

Abstract
The biological properties of molecules containing the trifluoromethoxy group have made these compounds important targets in pharmaceuticals and agrochemicals, yet their preparation is still a substantial challenge. Herein, we present a practical nucleophilic trifluoromethoxylation of alkyl halides with (E)-O-trifluoromethyl-benzaldoximes (TFBO) as a trifluoromethoxylation reagent in the absence of silver under mild reaction conditions. The trifluoromethoxylation reagent TFBO is easily prepared and thermally stable, and can release CF3O− species in the presence of a base. Furthermore, broad scope and good functional group compatibility are demonstrated by application of the method to the late-stage trifluoromethoxylation of alkyl halides in complex small molecules.
Similar content being viewed by others
Introduction
A growing number of fluorine-containing organic compounds have widespread application in the fields of pharmaceuticals, pesticides and materials because of irreplaceable properties of fluoride1,2,3,4,5. The incorporation of fluorine-containing groups has been an efficient strategy for the design of new drugs and agrochemical. In recent years, the late-stage and selective fluorination reaction of organic molecules has received significant attention, especially the trifluoromethoxylation reaction, which is one of the most important research hotspots, as the trifluoromethoxy group’s electron-withdrawing effects and high lipophilicity (Hansch parameter πx = 1.04)6,7,8,9,10. However, the trifluoromethoxylation reaction remain limitations and challenges, such as limited trifluoromethoxylation reagents and instability of trifluoromethoxide anion, which impede its development and application11,12,13,14.
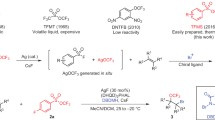
The synthesis of trifluoromethyl ethers can be achieved by indirect strategies and direct strategies. The indirect strategies include the nucleophilic fluorination of ether groups15,16,17 and electrophilic trifluoromethylation of hydroxyl groups18,19,20,21,22,23, which suffered from poor substrate scope, harsh reaction conditions and use of highly toxic reagents. The direct strategies are the direct introduction of the trifluoromethoxy group into organic compounds with trifluoromethoxylation reagents24. For example, tris(dimethylamino)sulfonium trifluoromethoxide (TASOCF3) was used as a trifluoromethoxylation reagent by Ritter’s group to achieve the direct trifluoromethoxylation of aryl stannanes and aryl boronic acids with equivalent silver25. Liu group reported a palladium-catalyzed intramolecular aminotrifluoromethoxylation of alkenes with AgOCF3 or CsOCF3 as the trifluoromethoxylation reagent26,27. The catalytic C(sp2)-H trifluoromethoxylation of arenes with the new N-OCF3 reagents under photocatalytic conditions had been reported by Ngai and Togni, respectively28,29,30. One of the simplest methods for the formation of the C(sp3)-OCF3 group is the nucleophilic substitution because of widely available leaving groups and inexpensive starting materials31,32,33,34,35,36,37. However, due to the poor nucleophilicity and instability of trifluoromethoxide anion, most of the known nucleophilic trifluoromethoxylation methods require to use activated electrophiles such as allylic halides, benzylic halides or α-halo carbonyl compounds with few exceptions, and available trifluoromethoxide anion (–OCF3) sources are scarce and usually suffere from disadvantages (Fig. 1a)38. For example, direct nucleophilic trifluoromethoxylation of alkyl iodides or bromides with trifluoromethyl triflate (TFMT)39,40, 2,4-dinitro(trifluoromethoxy)benzene (DNTFB)33 or trifluoromethyl benzoate (TFBz)37 were reported. However, less than 10% yield of desired products were obtained with unactivated alkyl iodide such as citronellyl iodide in the absence of silver, and trifluoromethoxide anion (CF3O−) was generated from these reagents under the activation of a fluoride salt, which might form the fluorinated byproduct. Furthermore, only one example of trifluoromethoxylation of benzyl chloride with AgOCF3 was reported to generate the desired product in 29% yield32, and no method is reported to achieve the trifluoromethoxylation of alkyl chloride in the absence of silver. Therefore, the development of an efficient method for nucleophilic trifluoromethoxylation of unactivated alkyl halides with a trifluoromethoxylation reagent in the absence of silver is highly desirable.

a Trifluoromethoxylation reagents. b Preparation of (E)-O-trifluoromethyl-benzaldoximes (TFBO). c The current method for nucleophilic trifluoromethoxylation of alkyl halides without silver.
We envisioned developing a trifluoromethoxylation reagent which is active enough to readily release CF3O− without the activation of nucleophiles such as fluoride salts. Inspired by alkoxy anion generated from the base-promoted elimination reactions of (E)-O-alkyl-benzaldoximes41, we were wondering whether (E)-O-trifluoromethyl-benzaldoximes (TFBO) can be prepared and used as a trifluoromethoxylation reagent if a suitable base is found to activate the reagent to generate trifluoromethoxide anion in situ, which would react with unactivated alkyl halides. (Fig. 1b) Herein, we report the development of (E)-O-trifluoromethyl-benzaldoximes (TFBO) as a trifluoromethoxylation reagent, which can be easily prepared from benzyl aldehydes and N-trifluoromethoxy phthalimide42 in modest yields. TFBO is shelf-stable and can be easily activated by the base to release CF3O− species (Fig. 1b). With (E)-O-trifluoromethyl-benzaldoximes (TFBO) as a trifluoromethoxylation reagent, an efficient nucleophilic trifluoromethoxylation of unactivated alkyl halides in the absence of silver is reported. This reaction is operationally simple, scalable, and which shows potential value in the field of drug synthesis (Fig. 1c).
Results
Investigations of reaction conditions and scope
The initial efforts were focused on the reaction of 5-iodopentyl 4-fluorobenzoate 2 with various (E)-O-trifluoromethyl-benzaldoximes (TFBO) in the presence of a base. As briefly illustrated in Fig. 2a, the use of a base was crucial for achieving an efficient transformation in the presence of TFBO, and Cs2CO3 was found to give the highest yield. Changing the base to other organic bases Et3N, DBU or inorganic bases KOtBu, CsF, Na2CO3, K2CO3 resulted in lower yields. Next, a thorough evaluation of different TFBOs revealed that substituents on the aromatic rings influenced the reaction yields, and the (E)-O-trifluoromethyl-4-tert-butyl-benzaldoximes 1a was found to be particularly effective. The control experiments were performed and no desired product was observed in the absence of a base. Monitoring of the reaction between TFBO (1a) and Cs2CO3 by 19F NMR spectroscopy indicated that CsOCF3 (–20.9 ppm) and aryl nitrile were generated in the reaction (Fig. 2b)31. After thoroughly optimizing the reaction conditions, reactions with 3.5 equiv. of Cs2CO3, 5.0 equiv. of TFBO (1a) in DMA under N2 atmosphere were found to produce high yields of the desired product.

a Optimization of the reaction conditions. Standard reaction conditions: alkyl halide (1.0 equiv.), base (3.5 equiv.), TFBO (5.0 equiv.), DMA, 70 °C, N2. #Yields were determined by 19F NMR with benzotrifluoride as a standard. b Monitoring of the reaction between TFBO (1a) and Cs2CO3 by 19F NMR spectroscopy.
With the optimized conditions in hand, we explored the substrate scope of the transformation (Fig. 3). First, a wide range of unactivated alkyl iodides was successfully converted to the corresponding trifluoromethyl ethers with yields ranging from 49 to 98% (3 to 34). Substrates bearing electron-donating and electron-withdrawing substituents on aryl rings proceeded well. This transformation was also compatible with excellent functionalities, such as ester, ether, ketone, aldehyde, imide, amide, cyano, nitro, aryl chloride, bromide, and iodide groups. Notably, heteroaromatic substrates and amino acid derivative were also successfully employed to provide the corresponding products17,18,30. Generally, the reactions of a variety of primary alkyl iodides gave rise to the desired trifluoromethoxylated products in high yields (3 to 30), while secondary alkyl iodides gave slightly lower yields (31 to 34). Next, we turned our attention to expanding the substrate scope to alkyl bromides and alkyl chlorides. To our great delight, the trifluoromethoxylation of alkyl bromides and alkyl chlorides proceeded smoothly with yields ranging from 30 to 97%. Furthermore, the allyl, propargyl, and benzyl halides were successfully converted into the desired trifluoromethoxylated products (35 to 39). It is worth mentioning that chemoselectivity trifluoromethoxylation of alkyl iodides or alkyl bromides in the presence of alkyl chlorides were observed. For example, the alkyl iodide or alkyl bromide was selectively converted into a trifluoromethoxy group while the alkyl chloride remained intact29. The yields of fluorination byproducts were less than 10% in all cases. In addition, the product 33 with 8% ee was observed when the chiral substrate (85% ee) was used, which suggested that the SN1-type nucleophilic trifluoromethoxylation might be involved in the reaction. Furthermore, other elecrophiles such as alkyl OMs, alkyl OTs were also successfully converted into the desired trifluoromethoxylated products in good yields (see the Supplementary Information for details). The limitation of this method was that no desired trifluoromethoxylated products were observed with tertiary halides. In addition, to demonstrate both scalability and practicality of this method, we prepared 28 on a gram scale under the standard reaction conditions in 89% isolated yield.

Standard reaction conditions: alkyl halide (1.0 equiv.), Cs2CO3 (3.5 equiv.), TFBO (5.0 equiv.), DMA, 70 °C, N2. #50 °C. †90 °C. ‡Cs2CO3 (4.0 equiv.), TFBO (6.0 equiv.), TBAI (3.0 equiv.), HMPA. §Cs2CO3 (4.0 equiv.), TFBO (6.0 equiv.), TBAI (0.2 equiv.), HMPA.*Yields were determined by 19F NMR with benzotrifluoride as a standard.
Due to the ubiquity of the trifluoromethoxy group in small-molecule drugs and preclinical candidates, it would be more significant to achieve the late-stage trifluoromethoxylation of complex small molecules with our trifluoromethoxylation reagents. To confirm this strategy, we selected ten meaningful small molecules as the substrates of trifluoromethoxylation.(Fig. 4). Each of these architecturally complex molecules underwent trifluoromethoxylation of alkyl halides to achieve the corresponding trifluoromethoxylated products in moderate to excellent yields (40 to 49, 40–97% yield). For example, pentacyclic diterpene gibberellic acid is a plant hormone that promotes growth and influences developmental processes, including cell germination and elongation. Cyclosporin A is an immunosuppressant medication and natural product, which is a macrocyclic peptide of 11 amino acids. To our delight, the trifluoromethoxylation reaction with the gibberellic acid derivative and cyclosporin A derivative proceeded smoothly to provide the corresponding products (46, 48) in good yields, which illustrates the ability to conduct the late-stage trifluoromethoxylation of complex structures.

Standard reaction conditions: alkyl halide (1.0 equiv.), Cs2CO3 (3.5 equiv.), TFBO (5.0 equiv.), DMA, 70 °C, N2. †Cs2CO3 (4.0 equiv.), TFBO (6.0 equiv.), Dibenzo-18-crown-6 (2.0 equiv.). ‡50 °C.
Discussion
In conclusion, we have developed (E)-O-trifluoromethyl-benzaldoximes (TFBO) as a trifluoromethoxylation reagent for nucleophilic trifluoromethoxylation of alkyl halides without silver. The method offers direct access to trifluoromethoxylated compounds from complex small molecules in the late stage. Compared to other trifluoromethoxylation reagents (see more detail in the Supplementary Table 12), TFBO can be easily activated by a base to release CF3O− species. Additionally, the reaction tolerates a various range of functional groups and is amenable to gram-scale synthesis. We expect that the operational simplicity, efficacy and broad scope of this method will find widespread use in pharmaceutical and agrochemical research.
Methods
General procedure for the synthesis of trifluoromethoxylation reagents
In a round-bottom flask, PhthNOCF3 (S1) (2-(trifluoromethoxy)isoindoline-1,3-dione) (1.00 g, 4.33 mmol, 1.00 equiv.), water (4.00 mL), HCl (0.720 mL ca. 6.00 M aq., 4.33 mmol, 1.00 equiv.) and aldehyde (1.50 equiv.) were added. The mixture was stirred at 80 °C overnight. Afterwards, the reaction mixture was extracted with CH2Cl2 (20.0 mL × 2). The combined organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel chromatography, to afford trifluoromethoxylation reagents.
General procedure for the synthesis of trifluoromethoxylated products
In a N2 glovebox, to alkyl halides (1.00 equiv.), (E)-O-trifluoromethyl-4-tert-butyl-benzaldoximes (1a) (307 mg, 1.25 mmol, 5.00 equiv.) in a 15.0 mL sealed vial were added DMA (2.00 mL). Cs2CO3 (285 mg, 0.875 mmol, 3.50 equiv.) was added to the reaction and the resulting mixture was stirred for overnight at 70 °C. After cooling to 50 °C, NMO (4-methylmorpholine N-oxide) (58.6 mg, 0.500 mmol, 2.00 equiv.) was added and the reaction mixture was stirred 2 h. Then the resulting mixture was filtered and concentrated in vacuo. The residue was purified by preparative TLC, to afford trifluoromethoxylated products.
Data availability
The authors declare that the data supporting this study are available within the article and supplementary information files. The X-ray crystallographic coordinates for compound (1d) reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 1907514. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Berger, R., Resnati, G., Metrangolo, P., Weber, E. & Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev. 40, 3496–3508 (2011).
Isanbor, C. & O’Hagan, D. Fluorine in medicinal chemistry: a review of anti-cancer agents. J. Fluor. Chem. 127, 303–319 (2006).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Liang, T., Neumann, C. N. & Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 52, 8214–8264 (2013).
Shimizu, M. & Hiyama, T. Modern synthetic methods for fluorine-substituted target molecules. Angew. Chem. Int. Ed. 44, 214–231 (2005).
Leroux, F., Jeschke, P. & Schlosser, M. α-Fluorinated ethers, thioethers, and amines: anomerically biased species. Chem. Rev. 105, 827–856 (2005).
Jeschke, P., Baston, E. & Leroux, F. R. alpha-fluorinated ethers as “exotic” entity in medicinal chemistry. Mini-Rev. Med. Chem. 7, 1027–1034 (2007).
Menteau, B., Pazenok, S., Vors, J. P. & Leroux, F. R. New trends in the chemistry of α-fluorinated ethers, thioethers, amines and phosphines. J. Fluor. Chem. 131, 140–158 (2010).
Landelle, G., Panossian, A. & Leroux, F. R. Trifluoromethyl ethers and-thioethers as tools for Medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 14, 941–951 (2014).
Tlili, A., Toulgoat, F. & Billard, T. Synthetic approaches to trifluoromethoxy-substituted compounds. Angew. Chem. Int. Ed. 55, 11726–11735 (2016).
Besset, T., Jubault, P., Pannecoucke, X. & Poisson, T. New entries toward the synthesis of OCF3-containing molecules. Org. Chem. Front. 3, 1004–1010 (2016).
Lee, J., Lee, K. & Ngai, M. Synthesis of tri- and difluoromethoxylated compounds by visible- light photoredox catalysis. Angew. Chem. Int. Ed. 58, 11171–11181 (2019).
Hardy, M. A., Chachignon, H. & Cahard, D. Advances in asymmetric di- and trifluoromethylthiolation and di- and trifluoromethoxylation reactions. Asian J. Org. Chem. 8, 591–609 (2019).
Yagupol’skii, L. M. Dokl. Akad. Nauk SSSR 105, 100–102 (1955).
Sheppard, W. A. α-fluorinated ethers. I. Aryl fluoroalkyl ethers. J. Org. Chem. 29, 1–11 (1964).
Kuroboshi, M., Suzuki, K. & Hiyama, T. Oxidative desulfurization-fluorination of xanthates. A convenient synthesis of trifluoromethyl ethers and difluoro (methylthio) methyl ethers. Tetrahedron Lett. 33, 4173–4176 (1992).
Umemoto, T. Electrophilic perfluoroalkylating agents. Chem. Rev. 96, 1757–1778 (1996).
Umemoto, T., Adachi, K. & Ishihara, S. CF3 oxonium salts, O-(trifluoromethyl)dibenzofuranium salts: In situ synthesis, properties, and application as a real CF3 + species reagent. J. Org. Chem. 72, 6905–6917 (2007).
Stanek, K., Koller, R. & Togni, A. Reactivity of a 10-I-3 hypervalent iodine trifluoromethylation reagent with phenols. J. Org. Chem. 73, 7678–7685 (2008).
Fantasia, S., Welch, J. M. & Togni, A. Reactivity of a hypervalent iodine trifluoromethylating reagent toward THF: ring opening and formation of trifluoromethyl ethers. J. Org. Chem. 75, 1779–1782 (2010).
Koller, R. et al. Zinc-mediated formation of trifluoromethyl ethers from alcohols and hypervalent iodine trifluoromethylation reagents. Angew. Chem. Int. Ed. 48, 4332–4336 (2009).
Liang, A. et al. Regioselective synthesis of N-heteroaromatic trifluoromethoxy compounds by direct O−CF3 bond formation. Chem. Eur. J. 22, 5102–5106 (2016).
Lee, K. N., Lee, J. W. & Ngai, M. Recent development of catalytic trifluoromethoxylation reactions. Tetrahedron 74, 7127–7135 (2018).
Huang, C., Liang, T., Harada, S., Lee, E. & Ritter, T. Silver-mediated trifluoromethoxylation of aryl stannanes and arylboronic acids. J. Am. Chem. Soc. 133, 13308–13310 (2011).
Chen, C., Chen, P. & Liu, G. Palladium-catalyzed intramolecular aminotrifluoromethoxylation of alkenes. J. Am. Chem. Soc. 137, 15648–15651 (2015).
Chen, C., Pflüger, P. M., Chen, P. & Liu, G. Palladium(II)-catalyzed enantioselective aminotrifluoromethoxylation of unactivated alkenes using CsOCF3 as a trifluoromethoxide source. Angew. Chem. Int. Ed. 58, 2392–2396 (2019).
Zheng, W., Morales-Rivera, C. A., Lee, J. W., Liu, P. & Ngai, M. Catalytic C−H trifluoromethoxylation of arenes and heteroarenes. Angew. Chem. Int. Ed. 57, 9645–9649 (2018).
Jelier, B. J. et al. Radical trifluoromethoxylation of arenes triggered by a visible-light-mediated N−O bond redox fragmentation. Angew. Chem. Int. Ed. 57, 13784–13789 (2018).
Zheng, W., Lee, J. W., Morales-Rivera, C. A., Liu, P. & Ngai, M. Redox-active reagents for photocatalytic generation of the OCF3 radical and (hetero)aryl C−H trifluoromethoxylation. Angew. Chem. Int. Ed. 57, 13795–13799 (2018).
Kolomeitsev, A. A., Vorobyev, M. & Gillandt, H. Versatile application of trifluoromethyl triflate. Tetrahedron Lett. 49, 449–454 (2008).
Marrec, O., Billard, T., Vors, J., Pazenok, S. & Langlois, B. R. A deeper insight into direct trifluoromethoxylation with trifluoromethyl triflate. J. Fluor. Chem. 131, 200–207 (2000).
Marrec, O. et al. A new and direct trifluoromethoxylation of aliphatic substrates with 2, 4-dinitro (trifluoromethoxy) benzene. Adv. Synth. Catal. 352, 2831–2837 (2010).
Chen, S. et al. Aryl-BIAN-ligated silver (I) trifluoromethoxide complex. Dalton Trans. 44, 19682–19686 (2015).
Guo, S., Cong, F., Guo, R., Wang, L. & Tang, P. Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes with a new trifluoromethoxylation reagent. Nat. Chem. 9, 546–551 (2017).
Jiang, X., Deng, Z. & Tang, P. Direct dehydroxytrifluoromethoxylation of alcohols. Angew. Chem. Int. Ed. 57, 292–295 (2018).
Zhou, M., Ni, C., Zeng, Y. & Hu, J. Trifluoromethyl benzoate: a versatile trifluoromethoxylation reagent. J. Am. Chem. Soc. 140, 6801–6805 (2018).
Zhang, X. & Tang, P. Recent advances in new trifluoromethoxylation reagents. Sci. Chi. Chem. 62, 525–532 (2019).
Noftle, R. E. & Cady, G. H. Preparation and properties of bis (trifluoromethylsulfuryl) peroxide and trifluoromethyl trifluoromethanesulfonate. Inorg. Chem. 4, 1010–1012 (1965).
Taylor, S. L. & Martin, J. C. Trifluoromethyl triflate: synthesis and reactions. J. Org. Chem. 52, 4147–4156 (1987).
Mauleón, D., Granados, R. & Minguillón, C. 4-alkoxybenzonitriles from O-alkyl-4-nitrobenzaldoximes: an elimination-aromatic substitution reaction. J. Org. Chem. 48, 3106–3108 (1983).
Matoušek, V., Pietrasiak, E., Sigrist, L., Czarniecki, B., Togni, A. O-Trifluoromethylation of N,N-Disubstituted Hydroxylamines with Hypervalent Iodine Reagents. Eur. J. Org. Chem. 2014, 3087–3092 (2014).
Acknowledgements
The authors are grateful for the financial support from the National Key Research and Development Program of China (2016YFA0602900), NFSC (21672110, 21925105), the Natural Science Foundation of Tianjin (Grant No. 18JCJQJC47000), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
Y.L., Y.Y., and J.X. performed the experiments and analyzed the data. P.T. designed and directed the project. P.T. wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Yang, Y., Xin, J. et al. Nucleophilic trifluoromethoxylation of alkyl halides without silver. Nat Commun 11, 755 (2020). https://doi.org/10.1038/s41467-020-14598-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14598-1
- Springer Nature Limited
This article is cited by
-
3,3-Difluoroallyl ammonium salts: highly versatile, stable and selective gem-difluoroallylation reagents
Nature Communications (2021)
-
Chemoselective desulfurization-fluorination/bromination of carbonofluoridothioates for the O-trifluoromethylation and O-bromodifluoromethylation of alcohols
Science China Chemistry (2021)
-
Selective C-H trifluoromethoxylation of (hetero)arenes as limiting reagent
Nature Communications (2020)