

Abstract
Charge-transfer excitons (CTEs) immensely enrich property-tuning capabilities of semiconducting materials. However, such concept has been remaining as unexplored topic within halide perovskite structures. Here, we report that CTEs can be effectively formed in heterostructured 2D perovskites prepared by mixing PEA2PbI4:PEA2SnI4, functioning as host and guest components. Remarkably, a broad emission can be demonstrated with quick formation of 3 ps but prolonged lifetime of ~0.5 μs. This broad PL presents the hypothesis of CTEs, verified by the exclusion of lattice distortion and doping effects through demonstrating double-layered PEA2PbI4/PEA2SnI4 heterostructure when shearing-away PEA2SnI4 film onto the surface of PEA2PbI4 film by using hand-finger pressing method. The below-bandgap photocurrent indicates that CTEs are vital states formed at PEA2PbI4:PEA2SnI4 interfaces in 2D perovskite heterostructures. Electroluminescence shows that CTEs can be directly formed with electrically injected carriers in perovskite LEDs. Clearly, the CTEs presents a new mechanism to advance the multifunctionalities in 2D perovskites.
Similar content being viewed by others

Introduction
Initiated by fast-developing thin-film photovoltaic functionalities1–5,2,3,4,, organic–inorganic metal halide hybrid perovskites have emerged as interesting light-emitting materials with high photoluminescence (PL) quantum yields6,7,8, large color tuning capabilities9,10,11, domain-size controllable exciton binding energies12,13 and bandgap engineering properties14,15,16. With these attractive optical characteristics, the hybrid perovskites have demonstrated high-performance light-emitting diodes (LEDs)7,17,18,19,20 based on solution-processed thin-film formation procedures. Undoubtedly, the intrinsic excitons formed within band structures function as the primary excited states responsible for developing high-performance light-emitting properties in such hybrid perovskites. Essentially, tuning the intrinsic excitons through the formation probability, binding energy, and radiative/nonradiative recombination can determine the light-emitting properties, as exampled by mixing different halides9,10,11, introducing nanostructures15,21,22, and passivating grain boundary defects18,23,24. On the other hand, it is interesting to note that CTEs have been introduced as artificially engineered excitons with delocalized wavefunctions, namely, spatially extended states, and widely observed in low-dielectric-constant organic materials through donor–acceptor design, to develop high-efficiency optoelectronic functionalities25,26,27,28,29. It has been recognized as an intriguing nature that CTEs can be conveniently tuned on energy, polarization, and spin parameters by physically changing the formulation of donor–acceptor heterostructures30,31. This leads to unique tuning capabilities to control optic, electric, and magnetic functionalities by using CTEs32,33,34,35. In general, CTEs can be formed at heterostructures where charge transfer occurs between different local structures to form electron–hole pairs36,37. The heterostructures can be both chemically and physically prepared by using two molecular structures where different local energies and electron negativities exist38,39. In general, the heterostructures can be naturally formulated between donor- and acceptor-type structures, exampled as highly efficient light-emitting exciplexes40,41,42. On the other hand, the heterostructures can be also formed between two same-type molecular structures where local energy disorders are occurred, shown as light-emitting excimers in organic materials43,44. In the past, the CTEs have been extensively explored in organic heterostructures prepared by chemical synthesis, physical mixing, and mechanical formation of film interfaces in light-emitting10,11,45, lasing46,47, photovoltaic38,39, sensing48, and magneto-optical49 applications. Recently, it has been shown that the perovskite–polymer bulk heterostructure, prepared by mixing quasi-2D/3D perovskite with wide-bandgap polymer, demonstrates a broad transient absorption (TA) signal, which is quickly appeared in 1 ps and slowly relaxed in nanoseconds19. This presents the possibility to form CTEs between perovskite and organic polymer, known as high- and low-dielectric materials. Essentially, the CTEs can be formed when a charge-transfer process is occurred with the consequence of forming electron–hole pairs between two adjacent structures with different electron negativities and local energies. It has also been observed that CTEs can be formed between 2D perovskite (BA2PbI4) and organic molecule (1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile) when the charge transfer at the interface is enhanced by orbital overlap50. Clearly, these published results provide the necessary condition to explore the CTEs within perovskite heterostructures.
In this work, we initially utilized the chemical method to prepare 2D perovskite heterostructures [(PEA2PbI4)x:(PEA2SnI4)1−x] by alternatively selecting the host and guest components when mixing two precursor (Pb and Sn perovskites) solutions with distinctly high and low concentrations. The heterostructures with alternatively switched host and guest components are both demonstrated with similar broad emission spectra and prolonged lifetime (~microseconds). This observation provides the first indication that the CTEs are formed at the [PEA2PbI4:PEA2SnI4] interfaces within heterostructures [(PEA2PbI4)0.999:(PEA2SnI4)0.001] and [(PEA2SnI4)0.999:(PEA2PbI4)0.001] to generate a broad emission in 2D perovskites. Remarkably, the broad emission can be reproduced when using hand–finger pressing method to mechanically shearing away the PEA2SnI4 film onto the PEA2PbI4 film surface to uniquely prepare the double-layered heterostructures without lattice distortion and doping effects. This hand–finger pressing method confirms that the broad emission represents the signature for CTEs formed in 2D perovskite heterostructures. The pump–probe TA studies present that the CTEs are quickly formed and essentially become metastable states, as compared to surface-trapped and intrinsic excitons in 2D perovskites.
Results
CTEs in heterostructured 2D perovskite thin film
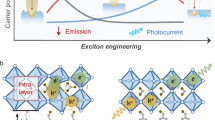
The 2D perovskite films were prepared by spin coating from mixed two precursor solutions (PEA2PbI4 and PEA2SnI4) and followed with thermal annealing at 100 °C for 10 min. The two precursor solutions were initially mixed with the volume ratio of 99.9:0.1 to formulate the heterostructures [(PEA2PbI4)0.999:(PEA2SnI4)0.001] in 2D perovskite films where the PEA2PbI4 and PEA2SnI4 act as host and guest components, respectively. Furthermore, the PEA2PbI4 and PEA2SnI4 were alternatively switched with distinctly low and high concentrations, serving as guest and host components, to formulate the heterostructures [(PEA2SnI4)0.999:(PEA2PbI4)0.001] in 2D perovskite films. The X-ray diffraction (XRD) data in Supplementary Fig. 1 indicate that all the 2D perovskite films (PEA2PbI4, [(PEA2PbI4)0.99:(PEA2SnI4)0.01], [(PEA2PbI4)0.95:(PEA2SnI4)0.05], [(PEA2PbI4)0.05:(PEA2SnI4)0.95], and PEA2SnI4) show excellent crystallinity with extremely narrow dominant peak at [0 0 2] direction. Furthermore, the peak of PEA2SnI4 film is slightly redshifted as compared with the PEA2PbI4 film due to smaller crystalline size. By increasing the ratio of the guest (PEA2SnI4) component in 2D perovskite heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film, the XRD peak is redshifted with decreased intensity towards that of pristine PEA2SnI4 film. This provides an evidence that, in the 2D perovskite heterostructured film, both PEA2PbI4 and PEA2SnI4 perovskites are formed. The scanning electron microscopy and atomic force microscopy results show that all the films are of high quality (Supplementary Figs. 2 and 3). Figure 1a shows that mixing 0.1% PEA2SnI4 into 99.9% PEA2PbI4 has little effect on the absorption characteristics in the heterostructures [(PEA2PbI4)0.999:(PEA2SnI4)0.001] where the bandgap is governed by the host (PEA2PbI4) component. Interestingly, a very broad PL spectrum is observed between 575 and 800 nm with the peak position located at 669 nm. When we further increase the guest Sn perovskite concentration to 1% or even 5%, broad emission can always be observed at same wavelength (Supplementary Fig. 4). As a comparison, the PEA2PbI4 and PEA2SnI4 perovskites give rise to their intrinsic PL at 524 and 621 nm, shown as spectral shoulders on the broad emission spectrum. The broad PL demonstrates a very broad spectral width of 127 nm, while the intrinsic emission from the PEA2PbI4 and PEA2SnI4 components give rise to much narrower spectral widths (15 and 37 nm). Remarkably, when the host and guest components are alternatively switched to shift the bandgap (derived from Supplementary Fig. 5) largely from 2.33 eV in the [(PEA2PbI4)0.999:(PEA2SnI4)0.001] to 1.95 eV in the [(PEA2SnI4)0.999:(PEA2PbI4)0.001], the broad emission spectrum is only slightly changed from 669 nm (1.85 eV) to 697 nm (1.78 eV) in peak position, leading to similar broad emission spectra between [(PEA2PbI4)0.999:(PEA2SnI4)0.001] and [(PEA2SnI4)0.999:(PEA2PbI4)0.001] heterostructures, as indicated in Fig. 1b. The similar broad emission observed by alternatively switching the host and guest components in 2D perovskite heterostructures [(PEA2PbI4)0.999:(PEA2SnI4)0.001] and [(PEA2SnI4)0.999:(PEA2PbI4)0.001] lead to the hypothesis that the CTEs are formed at the interfaces between local PEA2PbI4 and PEA2SnI4 structures, as illustrated in Fig. 1c. Here, the formation of CTEs requires that (a) the charge transfer is occurred from local host to guest structures and (b) the electron–hole pairs are formed at the interfaces in the 2D perovskite heterostructures.

a Absorption and PL spectra are shown for 2D perovskite heterostructured [(PEA2PbI4)0.999:(PEA2SnI4)0.001] film. The absorption spectrum indicates that the bandgap is governed by the host (PEA2PbI4) component in the heterostructures. The PL spectrum is dominated by a broad emission peaked at 669 nm with a spectral shoulder at 614 nm from the guest (PEA2SnI4) and the intrinsic emission (peaked at 531 nm) from the host (PEA2PbI4). b PL spectrum is shown for 2D perovskite heterostructured [(PEA2SnI4)0.999:(PEA2PbI4)0.001] film after switching host and guest components. The broad emission is observed with the peak position at 697 nm, in addition to intrinsic emission (peaked at 524 and 627 nm) from host (PEA2PbI4) and guest (PEA2SnI4) components. The PL spectra for pristine PEA2PbI4 and PEA2SnI4 films are also shown as reference. c Schematic diagram illustrates the formation of CTEs at the interfaces between PEA2PbI4 and PEA2SnI4 components in 2D perovskite heterostructures [(PEA2PbI4)1−x:(PEA2SnI4)x].
To further understand the origin of broad emission occurring in the 2D perovskite heterostructures, we measured the PL intensity-dependence and lifetime characteristics. We can see in Fig. 2a, b that the broad emission exhibits the intensity-dependence slope of 1.62, while the intrinsic emission from the host PEA2PbI4 component gives rise to the intensity-dependence slope of 1.14. We should note that germinate process dominates the radiative recombination of the intrinsic excitons in host/guest 2D perovskites due to large exciton binding energy, which leads to the power-dependence slope of ~1. On the other hand, when electrons and holes are paired to form electron–hole pairs, the pairing probability p is proportional to the product between the number (n) of electrons and the number (m) of holes: p ∝ m ⋅ n. In the situation where n = m, then p ∝ n2. Therefore, the non-germinate recombination can lead to the intensity-dependence slope of 2 in light emission. Here, the broad emission of our heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film demonstrates the power-dependence slope of 1.62, presenting the suggestion that the CTEs are responsible for the broad emission through non-germinate recombination. Furthermore, the broad emission peaked at 669 nm shows much prolonged lifetime of 0.5 μs relative to the short lifetime of 1 ns of the intrinsic emission peaked at 524 nm from the host PEA2PbI4 component, as shown in Fig. 2c. Detailed fittings of PL lifetimes of pure PEA2PbI4 and PEA2SnI4 films are shown in Supplementary Fig. 6. In general, the lifetime of CTEs is mainly determined by the electron–hole recombination rate governed by Coulomb attractive force. As a result, CTEs often exhibit an extended lifetime due to longer electron–hole separation distance as compared to intrinsic excitons in semiconducting materials. Here, the much prolonged PL lifetime further verifies that the broad emission originates from CTEs in heterostructured 2D perovskites. We should note that CTEs can still demonstrate various lifetimes when the Coulomb attraction between electron and hole located on different energetic structures is changed by local dielectric backgrounds, widely observed in organic–organic51,52,53, organic–inorganic35,54, and inorganic–inorganic mixtures34,55.

a Normalized PL spectra at different excitation intensities ranging from 5 to 94 mW/cm2. b PL-excitation intensity dependences for broad emission (peaked at 669 nm) as compared to intrinsic emission (524 nm) from host (PEA2PbI4). c PL lifetime results for broad emission (peaked at 669 nm) in 2D perovskite heterostructures and intrinsic emission (peaked at 524 nm, and 621 nm) from host (PEA2PbI4) and guest (PEA2SnI4).
CTEs in double-layered 2D perovskite heterostructures
We note that, in the mixed two precursor solutions with 99.9% host and 0.1% guest concentrations, the PEA2PbI4 and PEA2SnI4 have slower and faster crystallization rates to form heterostructured 2D perovskite [(PEA2PbI4)0.999:(PEA2SnI4)0.001] films. This is because the Pb and Sn perovskites have higher and lower Gibbs energies, respectively56. Consequently, the PEA2SnI4 domains can be quickly formed and followed with the formation of PEA2PbI4 domains, leading to the heterostructured 2D perovskite films. The heterostructures provide the necessary condition to initiate the charge transfer from the host to the guest domains towards the formation of CTEs at the host:guest interfaces. Essentially, the CTEs can be formed through Coulomb capture at the interfaces between PEA2PbI4 and PEA2SnI4 domains in 2D perovskite heterostructures. However, there is a question whether the low-concentration guest (PEA2SnI4) molecules dope into the high-concentration host (PEA2PbI4) domains, leading to the possibility of doping effect to generate a broad emission through the so-called self-trapped excitons caused by lattice deformation, which was proposed for doping-induced deformation in (1 0 0) 2D single crystals57 and corrugated (1 1 0) 2D structures58,59. To clarify this question, we designed a unique experimental method to mechanically prepare the double-layered PEA2PbI4/PEA2SnI4 heterointerface by shearing away the PEA2SnI4 film onto the surface of PEA2PbI4 film through hand–finger pressing procedure. Specifically, the PEA2SnI4 and PEA2PbI4 films were separately spin cast on glass substrates, followed by thermal annealing. Then, the PEA2SnI4 film coated on the glass substrate was made to hold onto the PEA2PbI4 film coated on another glass substrate with face-to-face contact under hand–finger pressing, as schematically illustrated in Fig. 3a. Upon hand–finger pressing with face-to-face contact, the PEA2SnI4 film was parallelly shearing away from the surface of PEA2PbI4 film. This hand–finger pressing method leaves the PEA2SnI4 film islands on the surface of PEA2PbI4 film (see the microscopic photos in Supplementary Fig. 7), forming the double-layered [PEA2PbI4/PEA2SnI4] interfaces, functioning as heterostructures clearly without doping effects and lattice deformation. Therefore, our hand–finger pressing method provides a unique experimental approach to clarify the origin of broad emission widely observed in hybrid metal halide perovskite heterostructures formed by mixing different precursor solutions or in nanocrystals formed with lattice strains. Here, it is very interesting to show that our broad emission can be reproduced with the peak position at 671 nm in the double-layered PEA2PbI4/PEA2SnI4 interfaces prepared by our hand–finger pressing method (Fig. 3b), very similar to the broad emission observed by mixing two precursor solutions. Because our hand–finger pressing method can only form a low density of double-layered PEA2PbI4/PEA2SnI4 heterostructures, the broad emission becomes an appreciable shoulder on the intrinsic emission peaked at 638 nm from the PEA2SnI4 perovskite. Clearly, the similar broad emission observed from our hand–finger pressing method as compared to the precursor-solution mixing method provides an unambiguous evidence to confirm that the broad emission indeed represents the CTEs formed at the interfaces between PEA2PbI4 and PEA2SnI4 domains in 2D perovskite heterostructures.

a Schematic diagram illustrates that PEA2SnI4 film is shearing away on PEA2PbI4 film surface to prepare double-layered [PEA2PbI4/PEA2SnI4] heterointerfaces. b PL spectrum was measured for double-layered [PEA2PbI4/PEA2SnI4] heterointerfaces. The broad emission is shown as an extended spectral tail in addition to intrinsic emission (530 and 637.5 nm) from PEA2PbI4 and PEA2SnI4 components. The dashed lines are shown for three spectral components from PEA2PbI4 component, PEA2SnI4 component, and [PEA2PbI4/PEA2SnI4] heterointerfaces.
Dynamic analysis of CTE formation
To explore the dynamic behaviors of the CTEs, we performed pump–probe TA studies on the heterostructured 2D perovskite [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film (Fig. 4a). The TA characteristics show an interesting phenomenon where a broad signal is observed between 600 and 800 nm (bottom panel in Fig. 4a). Obviously, the broad TA signal is consistent with the broad emission from CTEs in the heterostructured 2D perovskite film. Contrarily, the pristine PEA2PbI4 film does not show any broad TA signal between 600 and 800 nm, only leading to a photobleaching signal peaked at 514 nm related to intrinsic excitons, as shown in Fig. 4b. Clearly, the broad TA signal provides the dynamic information of CTEs formed in 2D perovskite heterostructures. Figure 4c summarizes the dynamic characteristics of CTEs with the broad TA signal between 600 and 800 nm occurring at the [PEA2PbI4:PEA2SnI4] interfaces in 2D perovskite heterostructures [(PEA2PbI4)0.95:(PEA2SnI4)0.05]. The CTEs shown as the broad TA signal between 600 and 800 nm are quickly formed in 3 ps and slowly generate a light emission after 230 ps. Interestingly, the broad photoinduced bleaching TA signal extends to a few nanoseconds without any appreciable decay. This indicates that the CTEs are essentially metastable states in 2D perovskite heterostructures. Furthermore, the TA characteristics show a spectral shoulder at 537.5 nm in the heterostructured 2D perovskite film, slightly below the emission from intrinsic excitons (peaked at 513.8 nm) in the PEA2PbI4 domains. This spectral shoulder (537.5 nm) is formed in 1 ps, quickly demonstrates a light emission after 2.2 ps, and decays within nanosecond time window. By considering the lower energy (51 meV below the band-to-band transition) and photobleaching developed after the band-to-band transition, we assigned the TA spectral shoulder (peaked at 537.5 nm) to surface-trapped excitons. Because the CTEs are developed into a bleaching signal after 230 ps, the surface-trapped excitons, which are quickly shown as a bleaching signal within 2.2 ps, can have an opportunity to contribute to the formation of CTEs. Furthermore, the surface-trapped excitons have a limited light emission in our 2D perovskite heterostructured film. It is shown in Supplementary Fig. 8 that the PL of pure PEA2PbI4 film contains a weak tail located around 540 nm, slightly below the emission (peaked at 520 nm) from intrinsic excitons with decreased film thickness (~20 nm), leading to an asymmetric spectrum. This weak PL tail at 540 nm coincides with the surface-trapped excitons at 537.5 nm shown in TA (Fig. 4a). With increasing the film thickness to ~150 nm, this weak tail becomes negligible, making the entire PL spectrum symmetric. We believe this weak tail on PL spectrum at thinner film provides a further indication to support the surface-trapped excitons. As a comparison, the intrinsic excitons shown as the band-to-band transition peaked at 513.8 nm are quickly developed into a light emission within ~1 ps. Therefore, the dynamics of CTEs include the following three processes. First, both intrinsic and surface-trapped excitons experience a quick charge transfer, leading to the formation of CTEs at the heterointerfaces between host and guest perovskite structures within 3 ps. Second, the CTEs formed at heterostructures gradually develop a broad light emission, showing a TA bleaching signal after 230 ps. Third, the light emission of CTEs exhibits an extended lifetime (~0.5 μs), indicating that the CTEs are metastable states in 2D perovskite heterostructured film. To further verify the formation of CTEs, we have also measured TA for the double-layered heterointerfaces prepared by our hand–finger pressing method (Supplementary Fig. 9). The double-layered heterointerfaces demonstrate a broad TA signal between 640 and 800 nm, in addition to the band-to-band transitions (peaked at 520 and 590 nm) in Pb and Sn perovskites. This broad TA signal observed from the double-layered heterointerfaces prepared by our hand–finger pressing method provides a further evidence to confirm the formation of CTEs.

a TA spectra for 2D perovskite heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film. b TA spectra for pristine PEA2PbI4 film. c TA dynamics plotted for band-to-band transition (513.8 nm), surface excitons (537.5 nm), and broad emission (657.3 nm) in 2D perovskite heterostructures.
Heterostructured 2D perovskite photodetectors and LEDs
Now we use below-bandgap photoexcitation to show that the CTEs are vital states formed in 2D perovskite heterostructures. Figure 5a illustrates the photocurrents for pristine (PEA2PbI4, PEA2SnI4) and heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] films with the device architecture of ITO/poly (3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS)/perovskite/phenyl-C61-butyric acid methyl (PC61BM)/PEI/Ag under the photoexcitation of 640 nm. We should note that the 2D heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] and the pristine PEA2PbI4 films have the same absorbance at the wavelength of 640 nm (selected as the below-bandgap photoexcitation) (Fig. 5b). Interestingly, the heterostructured [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film demonstrates a much enlarged ON–OFF ratio of 532 on the photocurrent when the 640 nm photoexcitation is applied and removed periodically. This below-bandgap photoexcitation-induced photocurrent directly indicates that the charge-transfer states are vital states formed in 2D perovskite heterostructures. In contrast, both pristine PEA2PbI4 and PEA2SnI4 films show significantly reduced ON–OFF ratios of 29.7 and 3.91 on photocurrent upon repeatedly applying and removing the 640 nm photoexcitation. Furthermore, we observed that the CTEs can be generated by electrically injected charge carriers in 2D perovskite heterostructures. Figure 5c shows the electroluminescence (EL) spectrum from the heterostructured 2D perovskite [(PEA2PbI4)0.95:(PEA2SnI4)0.05] film based on the device design of ITO/PEDOT:PSS/poly(9-vinylcarbazole) (PVK)/perovskite/2,2′,2″-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi)/lithium fluoride (LiF)/Ag. The EL spectrum combines the emission from both intrinsic excitons (peaked at 517 nm) from the host PEA2PbI4 component and the CTEs (peaked at 669 nm) formed at the interfaces between PEA2PbI4 and PEA2SnI4 components in 2D perovskite heterostructures [(PEA2PbI4)0.95:(PEA2SnI4)0.005]. The EL–voltage–current characteristics shown in Fig. 5d indicate the typical LED behaviors from CTEs under electrical injection. It should be pointed out that the injection current is quite low: (<1 mA/cm2) due to the lower conductance caused by insulating organic long-chain ligands in 2D perovskite film even when the applied bias reaches 13 V. Clearly, the CTEs allow the generation of broad EL in 2D perovskite heterostructures under electrical injection.

a The ON–OFF behaviors of heterostructured 2D perovskite [(PEA2PbI4)0.95:(PEA2SnI4)0.05] photodetector (red) as compared to pure PEA2PbI4 (black) and PEA2SnI4 (blue) devices when exciting at 640 nm (1000 mW/cm2). b Absorption spectra of two films. c EL spectrum containing broad CTE emission and host emission. d Current density–voltage–EL curve of doped device. The device structure is shown as the inset.
Discussion
In summary, we found that the CTEs can be formed at the interfaces between PEA2PbI4 and PEA2SnI4 components in 2D perovskite heterostructures [(PEA2PbI4)1−x:(PEA2SnI4)x] prepared by chemically mixing two precursor (PEA2PbI4 and PEA2SnI4) solutions. The PEA2PbI4 and PEA2SnI4 serve as host and guest components in the heterostructured 2D perovskite [(PEA2PbI4)0.999:(PEA2SnI4)0.001] films. A broad emission peaked at 669 nm with spectral range between 575 and 800 nm was observed with the intensity-dependence slope of 1.62 and the prolonged lifetime (~0.5 μs) in such 2D perovskite heterostructures. Particularly, when the host and guest components are alternatively switched to shift the bandgap largely from 2.33 eV in the [(PEA2PbI4)0.999:(PEA2SnI4)0.001] to 1.95 eV in the [(PEA2SnI4)0.999:(PEA2PbI4)0.001], the broad emission spectrum is only slightly changed from 669 nm (1.85 eV) to 697 nm (1.78 eV) in peak position, leading to similar broad emission between PEA2PbI4 and PEA2SnI4 based heterostructures. This leads to the hypothesis that the CTEs can be conveniently established in 2D perovskite heterostructures. This hypothesis was verified by the unique phenomenon: a similar broad emission can be reproduced when the PEA2SnI4 film was shearing away onto the surface of the PEA2PbI4 film by hand–finger pressing method. The pump–probe TA studies show that the CTEs are formed within 3 ps between PEA2PbI4 and PEA2SnI4 components and slowly developed into a photobleaching signal after 230 ps. Particularly, the photobleaching signal from CTEs extends a few nanoseconds without appreciable decay. Essentially, the CTEs become metastable states in 2D perovskite heterostructures. The photocurrent generated by below-gap photoexcitation shows that the charge-transfer states are vital states formed at the interfaces between PEA2PbI4 and PEA2SnI4 components in heterostructured 2D perovskite films. The EL indicates that the CTEs can be formed by electrically injected electrons and holes to generate a broad light emission in perovskite LEDs. Therefore, the CTEs present new opportunities to develop advanced optoelectronic properties based on heterostructural design in 2D perovskites.
Methods
Thin-film preparation
Lead iodide (PbI2), tin iodide (SnI2), PVK, TPBi, LiF, chlorobenzene, isopropyl alcohol (IPA), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. Phenethylammonium iodide (PEAI) was purchased from Greatcell Solar. PC61BM was purchased from 1-Materials. PEDOT:PSS was purchased from Clevios. All chemicals are used as received without further purification. The precursor solution of PEA2PbI4 was prepared by mixing PEAI (0.8 M) and PbI2 (0.4 M) in DMSO (1 mL). The precursor solution of PEA2SnI4 was made in the same way by replacing PbI2 with SnI2. Heterostructures of [(PEA2PbI4)1−x:(PEA2SnI4)x] were designed by mixing two precursor (PEA2PbI4 and PEA2SnI4) solutions with alternatively selected high and low volume ratios. All the perovskite films were fabricated by spin coating the precursor solutions on glass slides (1.5 × 1.5 cm2) at 3000 r.p.m. for 30 s, and then thermally annealed at 100 °C for 10 min.
Fabrications of LED and photodetector
The patterned ITO substrates were first cleaned by ultrasonic treatments with detergent, deionized water, acetone, and isopropanol, respectively, for 20 min in each step and dried with nitrogen gas, and then followed by ultraviolet treatment for 30 min. After that, the PEDOT:PSS hole-transport layer of 40 nm was spin coated on the precleaned ITO substrates at 4000 r.p.m. for 60 s, and then annealed at 150 °C for 30 min. For LED devices, a thin layer of PVK (16 mg/ml dissolved in chlorobenzene) was spin coated on the PEDOT:PSS layers with a speed of 5000 r.p.m. for 60 s, and treated with the thermal annealing at 120 °C for 20 min. The 2D perovskite precursor solutions were spin coated as described above. The TPBi was vacuum deposited with the thickness of 50 nm. Two nanometers of LiF and 90 nm silver were evaporated to prepare the perovskite LEDs with the architecture of ITO/PEDOT:PSS/PVK/perovskite/TPBi/LiF/Ag. For the fabrication of photodetector devices, the PC61BM (25 mg/ml dissolved in chlorobenzene) was spin coated at 2000 r.p.m. for 50 s, without thermal annealing, serving as an electron-transport layer, and then an interface modification layer of PEI (0.5 mg/ml, IPA) was spin coated at 5000 r.p.m for 60 s, followed by the evaporation of silver electrode of 90 nm. This leads to the photodetector device architecture of ITO/PEDOT:PSS/perovskite/PC61BM/PEI/Ag. The active area for both LED and photodetector devices is 0.06 cm2.
Mechanically preparing heterostructures
The double-layer 2D perovskite heterostructures were prepared by using hand–finger pressing method to avoid lattice distortion and doping effect. Specifically, the PEA2SnI4 film coated on glass substrate was held on the PEA2PbI4 film with the face-to-face contact by using hand–finger pressing method. Then, the PEA2SnI4 film was shearing on the surface of PEA2PbI4 film. This leaves the PEA2SnI4 film islands on the PEA2PbI4 film, leading to double-layer heterointerfaces (PEA2SnI4/PEA2PbI4). The PEA2SnI4 film islands can be visible as pale-yellow color on the PEA2PbI4 film. Therefore, our hand–finger pressing method can prepare the heterostructures without lattice distortion and doping effects to confirm the charge-transfer excitons responsible for broad emission observed in 2D perovskite heterostructures.
Characterizations and measurements
The steady-state and time-resolved PL characteristics were measured with Flouro Log III spectrometer. The absorption spectrum was measured with UV3600 (Shimadzu, Japan). TA spectra were collected by using a Helios Fire spectrometer (Ultrafast Systems LLC). The pump beam (346 nm, 5 µW) was generated through a harmonic generator (Ultrafast Systems LLC, third harmonic) pumped by a Pharos laser (Light Conversion, 1 kHz, 1030 nm, 290 fs). The probe beam was selected from the broad light emission generated by CaF2 crystal excited by 1030 nm fs primary beam.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Zhou, H. et al. Interface engineering of highly efficient perovskite solar cells. Science 345, 542–546 (2014).
Green, M. A., Ho-Baillie, A. & Snaith, H. J. The emergence of perovskite solar cells. Nat. Photonics 8, 506–514 (2014).
Jeon, N. J. et al. Compositional engineering of perovskite materials for high-performance solar cells. Nature 517, 476–480 (2015).
Li, Z. et al. Scalable fabrication of perovskite solar cells. Nat. Rev. Mater 3, 18017 (2018).
Bai, S. et al. Planar perovskite solar cells with long-term stability using ionic liquid additives. Nature 571, 245–250 (2019).
Woo Choi, J. et al. Organic-inorganic hybrid perovskite quantum dots with high PLQY and enhanced carrier mobility through crystallinity control by solvent engineering and solid-state ligand exchange. Nanoscale 10, 13356–13367 (2018).
Liu, Y. et al. Efficient blue light-emitting diodes based on quantum-confined bromide perovskite nanostructures. Nat. Photonics 13, 760–764 (2019).
Luo, J. et al. Efficient and stable emission of warm-white light from lead-free halide double perovskites. Nature 563, 541–545 (2018).
Yoon, H. C. et al. Study of perovskite QD down-converted LEDs and six-color white LEDs for future displays with excellent color performance. ACS Appl. Mater. Interfaces 8, 18189–18200 (2016).
Yoon, Y. J. et al. Full-color luminescence by post-treatment of perovskite nanocrystals. Joule 2, 2105–2116 (2018).
Smith, M. D., Connor, B. A. & Karunadasa, H. I. Tuning the luminescence of layered halide perovskites. Chem. Rev. 119, 3104–3139 (2019).
Tanaka, K. & Kondo, T. Bandgap and exciton binding energies in lead-iodide-based natural quantum-well crystals. Sci. Technol. Adv. Mater. 4, 599–604 (2004).
Galkowski, K. et al. Determination of the exciton binding energy and effective masses for methylammonium and formamidinium lead tri-halide perovskite semiconductors. Energy & Environ. Sci. 9, 962–970 (2016).
D'Innocenzo, V., Srimath Kandada, A. R., De Bastiani, M., Gandini, M. & Petrozza, A. Tuning the light emission properties by band gap engineering in hybrid lead halide perovskite. J. Am. Chem. Soc. 136, 17730–17733 (2014).
Hassan, Y. et al. Structure-tuned lead halide perovskite nanocrystals. Adv. Mater. 28, 566–573 (2016).
Yuan, M. et al. Perovskite energy funnels for efficient light-emitting diodes. Nat. Nanotechnol. 11, 872–877 (2016).
Cho, H. et al. Overcoming the electroluminescence efficiency limitations of perovskite light-emitting diodes. Science 350, 1222 (2015).
Lin, K. et al. Perovskite light-emitting diodes with external quantum efficiency exceeding 20 per cent. Nature 562, 245–248 (2018).
Zhao, B. et al. High-efficiency perovskite–polymer bulk heterostructure light-emitting diodes. Nat. Photonics 12, 783–789 (2018).
Yang, X. et al. Efficient green light-emitting diodes based on quasi-two-dimensional composition and phase engineered perovskite with surface passivation. Nat. Commun. 9, 570 (2018).
Mao, J. et al. Novel direct nanopatterning approach to fabricate periodically nanostructured perovskite for optoelectronic applications. Adv. Funct. Mater. 27, 1606525 (2017).
Ban, M. et al. Solution-processed perovskite light emitting diodes with efficiency exceeding 15% through additive-controlled nanostructure tailoring. Nature Commun 9, 3892 (2018).
Wang, H. et al. Trifluoroacetate induced small-grained CsPbBr3 perovskite films result in efficient and stable light-emitting devices. Nature Commun 10, 665 (2019).
Qin, J. et al. Enabling self-passivation by attaching small grains on surfaces of large grains toward high-performance perovskite LEDs. iScience 19, 378–387 (2019).
Zhu, X.-Y., Yang, Q. & Muntwiler, M. Charge-transfer excitons at organic semiconductor surfaces and interfaces. Acc. Chem. Res. 42, 1779–1787 (2019).
Jailaubekov, A. E. et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic photovoltaics. Nat. Mater. 12, 66–73 (2013).
Brédas, J.-L., Norton, J. E., Cornil, J. & Coropceanu, V. Molecular understanding of organic solar cells: the challenges. Acc. Chem. Res. 42, 1691–1699 (2009).
Goushi, K., Yoshida, K., Sato, K. & Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nat. Photonics 6, 253 (2012).
Scharber, M. C. et al. Design rules for donors in bulk‐heterojunction solar cells—towards 10% energy-conversion efficiency. Adv. Mater. 18, 789–794 (2006).
Wang, M. et al. Revealing the cooperative relationship between spin, energy, and polarization parameters toward developing high-efficiency exciplex light-emitting diodes. Adv. Mater. 31, 1904114 (2019).
Xu, H., Wang, M., Yu, Z.-G., Wang, K. & Hu, B. Magnetic field effects on excited states, charge transport, and electrical polarization in organic semiconductors in spin and orbital regimes. Adv. Phys. 68, 49–121 (2019).
Li, M. et al. Tuning emission color of electroluminescence from two organic interfacial exciplexes by modulating the thickness of middle gadolinium complex layer. Appl. Phys. Lett. 88, 091108 (2006).
Kawabe, Y. & Abe, J. Electron mobility measurement using exciplex-type organic light-emitting diodes. Appl. Phys. Lett 81, 493–495 (2002).
Hong, X. et al. Ultrafast charge transfer in atomically thin MoS2/WS2 heterostructures. Nat. Nanotechnol. 9, 682 (2014).
Zhu, T. et al. Highly mobile charge-transfer excitons in two-dimensional WS2/tetracene heterostructures. Sci. Adv. 4, eaao3104 (2018).
Rotkiewicz, K., Grellmann, K. H. & Grabowski, Z. R. Reinterpretation of the anomalous fluorescense of pn, n-dimethylamino-benzonitrile. Chem. Phys. Lett. 19, 315–318 (1973).
Grabowski, Z. R., Rotkiewicz, K. & Rettig, W. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 103, 3899–4032 (2003).
Dennler, G., Scharber, M. C. & Brabec, C. J. Polymer‐fullerene bulk‐heterojunction solar cells. Adv. Mater. 21, 1323–1338 (2009).
Heremans, P., Cheyns, D. & Rand, B. P. Strategies for increasing the efficiency of heterojunction organic solar cells: material selection and device architecture. Acc. Chem. Res. 42, 1740–1747 (2009).
Chandross, E. A. & Thomas, H. T. Intramolecular exciplex formation in naphthylalkylamines. Chem. Phys. Lett. 9, 393–396 (1971).
Bixon, M. & Jortner, J. Electron transfer—from isolated molecules to biomolecules. in Advances in Chemical Physics (eds Prigogine, I. & Rice, S. A.) 35–202 (John Wiley & Sons, Inc., 2007).
Gebler, D. D. et al. Exciplex emission in bilayer polymer light-emitting devices. Appl. Phys. Lett. 70, 1644–1646 (1997).
Liao, W.-M. et al. Tailoring exciton and excimer emission in an exfoliated ultrathin 2D metal-organic framework. Nat. Commun. 9, 2401 (2018).
Calvino, C., Guha, A., Weder, C. & Schrettl, S. Self‐calibrating mechanochromic fluorescent polymers based on encapsulated excimer‐forming dyes. Adv. Mater. 30, 1704603 (2018).
Li, W. et al. Employing∼100% Excitons in OLEDs by utilizing a fluorescent molecule with hybridized local and charge‐transfer excited state. Adv. Funct. Mater. 24, 1609–1614 (2014).
Dong, H. et al. Broadband tunable microlasers based on controlled intramolecular charge-transfer process in organic supramolecular microcrystals. J. Am. Chem. Soc. 138, 1118–1121 (2016).
Wei, C., Gao, M., Hu, F., Yao, J. & Zhao, Y. S. Excimer emission in self-assembled organic spherical microstructures: an effective approach to wavelength switchable microlasers. Adv. Opt. Mater. 4, 1009–1014 (2016).
Vijayakumar, C., Tobin, G., Schmitt, W., Kim, M.-J. & Takeuchi, M. Detection of explosive vapors with a charge transfer molecule: self-assembly assisted morphology tuning and enhancement in sensing efficiency. Chem. Commun. 46, 874–876 (2010).
Zang, H., Xu, Z. & Hu, B. Magneto-optical investigations on the formation and dissociation of intermolecular charge-transfer complexes at donor−acceptor interfaces in bulk-heterojunction organic solar cells. J. Phys. Chem. B 114, 5704–5709 (2010).
Zhao, L., Lin, Y. L., Kim, H., Giebink, N. C. & Rand, B. P. Donor/acceptor charge-transfer states at two-dimensional metal halide perovskite and organic semiconductor interfaces. ACS Energy Lett. 3, 2708–2712 (2018).
Verhoeven, J. W., van Ramesdonk, H. J., Groeneveld, M. M., Benniston, A. C. & Harriman, A. Long-lived charge-transfer states in compact donor–acceptor dyads. Chem. Phys. Chem. 6, 2251–2260 (2005).
Deotare, P. B. et al. Nanoscale transport of charge-transfer states in organic donor–acceptor blends. Nat. Mater. 14, 1130–1134 (2015).
Loi, M. A. et al. Charge transfer excitons in bulk heterojunctions of a polyfluorene copolymer and a fullerene derivative. Adv. Funct. Mater. 17, 2111–2116 (2007).
Bettis Homan, S. et al. Ultrafast exciton dissociation and long-lived charge separation in a photovoltaic pentacene–MoS2 van der Waals heterojunction. Nano Lett. 17, 164–169 (2017).
Wu, K. et al. Efficient and ultrafast formation of long-lived charge-transfer exciton state in atomically thin cadmium selenide/cadmium telluride type-II heteronanosheets. ACS Nano 9, 961–968 (2015).
Yokoyama, T. et al. Overcoming short-circuit in lead-free CH3NH3SnI3 perovskite solar cells via kinetically controlled gas–solid reaction film fabrication process. J. Phys. Chem. Lett. 7, 776–782 (2016).
Yu, J. et al. Broadband extrinsic self-trapped exciton emission in Sn-doped 2D lead-halide perovskites. Adv. Mater. 31, 1806385 (2018).
Hu, T. et al. Mechanism for broadband white light emission from two-dimensional (110) hybrid perovskites. J. Phys. Chem. Lett. 7, 2258–2263 (2016).
Li, X. et al. Small cyclic diammonium cation templated (110)-oriented 2D halide (X = I, Br, Cl) perovskites with white-light emission. Chem. Mater. 31, 3582–3590 (2019).
Acknowledgements
B.H. acknowledges the funding support from the National Science Foundation (NSF-1911659) in the United States. This research was partially conducted at the Center for Nanophase Materials Sciences based on user projects (CNMS2016-279, CNMS2016-R45), which is sponsored by Oak Ridge National Laboratory by the Division of Scientific User Facilities, US Department of Energy. The author X.X.Z. (visiting Ph.D. student) acknowledges the support from the National Natural Science Foundation of China (Grant Nos. 61634001, U1601651, 61475051, and 61604010).
Author information
Authors and Affiliations
Contributions
B.H. directed the research project and supervised the experimental design. J.Z. conceived and performed the experiment studies and analyzed the data. X.X.Z. and M.S.W. contributed to the fabrication of LEDs. The manuscript was prepared by both J.Z. and B.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Rui Zhu, Dawei Di and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Zhu, X., Wang, M. et al. Establishing charge-transfer excitons in 2D perovskite heterostructures. Nat Commun 11, 2618 (2020). https://doi.org/10.1038/s41467-020-16415-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16415-1
- Springer Nature Limited
This article is cited by
-
Automated design of hybrid halide perovskite monolayers for band gap engineering
npj Computational Materials (2024)
-
Quasi-two dimensional Ruddlesden-Popper halide perovskites for laser applications
Frontiers of Physics (2024)
-
Rational adjustment to interfacial interaction with carbonized polymer dots enabling efficient large-area perovskite light-emitting diodes
Light: Science & Applications (2023)
-
Optical properties of two-dimensional perovskites
Frontiers of Physics (2023)
-
Perovskite light-emitting diodes
Nature Electronics (2022)