of our knowledge, catalytic alkylations of any type of homoenolate, or their higher homologs, with unactivated alkyl halides remains a challenge. This is likely due to the relatively slow oxidative addition of alkyl electrophiles and facile β-hydride elimination of the resulting intermediates41,42,43,44.

a Palladium-catalyzed arylations of cyclopropanol derivatives. b Ni-catalyzed doubly stereoconvergent coupling. c Palladium-catalyzed functionalization of γ-C–H’s of carboxylic acid derivatives. d Ni-catalyzed alkylations with N-hydroxyphthalimides (NHPI).
We have been interested in developing new transformations of homoenolates45,46 and cross-electrophile coupling of cyclic anhydrides with aryl triflates to afford aryl ketoacids47. We were inspired by Yamamoto’s stoichiometric reactions of nickel(0) complexes with anhydrides and Rovis’s stoichiometric nickel promoted transformation with cyclic anhydrides and ZnPh2 (Fig. 2a)48,49. These latter reactions undergo insertion of nickel into the anhydride C–O bond followed by decarbonylation to generate a nickel homoenolate. Transmetallation from ZnPh2 is followed by reductive elimination to form the C–C bond. Herein, we report a nickel-catalyzed alkylation of homoenolates and their higher homologs via decarbonylation of monocyclic anhydrides (Fig. 2b). The in situ-generated nickel homoenolate species are successfully coupled with unactivated alkyl bromides, producing a diverse array of valuable functionalized acids. In addition to alkylation of nickel bound homoenolates, γ-alkylation of butyric acid derivatives can also be performed under the same conditions. Compared with Martín’s elegant carboxylation of alkyl bromides with CO2, our strategy provides an alternative synthesis of functionalized acids50,51. It is noteworthy that functionalized aliphatic acids are popular structures in soaps, dyes, plastics, and many chemicals52. They are also useful precursors in coupling reactions53,54,55.

a Rovis’s stoichiometric nickel complex with Ph2Zn. b This work: Ni-catalyzed functionalization with alkyl bromides.
Results
Reaction development and optimization
To initiate the development of the nickel catalyzed alkylation of homoenolates, we chose succinic anhydride 1a and 1-bromooctane 2a as model substrates. Ni(COD)2 and bipy were used as catalyst precursors in dimethylacetamide (DMA) at 80 °C for 12 h. As this is a cross-electrophile coupling reaction, a stoichiometric reducing agent is needed and zinc powder was chosen. After 12 h under these conditions, the desired decarbonylative product 3aa was obtained in 63% assay yield (AY) (Table 1, entry 1, AY, determined by GC analysis of the unpurified reaction mixture with dodecane as internal standard). It should be noted that the non-decarbonylated γ-keto acid 3aa’ was not observed under these conditions. A survey of solvents found DMA was a better choice than N,N-dimethylformamide (DMF), tetrahydrofuran (THF), THF, or acetonitrile (AY 23–42%, entries 2–4, see Supplementary Information for details). To increase the AY of 3aa, we next explored different nickel sources. Unfortunately, these nickel species afforded lower AY of 3aa (18–52%, entries 5–8).
To improve the decarbonylative cross-coupling, we turned to examination of nitrogen-based ligands. Under the conditions of entry 1, the parent bipy outperformed all others examined (the complete optimization data is presented in the Supplementary Information). We next examined the effect of concentration on the AY. Increasing the concentration from 0.4 to 2.0 M had a beneficial impact, affording the desired product in 87% AY at 1.3 M (entries 1 and 9–12). When 1-chloro- or 1-iodohexane were used in place of 1-bromohexane, the AY were lower than 40% (see Supplementary Information). Therefore, our optimized reaction conditions are 1.5 equiv of anhydride 1a, 1 equiv of alkyl bromide 2a, 2 equiv of zinc powder, 10 mol % Ni(COD)2, and 15 mol % bipy in DMA at 80 °C for 12 h.
Substrate scope
With the optimized reaction conditions in hand (Table 1, entry 11), we next explored the scope of the alkyl bromides. In general, a wide range of primary and secondary alkyl bromides participated in this transformation to afford the acid products in good to excellent yields with very good functional group tolerance (Table 2). We first chose the parent succinic anhydride 1a the homoenolate precursor to couple with a variety of primary alkyl bromides. Simple long-chain 1-bromooctane 2a underwent this coupling to afford 3aa in 84% isolated yield. More challenging functionalized alkyl bromides bearing benzodihydrofuran, ester, imide, and ether functional groups were compatible under our conditions, generating the corresponding products 3ab–3af in 68–91% yield.
We were also interested in using glutaric anhydride 1b. Fortunately, glutaric anhydride underwent the decarbonylative alkylation smoothly, providing access to γ-alkylated acids under mild conditions. Glutaric anhydride reacted with a diverse array of primary alkyl bromides, including functionalized substrates, exhibiting good reactivity and furnishing the desired acids in 68–92% yields (4ba–4bg). β-Branched alkyl bromides are expected to generate nickel alkyl intermediates that tend to undergo relatively fast β-hydride elimination56,57. Nonetheless, isobutyl bromide was compatible under the standard conditions, furnishing 4bh in 63% yield. Furthermore, alkyl bromides bearing halogens, such as fluoro and chloro reacted with excellent chemoselectivity, affording the corresponding products in 81 and 84% yield, respectively. Alkyl bromides containing ether, homobenzyl, and double bonds reacted efficiently, affording the products in 67–90% yields.
In addition to primary alkyl bromides, a variety of secondary alkyl bromides were compatible with the standard conditions. In general, both cyclic and acyclic secondary alkyl bromides, such as cyclopentyl-, cyclohexyl-, cyclobutyl-, isopropyl-, and sec-butyl- bromide, exhibited good reactivity, affording β- and γ-functionalized propionic and butyric acids, respectively (3an, 3ao, and 4bn–4bt, 56–83%). Unfortunately, when tertiary bromides were subjected to coupling with anhydrides, no products were obtained under the standard conditions.
We next explored the scope of the unsymmetrical anhydrides. It is observed that unsymmetrical succinic anhydrides such as 2-methylsuccinic anhydride 1c, 2-phenylsuccinic anhydride 1d and 2,2-dimethylsuccinic anhydride 1e could couple with 2l and 2f affording β-functionalized acids in 68–83% yield. It is noteworthy that the oxidative addition and decarbonylation processes took place at the less sterically hindered side of the anhydride. Disubstituted anhydrides, such as cis-2,3-dimethylsuccinic anhydride 1f is a competent coupling partner, affording 8fl and 8ff in 60% and 77% yield, respectively. It should be noted that 1f led to the corresponding products with high stereochemical fidelity (>20 : 1 dr). Furthermore, methyl and isobutyl substituted glutaric anhydrides 1g and 1h afforded γ-cross-coupled products in 72–83% yield. Unfortunately, bicyclic anhydrides, such as cis-1,2-cyclohexanedicarboxylic anhydride 1i and larger ring-sized anhydrides (1j and 1k), gave only trace coupling products under the standard conditions.
To test the scalability of this transformation, 15.0 mmol of glutaric anhydride 1b was coupled with 10.0 mmol of 1-bromo-4-chlorobutane 2j under the standard conditions. The decarbonylative cross-coupled product 4bj was isolated in 87% yield (1.55 g) (Fig. 3).

1-Bromo-4-chlorobutane reacted with 1b to give the desired product 4bj with 87% yield.
Mechanistic studies
Preliminary mechanistic studies demonstrated that nickel homoenolates are intermediates in this catalytic system. Ni-homoenolate H and its higher homolog G were synthesized independently58,59 (Fig. 4a). Exposure of H or G to alkyl bromides in the presence or absence of reducing agent generated 3aa and 4bg in good yields (Fig. 4b, c). These results demonstrate that Ni-bound homoenolate H and its derivative G are viable intermediates in these transformations. We have ruled out the possibility of cyclic anhydrides undergoing CO deinsertion followed by β-hydride elimination to generate acrylate intermediates and conjugate addition of radicals to these intermediates (see Supplementary Information). Notably, the stoichiometric reactions were successful in the absence of zinc powder (the source of electrons for this process may be the Ni(I) product reducing the alkyl bromide; see the catalytic cycle discussed below)60,61. These results lead to the hypothesis that zinc powder acts as a reductant to nickel to make these reactions catalytic62,63,64,65,66,67.

a Preparation of Ni-homoenolate and its higher homolog. b Alkylation of the independently synthesized homoenolate H. c Alkylation of the independently synthesized G with alkyl bromides.
To explore the selectivity in the oxidative addition to (L)Ni0, we examined the relative reactivities of glutaric anhydride (1b) and 1-bromohexane (2g) using stoichiometric Ni(COD)2 and bipy (Table 3) in a study that was inspired by the Weix group’s report of oxidative addition with nickel using aryl and alkyl halides68. After these reagents were stirred at room temperature for several hours, the reaction was quenched with 1 M HCl. The relative reactivities were determined by the loss of each starting material and formation of G and 4bg68 (Table 3). We found anhydride 1b was consumed much faster than alkyl bromide 2 g. These data indicate that the anhydride undergoes oxidative addition faster than the alkyl bromide in the presence of Ni(COD)2 and bipy, and that G is a likely intermediate in the catalytic reaction.
To further probe the reaction mechanism, radical trap and radical clock experiments were employed (Fig. 5a, b)66,69. Under otherwise standard conditions with alkyl bromide 2l, when the radical scavenger TEMPO was added, product formation was suppressed. If the alkyl bromide is activated through a radical mechanism, ring-opened products would be expected with cyclopropylmethyl bromide (2u). In the event, 2u led primarily to unsaturated acid 4bu’ (52% AY), arising from rapid ring opening of the cyclopropyl-methyl radical to the homoallylic radical.

a Inhibition of the reaction in the presence of TEMPO. b Radical clock reaction leads to ring opening, suggestive of radical intermediates. c Dependence of the ratio of 4bv/4bv’ on catalyst concentration, supporting a radical mechanism. d Demonstration that organozinc J is not a viable reaction intermediate.
Inspired by Weix’s 5-exo-trig experiment to evaluate ring-closed vs. ring-opened ratios as a function of catalyst loading68, we chose glutaric anhydride (1b) and 5-hexenyl bromide (2v) to examine the ratio of decarbonylative product 4bv and cyclized product 4bv’. By changing the catalyst loading of Ni(COD)2 from 5 mol% to 40 mol%, the ratio of 4bv/4bv’ was observed to increase linearly (Fig. 5c).
Finally, we probed the possibility of an organozinc intermediate in this cross-coupling, potentially formed from reaction of Zn0 with the alkyl bromide. Thus, we independently synthesized organozinc reagent J and subjected it to the metallocycle G under catalytically relevant conditions (Fig. 5d). No conversion to cross-coupled product was observed. Taken together, these experiments support single electron transfer (SET) processes70 and discount an organozinc intermediate.
Grounded in the experimental results presented above, a plausible mechanism is proposed (Fig. 6). Ni0 oxidatively adds cyclic anhydride 1 giving a NiII species (I)71,72,73,74. Next, decarbonylation of I with loss of CO forms the primary C(sp3)-Ni bond75 in II. The next step is generation of the alkyl radical via SET. This could take place from Ni(0) or Ni(I). We postulate that an intermediate Ni(I) performs the SET, because Table 3 suggests that reaction of Ni(0) with alkyl bromide is slow relative to oxidative addition of the anhydride. The alkyl radical is oxidatively trapped to provide the NiIII complex (III). Reductive elimination of intermediate III generates the C–C bond as well as the reactive NiI carboxylate species IV. As noted above, this Ni(I) species is proposed to reduce the alkyl bromide via SET giving BrNi(II)(carboxylate) (V) and an alkyl radical. Finally, the NiII product V is reduced by Zn0 to regenerate Ni0 and the zinc carboxylate. Acidic workup liberates the acid coupling product, 3.

Oxidative addition of anhydride (1) is followed by loss of carbon monoxide to generate homoenolate II. Oxidative capture of the alkyl radical formed Ni(III), which reductively eliminates to form Ni(I) intermediate IV. The Ni(I) intermediate is proposed to reduce the alkyl bromide 2 to furnish the alkyl radical and Ni(II) intermediate V. Zinc reduction of Ni(II) reforms Ni(0) and produces a zinc carboxylate, closing the catalytic cycle. Upon workup, the zinc carboxylate is protonated to give the observed carboxylic acid product 3.
Discussion
We have developed a nickel-catalyzed generation of homoenolates and their higher homologs via decarbonylation of monocyclic anhydrides. The in situ-formed nickel homoenolate derivatives were successfully coupled with unactivated alkyl bromides, generating a diverse array of functionalized aliphatic acids. Preliminary mechanistic studies demonstrated that the nickel(0) catalyst selectively reacts with anhydrides over alkyl bromides leading to the key homoenolates. Radical clock and related studies indicate that organozinc intermediates are unlikely and point to the involvement of alkyl radical species. Key advantages of this method include excellent functional group compatibility, mild reaction conditions, and avoidance of prefunctionalized organometallic reagents. This method enables construction of valuable carboxylic acids. Further studies using anhydrides as homoenolate precursors are ongoing in our team.
Methods
General procedure for the synthesis of aliphatic acids
To an oven-dried microwave vial (10 mL) equipped with a stir bar (10 × 5 mm) was added Ni(COD)2 (8.3 mg, 0.03 mmol) and bipy (7.0 mg, 0.045 mmol) under an argon atmosphere inside a glove box at 25 °C. Next, 0.225 mL of dry DMA was added via syringe to give a purple solution. After the catalyst/ligand solution was stirred for 1 h at 25 °C inside the glove box, Zn powder (39.2 mg, 0.6 mmol, 2.0 equiv) was added to the reaction vial followed by the monocyclic anhydride (0.45 mmol, 1.5 equiv) and alkyl bromide (0.3 mmol, 1.0 equiv). The microwave vial was sealed with a cap containing a rubber septum and removed from the glove box. The reaction mixture was stirred (~1000 r.p.m.) at 80 °C for 12 h. The resulting gray solution was cooled to room temperature, quenched by the addition of five drops of water via syringe through the septum and then the vial opened to air. The reaction mixture was passed through a short flash column chromatography in silica gel (200–300 mesh) and rinsed with 5 mL of ethyl acetate to afford a yellow solution. The solvent and volatile materials were removed by rotary evaporator. The crude residue was purified by flash column chromatography in silica gel to yield the corresponding product.
Data availability
Detailed experimental procedures and characterization of compounds can be found in the Supplementary Information. The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding authors.
References
Mills, L. R. & Rousseaux, S. A. L. Modern developments in the chemistry of homoenolates. Eur. J. Org. Chem. 2019, 8–26 (2019).
Murauski, K. J. R., Jaworski, A. A. & Scheidt, K. A. A continuing challenge: N-heterocyclic carbene-catalyzed syntheses of γ-butyrolactones. Chem. Soc. Rev. 47, 1773–1782 (2018).
Rosa, D., Nikolaev, A., Nithiy, N. & Orellana, A. Palladium-catalyzed cross-coupling reactions of cyclopropanols. Synlett 26, 441–448 (2015).
Menon, R. S., Biju, A. T. & Nair, V. Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon-carbon bond-forming reactions. Chem. Soc. Rev. 44, 5040–5052 (2015).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Grossmann, A. & Enders, D. N-heterocyclic carbene catalyzed domino reactions. Angew. Chem., Int. Ed. 51, 314–325 (2012).
Nithiy, N., Rosa, D. & Orellana, A. Carbon-carbon bond formation through palladium homoenolates. Synthesis 45, 3199–3210 (2013).
Nair, V., Vellalath, S. & Babu, B. P. Recent advances in carbon-carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 37, 2691–2698 (2008).
Nickon, A. & Lambert, J. L. Homoenolate anions. J. Am. Chem. Soc. 84, 4604–4605 (1962).
Nakamura, E. & Kuwajima, I. Homoenolate anion precursor. Reaction of ester homoenol silyl ether with carbonyl compounds. J. Am. Chem. Soc. 99, 7360–7362 (1977).
Seppi, M., Kalkofen, R., Reupohl, J., Froehlich, R. & Hoppe, D. Highly enantiomerically enriched ketone homoenolate reagents prepared by (-)-sparteine-mediated γ-deprotonation of achiral 1-alkenyl carbamates. Angew. Chem. Int. Ed. 43, 1423–1427 (2004).
Pippel, D. J., Curtis, M. D., Du, H. & Beak, P. Complex-induced proximity effects: stereoselective carbon-carbon bond formation in chiral auxiliary mediated β-lithiation-substitution sequences of β-substituted secondary carboxamides. J. Org. Chem. 63, 2–3 (1998).
Beak, P., Hunter, J. E. & Jun, Y. M. Dominance of the proximity effect of complexation over resonance and inductive effects in directing a metalation: regiospecific β lithiation of N,N-diisopropylcyclohex-3-enecarboxamide and of N,N-diisopropyl-2-methylpent-3-enecarboxamide. J. Am. Chem. Soc. 105, 6350–6351 (1983).
Mills, L. R., Barrera Arbelaez, L. M. & Rousseaux, S. A. L. Electrophilic zinc homoenolates: synthesis of cyclopropylamines from cyclopropanols and amines. J. Am. Chem. Soc. 139, 11357–11360 (2017).
Aspin, S., Goutierre, A.-S., Larini, P., Jazzar, R. & Baudoin, O. Synthesis of aromatic α-aminoesters: palladium-catalyzed long-range arylation of primary Csp3-H bonds. Angew. Chem. Int. Ed. 51, 10808–10811 (2012).
Renaudat, A. et al. Palladium-catalyzed β-arylation of carboxylic esters. Angew. Chem. Int. Ed. 49, 7261–7265 (2010).
Shen, Z.-L. et al. Synthesis of water-tolerant indium homoenolate in aqueous media and its application in the synthesis of 1,4-dicarbonyl compounds via palladium-catalyzed coupling with acid chloride. J. Am. Chem. Soc. 132, 15852–15855 (2010).
Nomura, K. & Matsubara, S. Preparation of zinc-homoenolate from α-sulfonyloxy ketone and bis(iodozincio)methane. Chem. Lett. 36, 164–165 (2007).
Yeh, M. C. P. & Knochel, P. 2-Cyanoethylzinc iodide: a new reagent with reactivity umpolung. Tetrahedron Lett. 29, 2395–2396 (1988).
Tamaru, Y., Ochiai, H., Nakamura, T. & Yoshida, Z. Synthesis of β, γ, δ-, ε-, and ζ-zinc ketones and their transition metal catalyzed reaction with carbon electrophiles. Angew. Chem. Int. Ed. 26, 1157–1158 (1987).
Park, H. S., Fan, Z., Zhu, R.-Y. & Yu, J.-Q. Distal γ-C(sp3)-H olefination of ketone derivatives and free carboxylic acids. Angew. Chem. Int. Ed. 59, 12853–12859 (2020).
Aoki, S., Fujimura, T., Nakamura, E. & Kuwajima, I. Palladium-catalyzed arylation of siloxycyclopropanes with aryl triflates. Carbon chain elongation via catalytic carbon-carbon bond cleavage. J. Am. Chem. Soc. 110, 3296–3298 (1988).
Rosa, D. & Orellana, A. Palladium-catalyzed cross-coupling of cyclopropanol-derived ketone homoenolates with aryl bromides. Chem. Commun. 49, 5420–5422 (2013).
Rosa, D. & Orellana, A. Palladium-catalyzed cross-coupling of cyclopropanols with aryl halides under mild conditions. Org. Lett. 13, 110–113 (2011).
Parida, B. B., Das, P. P., Niocel, M. & Cha, J. K. C-acylation of cyclopropanols: preparation of functionalized 1,4-diketones. Org. Lett. 15, 1780–1783 (2013).
Nithiy, N. & Orellana, A. Palladium-catalyzed cross-coupling of benzyl chlorides with cyclopropanol-derived ketone homoenolates. Org. Lett. 16, 5854–5857 (2014).
Wu, P., Jia, M., Lin, W. & Ma, S. Matched coupling of propargylic carbonates with cyclopropanols. Org. Lett. 20, 554–557 (2018).
Wright, T. B., Turnbull, B. W. H. & Evans, P. A. Enantioselective rhodium-catalyzed allylic alkylation of β,γ-unsaturated α-amino nitriles: synthetic homoenolate equivalents. Angew. Chem. Int. Ed. 58, 9886–9890 (2019).
Ye, Z., Cai, X., Li, J. & Dai, M. Catalytic cyclopropanol ring opening for divergent syntheses of γ-butyrolactones and δ-ketoesters containing all-carbon quaternary centers. ACS Catal. 8, 5907–5914 (2018).
Ye, Z., Gettys, K. E., Shen, X. & Dai, M. Copper-catalyzed cyclopropanol ring opening Csp3-Csp3 cross-couplings with (fluoro)alkyl halides. Org. Lett. 17, 6074–6077 (2015).
Das, P. P., Belmore, K. & Cha, J. K. \({\rm{S}}_{{\rm{N}}2^\prime}\) alkylation of cyclopropanols via homoenolates. Angew. Chem. Int. Ed. 51, 9517–9520 (2012).
Huo, H., Gorsline, B. J. & Fu, G. C. Catalyst-controlled doubly enantioconvergent coupling of racemic alkyl nucleophiles and electrophiles. Science 367, 559–564 (2020).
Uttry, A. & van Gemmeren, M. Direct C(sp3)-H activation of carboxylic acids. Synthesis 52, 479–488 (2020).
Le, K. K. A., Nguyen, H. & Daugulis, O. 1-Aminopyridinium Ylides as monodentate directing groups for sp3 C-H bond functionalization. J. Am. Chem. Soc. 141, 14728–14735 (2019).
Zhuang, Z. et al. Ligand-enabled β-C(sp3)-H olefination of free carboxylic acids. J. Am. Chem. Soc. 140, 10363–10367 (2018).
Shabashov, D. & Daugulis, O. Auxiliary-assisted palladium-catalyzed arylation and alkylation of sp2 and sp3 carbon-hydrogen bonds. J. Am. Chem. Soc. 132, 3965–3972 (2010).
Ghosh, K. K. et al. Ligand-enabled γ-C(sp3)-H olefination of free carboxylic acids. Angew. Chem. Int. Ed. 59, 12848–12852 (2020).
Wu, X., Zhao, Y. & Ge, H. Nickel-catalyzed site-selective alkylation of unactivated C(sp3)-H bonds. J. Am. Chem. Soc. 136, 1789–1792 (2014).
Chatani, N. Nickel-catalyzed C-H bond functionalization utilizing an N,N’-bidentate directing group. Top. Organomet. Chem. 56, 19–46 (2016).
Mills, L. R., Zhou, C., Fung, E. & Rousseaux, S. A. L. Ni-catalyzed β-alkylation of cyclopropanol-derived homoenolates. Org. Lett. 21, 8805–8809 (2019).
Iwasaki, T. & Kambe, N. Ni-catalyzed C-C couplings using alkyl electrophiles. Top. Curr. Chem. 374, 1–36 (2016).
Rudolph, A. & Lautens, M. Secondary alkyl halides in transition-metal-catalyzed cross-coupling reactions. Angew. Chem. Int. Ed. 48, 2656–2670 (2009).
Frisch, A. C. & Beller, M. Catalysts for cross-coupling reactions with non-activated alkyl halides. Angew. Chem. Int. Ed. 44, 674–688 (2005).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C-C bonds. Chem. Rev. 115, 9587–9652 (2015).
Yang, W. et al. Synergistic N-heterocyclic carbene/palladium-catalyzed umpolung 1,4-addition of aryl iodides to enals. Angew. Chem. Int. Ed. 59, 161–166 (2020).
Cheng, K. & Walsh, P. J. Arylation of aldehyde homoenolates with aryl bromides. Org. Lett. 15, 2298–2301 (2013).
Lin, T. et al. Nickel-catalyzed desymmetrizing cross-electrophile coupling of cyclic meso-anhydrides. Org. Lett. 20, 1191–1194 (2018).
O’Brien, E. M., Bercot, E. A. & Rovis, T. Decarbonylative cross-coupling of cyclic anhydrides: introducing stereochemistry at an sp3 carbon in the cross-coupling event. J. Am. Chem. Soc. 125, 10498–10499 (2003).
Yamamoto, T., Sano, K. & Yamamoto, A. Effect of ligand on ring contraction of six-membered nickel-containing cyclic esters, LnNiCH2CH2CH2COO, to their five-membered-ring isomers, LnNiCH(CH3)CH2COO. Kinetic and thermodynamic control of asymmetric induction by chiral diphosphines in the ring contraction. J. Am. Chem. Soc. 109, 1092–1100 (1987).
Julia-Hernandez, F., Moragas, T., Cornella, J. & Martin, R. Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 545, 84–88 (2017).
Borjesson, M., Moragas, T. & Martin, R. Ni-catalyzed carboxylation of unactivated alkyl chlorides with CO2. J. Am. Chem. Soc. 138, 7504–7507 (2016).
Landoni, M. F. & Soraci, A. Pharmacology of chiral compounds: 2-arylpropionic acid derivatives. Curr. Drug Metab. 2, 37–51 (2001).
Rodriguez, N. & Goossen, L. J. Decarboxylative coupling reactions: a modern strategy for C-C-bond formation. Chem. Soc. Rev. 40, 5030–5048 (2011).
Goossen, L. J., Rodriguez, N. & Goossen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 47, 3100–3120 (2008).
Stache, E. E., Ertel, A. B., Rovis, T. & Doyle, A. G. Generation of phosphoranyl radicals via photoredox catalysis enables voltage independent activation of strong C-O bonds. ACS Catal. 8, 11134–11139 (2018).
Yan, X.-B., Li, C.-L., Jin, W.-J., Guo, P. & Shu, X.-Z. Reductive coupling of benzyl oxalates with highly functionalized alkyl bromides by nickel catalysis. Chem. Sci. 9, 4529–4534 (2018).
Yonova, I. M. et al. Stereospecific nickel-catalyzed cross-coupling reactions of alkyl Grignard reagents and identification of selective anti-breast-cancer agents. Angew. Chem., Int. Ed. 53, 2422–2427 (2014).
Castan˜o, A. M. & Echavarren, A. M. Synthesis of protected 3-methylaspartic acids from glutamic anhydride via nickelacycles. Tetrahedron Lett. 34, 4361–4362 (1993).
Sano, K., Yamamoto, T. & Yamamoto, A. Preparation of Ni- or Pt-containing cyclic esters by oxidative addition of cyclic carboxylic anhydrides and their properties. Bull. Chem. Soc. Jpn. 57, 2741–2747 (1984).
Hegedus, L. S. & Thompson, D. H. P. The reactions of organic halides with (π-allyl)nickel halide complexes: a mechanistic study. J. Am. Chem. Soc. 107, 5663–5669 (1985).
Hegedus, L. S. & Miller, L. L. Reaction of π-allylnickel bromide complexes with organic halides. Stereochemistry and mechanism. J. Am. Chem. Soc. 97, 459–460 (1975).
Ye, Y., Chen, H., Sessler, J. L. & Gong, H. Zn-mediated fragmentation of tertiary alkyl oxalates enabling formation of alkylated and arylated quaternary carbon centers. J. Am. Chem. Soc. 141, 820–824 (2019).
Wang, J., Cary, B. P., Beyer, P. D., Gellman, S. H. & Weix, D. J. Ketones from nickel-catalyzed decarboxylative, non-symmetric cross-electrophile coupling of carboxylic acid esters. Angew. Chem. Int. Ed. 58, 12081–12085 (2019).
Chen, F. et al. Remote migratory cross-electrophile coupling and olefin hydroarylation reactions enabled by in situ generation of NiH. J. Am. Chem. Soc. 139, 13929–13935 (2017).
Huihui, K. M. M. et al. Decarboxylative cross-electrophile coupling of N-hydroxyphthalimide esters with aryl iodides. J. Am. Chem. Soc. 138, 5016–5019 (2016).
Zhao, C., Jia, X., Wang, X. & Gong, H. Ni-catalyzed reductive coupling of alkyl acids with unactivated tertiary alkyl and glycosyl halides. J. Am. Chem. Soc. 136, 17645–17651 (2014).
Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between vinyl and benzyl electrophiles. J. Am. Chem. Soc. 136, 14365–14368 (2014).
Biswas, S. & Weix, D. J. Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 135, 16192–16197 (2013).
Huang, L., Olivares, A. M. & Weix, D. J. Reductive decarboxylative alkynylation of N-hydroxyphthalimide esters with bromoalkynes. Angew. Chem. Int. Ed. 56, 11901–11905 (2017).
Correa, A., Leon, T. & Martin, R. Ni-catalyzed carboxylation of C(sp2)- and C(sp3)-O bonds with CO2. J. Am. Chem. Soc. 136, 1062–1069 (2014).
Diccianni, J., Lin, Q. & Diao, T. Mechanisms of nickel-catalyzed coupling reactions and applications in alkene functionalization. Acc. Chem. Res. 53, 906–919 (2020).
Diccianni, J. & Diao, T. Mechanisms of nickel-catalyzed cross-coupling reactions. Trends Chem. 1, 830–844 (2019).
Johnson, J. B., Bercot, E. A., Rowley, J. M., Coates, G. W. & Rovis, T. Ligand-dependent catalytic cycle and role of styrene in nickel-catalyzed anhydride cross-coupling: evidence for turnover-limiting reductive elimination. J. Am. Chem. Soc. 129, 2718–2725 (2007).
Bercot, E. A. & Rovis, T. A mild and efficient catalytic alkylative monofunctionalization of cyclic anhydrides. J. Am. Chem. Soc. 124, 174–175 (2002).
Guo, L. & Rueping, M. Decarbonylative cross-couplings: nickel catalyzed functional group interconversion strategies for the construction of complex organic molecules. Acc. Chem. Res. 51, 1185–1195 (2018).
Acknowledgements
We acknowledge the National Natural Science Foundation of China (21801128 and 22071107 to J.M.), Natural Science Foundation of Jiangsu Province, China (BK20170965 to J.M.), and Nanjing Tech University (39837112) for financial support. P.J.W. thanks the US National Science Foundation (CHE-1902509).
Author information
Authors and Affiliations
Contributions
T.L. performed most of the experiments and mechanistic study with the help of Y.G., P.Q., and H.G. The project conceived J.M. and T.L. with help from P.J.W. The project was directed by J.M. and the manuscript was written by T.L., J.M., and P.J.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Naohiko Yoshikai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, T., Gu, Y., Qian, P. et al. Nickel-catalyzed reductive coupling of homoenolates and their higher homologues with unactivated alkyl bromides. Nat Commun 11, 5638 (2020). https://doi.org/10.1038/s41467-020-19194-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19194-x
- Springer Nature Limited