
Abstract
G protein-coupled receptors (GPCRs) mediate cellular responses to a myriad of hormones and neurotransmitters that play vital roles in the regulation of physiological processes such as blood pressure. In organs such as the artery and kidney, hormones or neurotransmitters, such as angiotensin II (Ang II), dopamine, epinephrine, and norepinephrine exert their functions via their receptors, with the ultimate effect of keeping normal vascular reactivity, normal body sodium, and normal blood pressure. GPCR kinases (GRKs) exert their biological functions, by mediating the regulation of agonist-occupied GPCRs, non-GPCRs, or non-receptor substrates. In particular, increasing number of studies show that aberrant expression and activity of GRKs in the cardiovascular system and kidney inhibit or stimulate GPCRs (e.g., dopamine receptors, Ang II receptors, and α- and β-adrenergic receptors), resulting in hypertension. Current studies focus on the effect of selective GRK inhibitors in cardiovascular diseases, including hypertension. Moreover, genetic studies show that GRK gene variants are associated with essential hypertension, blood pressure response to antihypertensive medicines, and adverse cardiovascular outcomes of antihypertensive treatment. In this review, we present a comprehensive overview of GRK-mediated regulation of blood pressure, role of GRKs in the pathogenesis of hypertension, and highlight potential strategies for the treatment of hypertension.

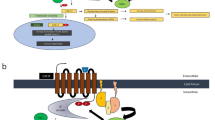
Schematic representation of GPCR desensitization process. Activation of GPCRs begins with the binding of an agonist to its corresponding receptor. Then G proteins activate downstream effectors that are mediated by various signaling pathways. GPCR signaling is halted by GRK-mediated receptor phosphorylation, which causes receptor internalization through β-arrestin.
Similar content being viewed by others

Essential hypertension, also known as primary hypertension, is a major contributing risk factor for cardiovascular and cerebrovascular diseases, and causes damage of target organs, such as the kidney, heart and brain [1]. Worldwide data from the Noncommunicable Disease (NCD) Risk Factor Collaboration show that the number of people 30–79 years old with hypertension has doubled from 1990 to 2019, with most of the increase occurring in low-income and middle-income regions [2]. In the United States, 46.7% (122.4 million) of adults ≥20 years of age has hypertension, and the number of deaths attributable to high blood pressure increased 90.1% from 2010 to 2020 [3]. Hypertension is a major public health problem and challenges worldwide [4].
Cells communicate with each other by receiving and sending through a myriad of extracellular proteins/molecules, such as neurotransmitters, chemoattractants, and hormones, which bind to receptors and drive the initiation of multiple intracellular signaling routes. G protein-coupled receptors (GPCRs) are the largest superfamily of cell-surface receptors and the most diverse group of proteins involved in transmembrane signal transduction [5, 6]. They share conserved seven α-helical structured transmembrane domains, extracellular ligand-binding domains, and carboxy-terminal intracellular domains. GPCRs, including adrenergic receptors, angiotensin II (Ang II) receptors, and dopamine receptors, control a variety of physiological processes involved in the regulation of blood pressure [7, 8]. Cell dysfunction, induced by aberrant GPCRs, including abnormal phosphorylation, trafficking, and expression, causes hypertension by various mechanisms [8,9,10].
The phosphorylation and desensitization of agonist-occupied GPCRs are primarily mediated by GPCR kinases (GRKs) [11]. As their name implies, GRKs are defined by their actions on GPCR phosphorylation, receptor internalization, and subsequent signal termination (Fig. 1). Abnormalities of GRKs are associated with cardiovascular diseases, including hypertension [12, 13]. A number of studies have shown that GRKs play a vital role in the regulation of blood pressure; aberrant expression and/or activity of GRKs cause hypertension and are associated with impaired response to antihypertensive treatment [14,15,16,17]. There are abundant epidemiological and animal studies reinforcing the role of GRKs and their underlying mechanisms in the regulation of blood pressure. Therefore, in this article, we review our evolving understanding of GRK-mediated regulation of blood pressure and highlight potential strategies for targeting GRKs in the prevention and treatment of hypertension.

Schematic representation of GPCR desensitization process. Activation of GPCRs begins with the binding of an agonist to its corresponding receptor. Then G proteins activate downstream effectors that are mediated by various signaling pathways. GPCR signaling is halted by GRK-mediated receptor phosphorylation, which causes receptor internalization through β-arrestin. ATP adenosine triphosphate, cAMP cyclic adenosine monophosphate, GAP GTPase activating protein, GDP Guanosine-5’-diphosphate, GEF guanine nucleotide exchange factor; GPCR G protein-coupled receptor, GRK G protein–coupled receptor kinase, GTP guanosine triphosphate, PI3K phosphatidylinositol-3 kinase, PKC protein kinase C, PLC-β phospholipase C-β; RGS regulators of G protein signaling
The Grk family
GRK-mediated receptor phosphorylation is one of the well-characterized mechanisms for GPCR desensitization. There are more than 800 distinct GPCRs in the human genome but of the vital regulators of agonist-induced phosphorylation of GPCRs, only seven GRKs (GRK1–7) have been identified [18]. Based on their sequence similarity and gene structure, the serine/threonine kinase GRK family encompasses seven protein isoforms that are classified into three subfamilies. GRK1 and GRK7 belong to the GRK1-like subfamily, also known as visual or rhodopsin-specific GRKs. GRK2 (β-adrenergic receptor kinase 1, β-ARK1) and GRK3 (β-ARK2) belong to the GRK2-like subfamily, also known as β-adrenergic receptor (β-AR) kinase subfamily. GRK4, GRK5, and GRK6 belong to the GRK4-like subfamily [17, 18].
The seven GRKs share a common general protein structure with a central catalytic domain (~270 amino acids), flanked by an amino-terminal (N-terminal) domain (~185 amino acids) and a carboxyl terminal (C- terminal) domain (~105 to 230 amino acids) [19]. The N-terminus, a well-conserved domain between GRK members, is vital for the selective recognition of the activated receptor and also contains a region termed as a regulator of G protein signaling homology (RH) domain (~120 amino acids). However, the C-terminus is divergent among GRK subfamilies and contributes to their subcellular membrane localization and agonist-dependent translocation [19, 20].
In addition to GPCRs, GRKs also regulate the phosphorylation or expression of other non-GPCR receptors, such as adiponectin receptor 1 (AdipoR1) and insulin-like growth factor-1 receptor [21, 22]. Moreover, GRKs regulate non-receptor substrates, including signaling proteins, such as insulin receptor substrate-1, nuclear proteins, such as histone deacetylase 5, and transcription factors, such as IκBα, among others [23,24,25,26]. We also reported that GRK4 increases the phosphorylation of signal transducer and activator of transcription 1 and HDAC4 [27, 28]. It should also be noted that kinase activity is not necessary for some GRK-mediated functions. For example, GRK4 regulates cilia and kidney development independent of its kinase function [29].
All GRKs are primarily localized in the cytosol and plasma membrane. However, the tissue distribution of the seven GRK subtypes is different from each other. GRK2, GRK3, GRK5, and GRK6 are ubiquitously distributed among mammalian tissues [17]. GRK1, GRK4, and GRK7 are expressed in specific tissues. GRK1 and GRK7 are expressed exclusively in the retina, whereas GRK4 is expressed in a few organs [17, 30]. These indicate that GRKs may exert different physiological functions based on their tissue distribution.
Role of Grks on blood pressure regulation
Many studies have shown that GRKs exert their biological functions by mediating the regulation of agonist-occupied GPCRs, non-GPCR receptors, and non-receptor substrates (Table 1). Several organs, including the artery, heart, and kidney, are involved in GRK-mediated blood pressure regulation (Fig. 2). The expression and/or activity of GRKs are aberrant in the hypertensive humans and animal models of hypertension. Because GRK1 and GRK7 are expressed only in the retina, only the role of the other five GRKs in the regulation of blood pressure have been studied.

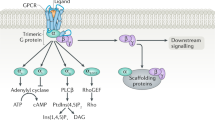
Regulation of blood pressure by GRKs. GRKs exert their functions in organs, including kidney, artery, and heart, by regulating the expression or phosphorylation of various GPCRs to modulate blood pressure. Pink color: various substrates regulated by GRKs. α-1DAR α-1D adrenergic receptor, adipoR1 adiponectin receptor 1, Ang II angiotensin II, AT1R angiotensin II type 1 receptor, AT2R angiotensin II type 2 receptor, β-AR β-adrenergic receptor, D1R dopamine D1 receptor, D3R dopamine D3 receptor, ETBR endothelin receptor type B, GRK G protein-coupled receptor kinase, P2Y2R P2Y2 receptor
GRK2
Role of GRK2 in the regulation of blood pressure
GRK2 is ubiquitously expressed in mammalian organs and tissues, including the artery, heart, and kidney. GRK2 is not only a key regulator of vascular responsivity in resistance arteries but also regulates myocardial contractility [31, 32], and renal function (vide infra).
GRK2 regulates GPCR-mediated vascular signaling, which participates in the regulation of blood pressure. Global knockdown of GRK2 [33] or vascular smooth muscle (VSM)-targeted overexpression of GRK2 in mice increases resting blood pressure [34]. This apparent inconsistent effect on blood pressure may be related to the fact that the alteration in GRK2 expression (increased or decreased) can lead to the imbalance of vasoconstrictor and vasodilator effects, resulting in hypertension [33]. GRK2 negatively regulates GPCRs such as endothelin type A receptor [35], P2Y2 receptor [36, 37], angiotensin II type 1 receptor (AT1R) [38] and α1D-adrenergic receptor [39], which should result in vasodilation. By contrast, GRK2 also negatively regulates β2-adrenergic receptor [40] and dopamine D1 receptor (D1R)/adenylyl cyclase signaling [41], which should result in vasoconstriction. The rebound hypertension in mice that occurs after withdrawal of clonidine, an α1D, α2A-C adrenergic receptor agonist, is mitigated by an increase in nitric oxide bioavailability, an effect that is prevented by suppression of GRK2 activity [42]. However, one group reported that global GRK2 knockdown impaired kidney development, and increased blood pressure [43] while another group reported that Grk2+/− mice have normal blood pressure and attenuated response to the hypertensive effect of Ang II infusion [31]. Therefore, the overall effect of GRK2 on blood pressure is difficult to predict (vide infra).
GRK2 directly interacts with the epithelial sodium channel (ENaC) [44], which increases the reabsorption of filtered sodium across the luminal membrane of the principal cells of the aldosterone-sensitive distal nephron [45]. GRK2 positively regulates ENaC activity by phosphorylating Nedd4 and Nedd4-2, two ubiquitin-protein ligases binding with the channels, to prevent their ability to inhibit epithelial channel activity [46]. The dopaminergic system plays a vital role in the regulation of sodium excretion. The activation of GRK2 by some factors such as oxidative stress and insulin impairs the ability of renal D1R to inhibit sodium transport [47, 48] that leads to the development of hypertension [8]. We have also reported that exposure to maternal diabetes mellitus causes renal D1R dysfunction and hypertension in adult rat offspring by increasing GRK2 activity [49]. Thus, GRK2 can increase blood pressure by increasing renal sodium transport.
GRK2 also regulates receptor-mediated signaling in the heart. GRK2 phosphorylates cardiac β2-adrenergic receptor and increases Gi-biased signaling, which contributes to heart failure [50]. However, GRK2 inhibition increases β-adrenergic receptor-dependent cardiomyocyte and cardiac contractility and reverses cardiac dysfunction in heart failure models [51]. GRK2 also regulates the desensitization of other cardiac receptors, such as AdipoR1 and κ-opioid receptor [52, 53]. In addition, GRK2 participates in the regulation of other physiological or pathological functions in the heart, including fatty acid metabolism, production of NLRP3 inflammasome and oxidative stress, and modulation of insulin signaling [54,55,56]. How, these GRK2-mediated effects in the heart regulate blood pressure need to be studied further.
Blood pressure phenotypes in mice with modified Grk2 expression
As aforementioned, the role of GRK2 on the regulation of blood pressure has been demonstrated in mice with genetically modified Grk2 expression. Since germline deletion of Grk2 is lethal, Grk2 hemizygous mice (GRK2+/−), shRNA-GRK2 transgenic mice, or knockdown in local organs have been used to investigate its effects on the regulation of blood pressure. shRNA-induced total body knockdown of GRK2 causes spontaneous hypertension and impairs vascular reactivity [33]. This is similar to the findings of another study in which Grk2 knockdown in mice exacerbates kidney injury and causes spontaneous hypertension, accompanied with over-activation of the renal renin-angiotensin system (RAS) and AT1R signaling [43]. However, Grk2 deficiency in global adult hemizygous mice does not cause hypertension but impairs the increase in blood pressure and prevents vascular remodeling secondary to Ang II infusion [31]. By contrast, inhibition of VSM-Grk2 by overexpression of the C-terminal portion of Grk2 or VSM-specific ablation of Grk2 protein expression in mice has no effect on the increased blood pressure in the two-kidney, one-clip (2K1C) model, which may be due to a balance between the increased β-AR-mediated dilation and the enhancement of α1D-AR-stimulated vasoconstriction [39].
The reason for the inconsistent reports on the effect of Grk2 knockdown on blood pressure remains unclear. We speculate some possibilities: 1) Different methods of Grk2 knockdown may produce different effects. Global knockdown of GRK2 using a small hairpin (sh) RNA leads to hypertension [33, 43], a partial deficiency of GRK2 protects against Ang II-induced hypertension [31]; VSM-specific Grk2 knockdown has no effect on blood pressure [39]. 2) GRK2 regulates both vasoconstriction and dilation. For example, GRK2 deficiency increases phenylephrine, but not KCl-mediated constriction and isoproterenol-mediated vasodilation [33]. The dominant effect induced by GRK2 knockdown may determine the physiological or pathological phenotype. 3) Duration of Grk2 silencing. There is a balance between vasoconstriction and vasodilation in Grk2 knockdown mice. Twelve to 14 weeks of Grk2 knockdown using small hairpin RNA causes a shift toward vasoconstriction and subsequent hypertension [33]. Additionally, activated renal AT1R-mediated signaling also plays an important role in the development of hypertension after six months of Grk2 knockdown [43]. However, a longer (nine months) duration of Grk2 knockdown was reported to lead to a nitric oxide-dependent vasodilation which overcomes vasoconstriction, making Grk2+/− mice resistant to the development of hypertension [31]. 4) It is possible that the different effects on blood pressure induced by Grk2 knockdown may be also attributed to experimental conditions such as animal species and shRNA induction.
The depletion of Grk2 expression in aortic smooth muscle cells also impairs the proliferative effect of endothelin and Ang II [57], that should prevent the increase in blood pressure. Grk2 knockdown in the endothelial cells of mesenteric arterioles promotes insulin-induced vasodilation and insulin-stimulated signaling pathway in SHRs; this effect is prevented by upregulation of Grk2 [58]. Selective deletion of Grk2 in vascular endothelium of mice also blunts the vasoconstriction induced by the α1-adrenergic receptor agonist phenylephrine and other vasoconstrictors (serotonin, oxytocin, and KCl) [59].
By contrast, transgenic mice with VSM-targeted Grk2 over-expression have higher resting blood pressure than their wild-type littermates [34]. This is consistent with another study, which found that VSM Grk2 over-expression leads to increased systolic blood pressure (SBP). Moreover, epinephrine injection increased SBP to a greater extent in the VSM-Grk2 over-expressing mice than wild-type control mice [60]. However, cardiac-specific Grk2 expression attenuates Ang II-induced increase in contractility and blood pressure [32], consistent with the pro- and anti-hypertensive effects of GRK2 that are tissue-dependent.
GRK2 expression is increased in animal models of hypertension
The expression of Grk2 mRNA is higher in the aorta but lower in the kidney of spontaneously hypertensive rats (SHRs) than Wistar-Kyoto (WKY) rats [61, 62]. GRK2 protein expressions in lymphocytes and VSMs are also higher in SHRs than Wistar- and WKY rats; vascular GRK2 protein expression is also higher in hypertensive Dahl salt-sensitive than normotensive Dahl salt-resistant rats [63]. The expressions of GRK2 and adrenergic receptors are decreased in the aorta but not in mesenteric arteries of NG-nitro-L-arginine methyl ester (L-NAME)-induced hypertensive rats, that is accompanied by decreased α1-adrenergic receptor-induced vasoconstriction along with increased β2-adrenergic receptor-mediated vasodilation; these changes are the opposite to those found in the aortas of SHRs [64]. In addition, renal GRK2 expression is increased in L-NAME-induced hypertensive rats. However, renal but not left ventricular GRK2 expression is decreased in SHRs [62].
GRK2 and human hypertension
Studies have shown abnormal GRK2 expression and/or activity in the hypertensive state. GRK activity and GRK2 expression in lymphocytes are increased in hypertensive relative to normotensive humans [14]. The increase in GRK2 protein expression in lymphocytes from hypertensive subjects is positively associated with blood pressure and inversely associated with β-adrenergic agonist-mediated increase in adenylyl cyclase activity [14, 65]. Izzo et al. also reported that GRK2 expression is increased in blood lymphocytes of hypertensive patients and the impaired β-AR-mediated vasorelaxation in the brachial artery is restored by treatment with heparin, a nonspecific GRK inhibitor [66]. Further studies showed that in Black Americans, GRK2 but not GRK5 protein expression is increased 2-fold and GRK activity is increased >40% in the lymphocytes, which correlated with increasing blood pressure [67]. A case-control study in Han Chinese showed that the dominant model (CC vs. CT + TT) of rs1894111 polymorphism in the ADRBK1 (GRK2) gene is associated with low-renin hypertension [68]. In addition, abnormal GRK2 expression or activity is associated with other types of hypertension. For example, elevated GRK2 levels in the umbilical vasculature are associated with elevated blood pressure in subjects with gestational hypertension and preeclampsia [69], but this may be a compensatory rather than a causative effect. Insufficient GRK2 activity compromises spiral artery remodeling and initiates necrotic events in the placenta, thereby causing preeclampsia [70].
GRK3
GRK3 is ubiquitously expressed in mammalian organs and tissues. There are limited studies on the association between GRK3 and hypertension. GRK3 expression in cardiac myocytes progressively increases in 6-, 12-, and 24-month-old spontaneously hypertensive heart failure rats and is significantly higher than in control rats [71]. Grk3 mRNA levels in human lymphocytes are inversely associated with SBP and diastolic blood pressure (DBP) [72], suggesting that GRK3 may play a beneficial role in the regulation of blood pressure. This is, at least in part, supported by findings in transgenic mice. Cardiac myocyte-restricted expression of the carboxyl-terminal fragment of Grk3, a competitive inhibitor of Grk3, results in a phenotype with elevated SBP and DBP but without alterations of heart rate in conscious, unrestrained mice [73]. Further studies found that the cardiomyocyte-restricted inhibition of Grk3 in mice does not alter the hypertrophic response but attenuates cardiac dysfunction and heart failure with chronic pressure overload [74]. It should be noted that some GPCRs that regulate blood pressure, such as dopamine receptors and β-ARs are also regulated by GRK3 [75, 76]. Grk3 expressed in HEK293 cells augments the agonist-induced AT1R or D1R phosphorylation, causing their desensitization [41]. However, the physiological consequences of altered GRK3 expression is unclear. Renal GRK3 expression is not different between WKY rats and SHRs but renal GRK3 mRNA is increased in the kidneys of rats made hypertensive with L-NAME [62]. The role of GRK3 in the regulation of blood pressure needs to be studied further.
GRK4
Effect of GRK4 on blood pressure
GRK4 is expressed to a greater extent in the testes and myometrium and to a lesser extent in other tissues such as the brain and kidney [13]. We also reported its expression in the artery and heart [28, 77], and found that renal and cardiac GRK4 levels are elevated by kidney ischemia-reperfusion injury and myocardial infarction, respectively [27, 28].
The role of GRK4 in the regulation of blood pressure is confirmed in several animal models of hypertension. Global Grk4 knockout mice have lower SBP and DBP than Grk4 wild-type (WT) mice [78]. Selective renal Grk4 knockdown, via chronic renal cortical interstitial-selective infusion of Grk4 antisense oligodeoxynucleotides, increases urine flow and ameliorates the sodium retention and elevated blood pressure of SHRs [79, 80]. We have reported that down-regulation of renal GRK4 expression, via ultrasound-targeted microbubble destruction-delivered Grk4 small interfering RNA (siRNA), decreases blood pressure, accompanied with increased sodium excretion in SHRs [21, 81].
In contrast to the lowering of blood pressure caused by the silencing of Grk4 [78,79,80,81,82], the over-expression of human Grk4 gene variants (hGrk4γ 65L, hGrk4γ 142V, or hGrk4γ 486V) in mice has the opposite effect on blood pressure. On normal salt diet, transgenic mice with over-expression of human GRK4γ (hGRK4γ) variant 142V have higher blood pressure than WT transgenic mice [15, 77, 78, 82]. However, hGrk4γ 486V [83] and hGrk4γ 65L transgenic mice have normal blood pressure on a normal-salt diet but have elevated blood pressure on a high-salt diet, indicating that hGRK4γ 486V and hGRK4γ 65L transgenic mice have salt-sensitive hypertension while hGrk4γ 142V transgenic mice have salt-resistant hypertension.
The reason why different GRK4 variants exhibit different phenotypes in blood pressure regulation is still unknown. Our previous study found that the salt sensitivity of hGRK4γ 486V mice is due, in part, to increased renal reactive oxygen species production, which can be attributed to defective renal antioxidant mechanisms [83]. However, the main reason for the different phenotypes of different GRK4 variants could be due to the different structures of the GRK4 variants. We presume that the polymorphisms alter the structure of GRK4, that modulate its kinase activity, and affect substrate-regulated biological responses that subsequently cause hypertension. Allen et al. reported the overall crystal structure of GRK4α A486V and differences in the structure of GRK4α A486V from other GRKs. They found that relative to WT GRK4α, GRK4α A486V has an increased rate of autophosphorylation of a number of residues, which is due to a defective lag in the absence of preincubation with ATP [84]. However, the mechanism by which different GRK4 polymorphisms affect enzymatic activity needs more investigation in future studies.
The control of blood pressure by GRK4 is due in part to its regulation of renal dopamine receptors. In human renal proximal tubule cells, GRK4 constitutively phosphorylates the D1R, even in the absence of agonist activation, causing its internalization and constitutive desensitization [85]. Treatment with heparin, an inhibitor of GRK activity, or antisense oligonucleotides (GRK4 > GRK2) blunt the agonist-induced D1R desensitization [86]. In human essential hypertension, single nucleotide polymorphisms (SNPs) in GRK4γ increase GRK activity and D1R serine phosphorylation, causing the uncoupling of D1R from its G protein/effector enzyme complex in the renal proximal tubule [15]. GRK4-mediated regulation of D1R expression or activity has been confirmed in several animal models of hypertension. We have reported that GKR4 impairs renal tubular and arterial D1R expression and D1R-mediated sodium excretion and vasodilation in offspring with hypertension related to in utero exposure to some factors such as fine particulate matter, e.g., PM2.5, lipopolysaccharide, and cold stress [87,88,89]. The impairment of the D1R function as part of the insulin resistance in type 2 diabetes mellitus in mice has also been related to increased GRK4 expression [90]. GRK4 also regulates the phosphorylation and function of the dopamine D3 receptor (D3R) in human proximal tubule cells [91]. However, the role of GRK4 in D3R-mediated regulation of blood pressure remains unclear.
GRK4 can also control blood pressure by interacting with the AT1R. hGrk4γ 142V phosphorylates HDAC1 and promotes its export from the nucleus to the cytoplasm resulting in an increase in the transcription of AT1R, presumably by decreasing its deacetylation [78]. AT1R blockade or the deletion of the Agtr1a gene normalizes hypertension in Grk4γ 142V mice [78]. We have reported that GRK4 is expressed in VSMCs of the aorta. AT1R expression is higher in the aorta of Grk4γ 142V transgenic mice than WT mice. Ang II-mediated vasoconstriction of the aorta is greater in Grk4γ 142V mice than in WT mice [77]. GRK4-positive regulation of AT1R expression and function is also found in other hypertensive animal models, including the male hypertensive offspring of male Sprague-Dawley rats that were exposed to fine particulate matter with an aerodynamic diameter of ≤2.5 μm [92]. We have also recently reported that GRK4 increases the phosphorylation of angiotensin II type 2 receptor (AT2R) which impairs renal AT2R-mediated diuresis and natriuresis, which participate in the GRK4-mediated regulation of blood pressure [93].In addition to dopamine and Ang II receptors, GRK4 also regulates other receptors such as the endothelin type B receptor (ETBR) and AdipoR1. Compared with WT mice, hGRK4γ 142V mice have increased phosphorylation of renal ETBR and AdipoR1 and impaired ETBR- and AdipoR1-induced diuresis and natriuresis [21, 82]. By contrast, renal-selective GRK4 knockdown normalizes the increased ETBR and AdipoR1 phosphorylation, and partially or fully restores the ETBR- and AdipoR1-mediated increase in sodium excretion and reduction of blood pressure in SHRs [21, 82].
GRK4 in hypertensive animal models
Basal GRK4 expression and activity in the kidney and artery are higher in SHRs than in WKY rats [61, 79, 80, 82, 93]. The increased GRK4 expression in the renal and artery in SHRs is organ-specific because there is no difference in cardiac GRK4 expression between WKY and SHRs [79]. Similar results are found in kidneys of humans with essential hypertension. Moreover, the increased activity of GRK4 in the kidneys of hypertensive subjects is caused not by increased renal GRK4 protein expression but rather by constitutively active variants of GRK4 [15]. The role of GRK4 in hypertension is further confirmed by studies involving renal Grk4 depletion, which efficiently reduces GRK4 expression and lowers blood pressure in SHRs, accompanied with increased urine volume and sodium excretion [21, 79,80,81,82]. In addition, GRK4 expression and activity in the kidney and artery are increased in other animal models of hypertension [87,88,89, 94,95,96].
GRK4 in human essential hypertension
The GRK4 locus on human chromosome 4p16.3 is associated with essential hypertension and salt sensitivity [13]. Several GRK4 gene variants, e.g., R65L, A142V, and A486V, are positively associated with essential hypertension in several ethnic populations [97,98,99,100,101,102,103,104,105,106,107,108,109,110] [Table 2]. However, the association between the GRK4 variants and hypertension was not found in other studies, which may be age/sex/race/ethnicity-related, or a failure to study all the GRK4 variants [103, 109, 111,112,113,114]. It should be noted that recent studies reported that some new GRK4 variants are associated with hypertension (GRK4 rs1644731), both hypertension and diabetes (GRK4 rs1557213), or cardiovascular disease risk and diabetes (GRK4 rs60314379) [104, 105, 115]. In addition, an earlier meta-analysis showed that GRK4 rs1024323 is associated with hypertension in Caucasians, but not in East Asians and Africans [106].
GRK4 variants are also associated with salt sensitivity. In a Japanese cohort, the genotype and allele frequencies of GRK4 SNPs, including R65L, A142V, and A486V, are higher in salt-sensitive than in salt-resistant hypertensive subjects. Sodium excretion was reported to be inversely related to the number of GRK4 variants in hypertensive persons, and the natriuretic response to dopaminergic stimulation is impaired in normotensive persons having three or more GRK4 gene variants [99]. A large genome-wide association study in 15,034 Korean adults investigated the correlation of 1282 candidate SNPs with dietary sodium intake and found that the GRK4 R65L TT genotype is inversely associated with salt-sensitive hypertension risk [103]. Moreover, GRK4 polymorphisms are also associated with salt sensitivity in normotensive subjects [107, 116]. The GRK4 65L allele is associated with impaired renal sodium handling in response to stress in Black normotensive adolescents [117].
GRK5
GRK5 is ubiquitously expressed in mammalian organs and tissues, including the heart, lung, and retina [118]. GRK5 is involved in cardiovascular diseases, including cardiac fibrosis, heart failure, and hypertension [12, 119,120,121,122,123,124]. GRK5 mRNA and protein levels are increased in the aorta of Ang II- and norepinephrine-induced hypertensive rats [121]. Gkr5 mRNA expression is also increased in the left ventricle and kidney of rats with hypertension induced by L-NAME [62]. However, in this report, the mRNA levels of renal GRK3 and GRK5 are not different between WKY and SHRs [62].
The role of GRK5 in the regulation of blood pressure is verified in transgenic mice. VSM-specific overexpression of Grk5 increases blood pressure, which is sex-modified because male mice with VSM-Grk5 overexpression have a much greater increase in blood pressure (~26 mm Hg, mean arterial pressure) than female Grk5 mice [122]. However, the intracardiac injection of adenovirus that encodes for the RH domain within the amino terminus of Grk5 does not reduce blood pressure levels in SHRs or in WKY rats [123]. It should be noted that the role of GRK5 in the regulation of blood pressure is complicated. Mice with global Grk5 ablation have impaired glucose tolerance and insulin sensitivity compared with their WT littermates [124]. Therefore, GRK5 may be a positive regulator of insulin resistance, which contributes to the pathogenesis of hypertension [125]. In addition, Grk5 overexpression augments the agonist-induced AT1R or D1R phosphorylation, leading to their desensitization; desensitization of AT1R should favor a lower blood pressure while the opposite is true for D1R and therefore, these two effects by themselves may not lead to a change in blood pressure [38, 41]. Cardiac-specific Grk5 expression decreases basal left ventricle systolic pressure, slows left ventricle relaxation, and increases β-AR desensitization, but has no effect on Ang II-induced contractile response [32]. Thus, defining the role of GRK5 in the regulation of blood pressure needs to be studied further.
GRK5 is also associated with other blood pressure regulation-related cardiovascular diseases such as myocardial infarction or cardiometabolic traits such as hyperlipidemia [119, 126]. Grk5 SNPs are also associated with the race-related therapeutic efficacy of some drugs, including β-adrenergic blockers [127, 128].
GRK6
GRK6 is ubiquitously expressed in mammalian organs and tissues, including the heart and intestines [71, 129,130,131]. There is no direct proof of the role of GRK6 in the regulation of blood pressure. However, there are some reports on the association between GRK6 and hypertension [71]. Compared with control rats, total GRK6 expression is increased, and its distribution is reduced in the intercalated disks but increased in the cytoplasm of cardiac myocytes in spontaneously hypertensive heart failure rats [71]. This may be involved in abnormal remodeling of cardiac myocytes in hypertensive hypertrophy and failure. The cardiomyocytes of GRK6 but not GRK5 knockout mice have an augmented fractional shortening in response to Ang II [132].
Abnormal proliferation of endothelial cells is part of the vascular remodeling in hypertension that may be related to a decrease in vascular endothelial GRK6 expression caused by miR-27a [133]. Cyclic stretching of VSMs, to mimic the pathologically increased stretch in hypertension, causes the transfer of miR-27a via VSMC microparticles to the endothelial cells [133].
As aforementioned, sodium retention, caused by impaired renal sodium excretion, is part of the pathological process in hypertension [134]. An increase in intestinal sodium absorption is also part of the increased sodium balance in hypertension, which may be related to increased activity of NHE3 and Na+-K+-ATPase [135, 136]. Inhibition of GRK6, but not GRK4, prevents the loss of dopamine D1-like receptor-mediated inhibition of Cl/HCO3- exchanger and NHE3 activity in rat intestinal epithelial cells [130, 131]. In addition, GRK6 regulates insulin homeostasis, an abnormality which participates in the pathogenesis of hypertension [137]. GRK6 inhibition increases insulin secretion but reduces insulin processing; Grk6 knockdown reduces cellular insulin levels, and attenuates insulin secretion, but enhances proinsulin secretion consistent with decreased processing [138]. However, the proof of the association between GRK6 and hypertension is still weak and needs further exploration.
Modulation of Grks in the regulation of blood pressure
Most studies have focused on the role of GRKs in the regulation of GPCR expression and activity. However, there are studies reporting on how GRKs are regulated. In fact, there are some studies reporting that GRKs per se are modulated by factors, which subsequently regulate blood pressure. For example, long-term exercise in SHRs, beginning at the prehypertensive stage (four weeks old), improves vascular insulin sensitivity via down-regulation of vascular GRK2, which limits the progression of hypertension [58]. Activation of the TGF-β signaling cascade in VSMCs increases GRK2 expression, consequently inhibiting Ang II-induced VSMC proliferation and migration [139]. We have reported that the expression and activity of GRKs are regulated by several factors, including long-term PM2.5 exposure, prenatal and paternal PM2.5 exposure, in utero lipopolysaccharide or cold stress exposure, and maternal diabetes mellitus [49, 87,88,89, 92, 94].
The expression of GRKs is regulated by transcription factors and signaling molecules. Others and we have reported that renal GRK4 expression is positively regulated by the activation of the transcription factor c-Myc, which binds to the promoter of Grk4 gene [87, 90, 140]. Other transcription factors, such as Foxo1, NF-κB, and cAMP-responsive element binding protein, are also involved in the regulation of transcription of GRKs (GRK2, GRK5, GRK6) [141,142,143]. Some key signaling molecules such as protein kinases participate in the modulation of GRK expression. We reported that inhibition of PKC reduces the increased GRK2 expression and normalizes D1R function in renal proximal tubule cells from diabetic mother offspring, which may be beneficial in the treatment of maternal diabetes mellitus-programmed hypertension [49]. Activation of PKC increases GRK2 protein and activity, causing the dysfunction of endothelium-dependent relaxation to insulin in mouse aorta [144]. By contrast, activation of PKC decreases GRK6 promoter activity and expression [143]. Post-transcriptional regulation, including ubiquitination, also plays an important role in the regulation of GRKs [145, 146].
The regulation of GRK activity or cellular location, independent of its expression, is also important in disease states. Insulin or oxidative stress causes renal D1R desensitization and impairs its ability to inhibit Na+-K+-ATPase activity by promoting GRK2 translocation to the plasma membrane [48, 147]. The increased blood pressure of SHRs is caused, in part, by impaired renal D1R function [148], that is due to increased GRK4 activity and sorting nexins (e.g., SNX5 and SNX19) [15, 149, 150]. SNXs play a vital role in receptor trafficking [151]. SNX5 directly interacts with renal GRK4 and inhibits its ability to phosphorylate and desensitize D1R [149].The nuclear translocation of GRK5 participates in fibroblast activation and cardiac fibrosis; Ang II induces the nuclear translocation of cardiac GRK5 [12]. It should be noted that SNPs of GRKs also affect the activity of GRKs. GRK4 variants can increase the activity of GRK4 [28, 77,78,79, 93]. As aforementioned, increased renal GRK4 activity, caused by GRK4 gene variants, causes hypertension [15, 77, 78, 82, 83]. The GRK4 variant A486V has been shown to increase the impairment of cardiac function after myocardial infarction [28]. A functional GRK2 SNP which inhibits its activity potentiates β-adrenergic receptor-mediated cAMP accumulation and potentially can be used in the treatment of heart failure [152].
Grks and treatment of hypertension
GRK inhibition
Because GRKs play a role in the pathogenesis of cardiovascular diseases, including hypertension, many studies have focused on searching selective inhibitors of GRKs, especially GRK2 and GRK5. Some existing drugs have been verified as potential GRK2 inhibitors. For example, paroxetine directly inhibits the activity of GRK2 by binding its active site which stabilizes the kinase domain [153]. In hypertension, paroxetine increases β1-AR sensitivity and attenuates cardiac hypertrophy by blocking the interaction between GRK2 and β1-AR [154]. The actions of some GRK inhibitors have been related to their crystal structure. For example, based on the conformation of the HJ loop within the X-ray structure of GRK2, cyclic peptide 7 was found to suppress selectively and potently GRK2 activity [155]. The pharmacological inhibition of GRK2 by a cyclic peptide C7 improves cardiac metabolism and function in experimental heart failure [156]. Small molecule GRK inhibitors, including paroxetine, Takeda compound 101, and CCG224063, attenuate GRK2-mediated desensitization of vasoconstrictor-induced arterial contractions, highlighting a potential strategy for blood pressure regulation by targeting GRK2 function [157].
Most of the current studies on the inhibition of GRK function have focused on the direct interaction between the inhibitor and the kinase domain of the protein. However, inhibitors of the RH domain of GRK2 have also been reported to reduce the RH-mediated receptor desensitization and GRK2 translocation to the plasma membrane [158, 159]. These novel inhibitors of phosphorylation-independent actions of GRK2 provide new strategies for the development of innovative pharmacologic therapy for hypertension.
Some studies have also explored the possibility of simultaneous inhibition of two types of GRKs by one inhibitor. For example, analyses of the structures and chemical properties of key residues in the protein crystal structures of GRK2 and GRK5 have determined the specific amino acid distribution within the GRK2/GRK5 target site [160, 161]. CCG215022, a GRK2 inhibitor, has nanomolar IC50 values against both GRK2 and GRK5. A 2.4 Å crystal structure of the GRK5·CCG215022 complex revealed that the inhibitor binds in the active site similarly to its parent compound [162]. The structure and function of GRK4 486V have also been reported; Ser-485 in the kinase C-tail is one of the residues that is responsible for the increased rate of autophosphorylation of GRK4 486V [84]. These are promising reports on the generation of selective GRK inhibitors in the treatment of hypertension.
GRKs and pharmacogenomics of hypertension
Over the past decade, pharmacogenetics has been used to guide cardiovascular drug therapy [163]. The gene variants of GRKs may be important to guide antihypertensive strategies. The African American Study of Kidney Disease and Hypertension Study showed that in African-American men, but not women, the response to the β1-AR blocker metoprolol was more rapid in carriers of GRK4 142V than carriers of GRK4 A142 but the response was decreased in the presence of GRK4 65L [111]. The Pharmacogenomic Evaluation of Antihypertensive Responses trial showed that the response of hypertensive patients to β-blockers is influenced by GRK4 variants. In African-Americans and Caucasians, increasing number of copies of the GRK4 variant 65L-142V haplotype is associated with reduced response to β-blocker monotherapy with atenolol [164]. Moreover, all three GRK4 variants (65L, 142V, and 486V) are associated with increased risk for the primary outcome of first occurrence of all cause death, nonfatal myocardial infarction, or nonfatal stroke in American whites and Hispanics [164]. Another study in hypertensive Japanese showed that patients with hGRK4 142V have a greater decrease in SBP in response to angiotensin receptor blockers (ARBs) than non-carrier subjects. However, those with GRK4 486V are less likely to achieve the blood pressure goal in response to ARBs than those without GRK4 variants [165]. Our study also found that non-dipper circadian rhythm is positively associated with the presence of GRK4 variants. Moreover, patients with these three GRK4 variants have a better antihypertensive response to candesartan, an ARB, than those with GRK4 WT gene [166].
Regarding the antihypertensive effect of diuretics, one study showed that subjects with more than three GRK4 gene variants (A142V, A486V, and R65L) have a greater increase in potassium and sodium excretion in response to hydrochlorothiazide than those with three or less than three GRK4 gene variants [167]. In another study, hypertensive subjects with GRK4 65L and 142V needed more antihypertensive therapy, especially diuretics, to reach the same mean arterial pressure as did the heterozygous and wild-type carriers of GRK4 [168]. Other GRK variants are reported to be associated with therapeutic effect of antihypertensive drugs. African-American carriers of GRK2 rs4930416 have a greater decrease in both SBP and DBP in response to hydrochlorothiazide while European-Americans with GRK2 rs1894111 have a greater decrease in both SBP and DBP in response to hydrochlorothiazide [16]. African-American or European-American carriers of GRK5Gln41, relative to homozygous carriers of GRK5Leu41, do not respond differently to hydrochlorothiazide or atenolol but those with GRK5Leu41 have a 46.5% risk reduction for adverse cardiovascular outcomes compared with GRK5 Gln41 homozygotes, independent of treatment [16].
GRK4 variants are also associated with the blood pressure response to lifestyle modification. For example, Rayner et al. found that in South African Blacks 50-75 years old with mild-to-moderate hypertension, GRK4 polymorphisms, A142V and R65L, can predict blood pressure response to dietary reduction of dietary salt intake. Hypertensive subjects with wild-type GRK4 A142 and GRK4 R65 or heterozygous GRK4 A142V and GRK4 R65L but no homozygous GRK4 142V and GRK4 65L have a reduction in their blood pressures in response to a low salt diet [169].
Limitations of Grks research
There are several limitations in the current GRK research. First, there are only a few reports using organ-specific knockout or overexpression GRK mice, which are better to study the role of GRKs in the regulation of blood pressure. Second, the mechanism by which different GRKs polymorphisms affect enzymatic activity remains still unknown, which may be related with the crystal structure of GRKs polymorphisms. Especially, the reason why different GRK4 variants exhibit different salt-sensitive phenotypes in blood pressure regulation needs to be studied in the future. Third, GRK2-mediated effects in the regulation of blood pressure is complicated, and its role in hypertension needs to be clarified more clearly. Fourth, the regulatory mechanisms of GRK remain unclear.
Conclusions
In summary, increasing pieces of evidence demonstrate that GRKs, by regulating different substrates, play important roles in the regulation of blood pressure. Various genetically modified animal models confirm the key role of GRKs in the regulation of blood pressure (Table 3). Aberrant expression and/or activity of GRKs in the cardiovascular system and kidney participate in the pathogenesis of hypertension. Genetic association studies have found associations between GRK gene variants and hypertension. Moreover, individual GRK variants also affect the response to antihypertensive treatment. Increased understanding of the role of GRKs in the regulation of blood pressure may provide novel insights into the pathogenesis of hypertension and new strategies for the prevention and treatment of hypertension.
References
Brouwers S, Sudano I, Kokubo Y, Sulaica EM. Arterial hypertension. Lancet. 2021;398:249–61.
NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet. 2021;398:957–80.
Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93–e621.
Carey RM, Moran AE, Whelton PK. Treatment of hypertension: a review. JAMA. 2022;328:1849–61.
Grogan A, Lucero EY, Jiang H, Rockman HA. Pathophysiology and pharmacology of G protein-coupled receptors in the heart. Cardiovasc Res. 2023;119:1117–29.
Jiang H, Galtes D, Wang J, Rockman HA. G protein-coupled receptor signaling: transducers and effectors. Am J Physiol Cell Physiol. 2022;323:C731–C748.
Vaz de Castro PAS, Jose PA, Simões E Silva AC. Interactions between the intrarenal dopaminergic and the renin-angiotensin systems in the control of systemic arterial pressure. Clin Sci (Lond). 2022;136:1205–27.
Yang J, Villar VAM, Jose PA, Zeng C. Renal dopamine receptors and oxidative stress: role in hypertension. Antioxid Redox Signal. 2021;34:716–35.
Banday AA, Lokhandwala MF. Transcriptional regulation of renal dopamine D1 receptor function during oxidative stress. Hypertension. 2015;65:1064–72.
Kemp BA, Howell NL, Gildea JJ, Keller SR, Brautigan DL, Carey RM, et al. 2 receptors mediate natriuresis via protein phosphatase PP2A. Circ Res. 2022;130:96–111.
Pfleger J, Gresham K, Koch WJ. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat Rev Cardiol. 2019;16:612–22.
Eguchi A, Coleman R, Gresham K, Gao E, Ibetti J, Chuprun JK, et al. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc Natl Acad Sci USA. 2021;118:e2012854118.
Yang J, Hall JE, Jose PA, Chen K, Zeng C. Comprehensive insights in GRK4 and hypertension: from mechanisms to potential therapeutics. Pharm Ther. 2022;239:108194.
Gros R, Benovic JL, Tan CM, Feldman RD. G-protein-coupled receptor kinase activity is increased in hypertension. J Clin Investig. 1997;99:2087–93.
Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci USA. 2002;99:3872–7.
Lobmeyer MT, Wang L, Zineh I, Turner ST, Gums JG, Chapman AB, et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharmacogenet Genom. 2011;21:42–49.
Yang J, Villar VA, Armando I, Jose PA, Zeng C. G protein-coupled receptor kinases: crucial regulators of blood pressure. J Am Heart Assoc. 2016;5:e003519.
Sulon SM, Benovic JL, Targeting G. protein-coupled receptor kinases (GRKs) to G protein-coupled receptors. Curr Opin Endocr Metab Res. 2021;16:56–65.
Benovic JL. Historical perspective of the G protein-coupled receptor kinase family. Cells. 2021;10:555.
Chaudhary PK, Kim S. The GRKs reactome: role in cell biology and pathology. Int J Mol Sci. 2021;22:3375.
Zhang Y, Wang S, Huang H, Zeng A, Han Y, Zeng C, et al. GRK4-mediated adiponectin receptor-1 phosphorylative desensitization as a novel mechanism of reduced renal sodium excretion in hypertension. Clin Sci. 2020;134:2453–67.
Crudden C, Shibano T, Song D, Dragomir MP, Cismas S, Serly J, et al. Inhibition of G protein-coupled receptor kinase 2 promotes unbiased downregulation of IGF1 receptor and restrains malignant cell growth. Cancer Res. 2021;81:501–14.
Usui I, Imamura T, Babendure JL, Satoh H, Lu JC, Hupfeld CJ, et al. G protein-coupled receptor kinase 2 mediates endothelin-1-induced insulin resistance via the inhibition of both G alpha q/11 and insulin receptor substrate-1 pathways in 3T3-L1 adipocytes. Mol Endocrinol. 2005;19:2760–8.
Martini JS, Raake P, Vinge LE, DeGeorge BR Jr, Chuprun JK, Harris DM, et al. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc Natl Acad Sci USA. 2008;105:12457–62.
Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N. G-protein-coupled-receptor kinases mediate TNFα-induced NFκB signalling via direct interaction with and phosphorylation of IκBα. Biochem J. 2009;425:169–78.
Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharm Ther. 2012;133:40–69.
Yang D, Tang M, Zhang M, Ren H, Li X, Zhang Z, et al. Downregulation of G protein-coupled receptor kinase 4 protects against kidney ischemia-reperfusion injury. Kidney Int. 2023;103:719–34.
Li L, Fu W, Gong X, Chen Z, Tang L, Yang D, et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J. 2021;42:1415–30.
Gerhards J, Maerz LD, Matthees ESF, Donow C, Moepps B, Premont RT, et al. Kinase activity is not required for G protein-coupled receptor kinase 4 restraining mTOR signaling during cilia and kidney development. J Am Soc Nephrol. 2023;34:590–606.
Yang J, Villar VA, Jones JE, Jose PA, Zeng C. G protein-coupled receptor kinase 4: role in hypertension. Hypertension. 2015;65:1148–55.
Avendaño MS, Lucas E, Jurado-Pueyo M, Martínez-Revelles S, Vila-Bedmar R, Mayor F Jr, et al. Increased nitric oxide bioavailability in adult GRK2 hemizygous mice protects against angiotensin II-induced hypertension. Hypertension. 2014;63:369–75.
Rockman HA, Choi DJ, Rahman NU, Akhter SA, Lefkowitz RJ, Koch WJ. Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc Natl Acad Sci USA. 1996;93:9954–9.
Tutunea-Fatan E, Caetano FA, Gros R, Ferguson SSG. GRK2 targeted knock-down results in spontaneous hypertension, and altered vascular GPCR signaling. J Biol Chem. 2015;290:5141–55.
Eckhart AD, Ozaki T, Tevaearai H, Rockman HA, Koch WJ. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Mol Pharm. 2002;61:749–58.
Morris GE, Nelson CP, Standen NB, Challiss RA, Willets JM. Endothelin signalling in arterial smooth muscle is tightly regulated by G protein-coupled receptor kinase 2. Cardiovasc Res. 2010;85:424–33.
Morris GE, Nelson CP, Everitt D, Brighton PJ, Standen NB, Challiss RA, et al. G protein-coupled receptor kinase 2 and arrestin2 regulate arterial smooth muscle P2Y-purinoceptor signalling. Cardiovasc Res. 2011;89:193–203.
Morris GE, Nelson CP, Brighton PJ, Standen NB, Challiss RA, Willets JM. Arrestins 2 and 3 differentially regulate ETA and P2Y2 receptor-mediated cell signaling and migration in arterial smooth muscle. Am J Physiol Cell Physiol. 2012;302:C723–C734.
Oppermann M, Freedman NJ, Alexander RW, Lefkowitz RJ. Phosphorylation of the type 1A angiotensin II receptor by G protein-coupled receptor kinases and protein kinase C. J Biol Chem. 1996;271:13266–72.
Cohn HI, Harris DM, Pesant S, Pfeiffer M, Zhou RH, Koch WJ, et al. Inhibition of vascular smooth muscle G protein-coupled receptor kinase 2 enhances alpha1D-adrenergic receptor constriction. Am J Physiol Heart Circ Physiol. 2008;295:H1695–H1704.
Nash CA, Nelson CP, Mistry R, Moeller-Olsen C, Christofidou E, Challiss RAJ, et al. Differential regulation of β2-adrenoceptor and adenosine A2B receptor signalling by GRK and arrestin proteins in arterial smooth muscle. Cell Signal. 2018;51:86–98.
Tiberi M, Nash SR, Bertrand L, Lefkowitz RJ, Caron MG. Differential regulation of dopamine D1A receptor responsiveness by various G protein-coupled receptor kinases. J Biol Chem. 1996;271:3771–8.
Taguchi K, Bessho N, Hasegawa M, Narimatsu H, Matsumoto T, Kobayashi T. Co-treatment with clonidine and a GRK2 inhibitor prevented rebound hypertension and endothelial dysfunction after withdrawal in diabetes. Hypertens Res. 2018;41:263–74.
Tutunea-Fatan E, Abd-Elrahman KS, Thibodeau JF, Holterman CE, Holleran BJ, Leduc R, et al. GRK2 knockdown in mice exacerbates kidney injury and alters renal mechanisms of blood pressure regulation. Sci Rep. 2018;8:11415.
Dinudom A, Fotia AB, Lefkowitz RJ, Young JA, Kumar S, Cook DI. The kinase Grk2 regulates Nedd4/Nedd4-2-dependent control of epithelial Na+ channels. Proc Natl Acad Sci USA. 2004;101:11886–90.
Pitzer AL, Van Beusecum JP, Kleyman TR, Kirabo A. ENaC in salt-sensitive hypertension: kidney and beyond. Curr Hypertens Rep. 2020;22:69.
Sanchez-Perez A, Kumar S, Cook DI. GRK2 interacts with and phosphorylates Nedd4 and Nedd4-2. Biochem Biophys Res Commun. 2007;359:611–5.
Asghar M, Banday AA, Fardoun RZ, Lokhandwala MF. Hydrogen peroxide causes uncoupling of dopamine D1-like receptors from G proteins via a mechanism involving protein kinase C and G-protein-coupled receptor kinase 2. Free Radic Biol Med. 2006;40:13–20.
Banday AA, Fazili FR, Lokhandwala MF. Insulin causes renal dopamine D1 receptor desensitization via GRK2-mediated receptor phosphorylation involving phosphatidylinositol 3-kinase and protein kinase C. Am J Physiol Ren Physiol. 2007;293:F877–F884.
Luo H, Chen C, Guo L, Xu Z, Peng X, Wang X, et al. Exposure to maternal diabetes mellitus causes renal dopamine D1 receptor dysfunction and hypertension in adult rat offspring. Hypertension. 2018;72:962–70.
Zhu W, Petrashevskaya N, Ren S, Zhao A, Chakir K, Gao E, et al. Gi-biased β2AR signaling links GRK2 upregulation to heart failure. Circ Res. 2012;110:265–74.
Guo S, Carter RL, Grisanti LA, Koch WJ, Tilley DG. Impact of paroxetine on proximal β-adrenergic receptor signaling. Cell Signal. 2017;38:127–33.
Wang Y, Gao E, Lau WB, Wang Y, Liu G, Li JJ, et al. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation. 2015;131:1392–404. Apr 21
Chen X, Zhao S, Xia Y, Xiong Z, Li Y, Tao L, et al. G protein coupled receptor kinase-2 upregulation causes κ-opioid receptor desensitization in diabetic heart. Biochem Biophys Res Commun. 2017;482:658–64.
Pfleger J, Gross P, Johnson J, Carter RL, Gao E, Tilley DG, et al. G protein-coupled receptor kinase 2 contributes to impaired fatty acid metabolism in the failing heart. J Mol Cell Cardiol. 2018;123:108–17.
Liu J, Li X, Ding L, Li W, Niu X, Gao D. GRK2 participation in cardiac hypertrophy induced by isoproterenol through the regulation of Nrf2 signaling and the promotion of NLRP3 inflammasome and oxidative stress. Int Immunopharmacol. 2023;117:109957.
Bledzka KM, Manaserh IH, Grondolsky J, Pfleger J, Roy R, Gao E, et al. A peptide of the amino-terminus of GRK2 induces hypertrophy and yet elicits cardioprotection after pressure overload. J Mol Cell Cardiol. 2021;154:137–53.
Alonazi ASA, Willets JM. G protein-coupled receptor kinase 2 is essential to enable vasoconstrictor-mediated arterial smooth muscle proliferation. Cell Signal. 2021;88:110152.
Xing W, Li Y, Zhang H, Mi C, Hou Z, Quon MJ, et al. Improvement of vascular insulin sensitivity by downregulation of GRK2 mediates exercise-induced alleviation of hypertension in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2013;305:H1111–H1119.
Ciccarelli M, Sorriento D, Franco A, Fusco A, Del Giudice C, Annunziata R, et al. Endothelial G protein-coupled receptor kinase 2 regulates vascular homeostasis through the control of free radical oxygen species. Arterioscler Thromb Vasc Biol. 2013;33:2415–24.
Yano H, Onoue K, Tokinaga S, Ioka T, Ishihara S, Hashimoto Y, et al. Overexpression of GRK2 in vascular smooth muscle leads to inappropriate hypertension and acute heart failure as in clinical scenario 1. Sci Rep. 2023;13:7707.
Zhao Y, Vanhoutte PM, Leung SW. α1-Adrenoceptor activation of PKC-ε causes heterologous desensitization of thromboxane receptors in the aorta of spontaneously hypertensive rats. Br J Pharm. 2015;172:3687–701.
Montó F, Oliver E, Vicente D, Buendía F, Rueda J, Agüero J, et al. β2- and β1-adrenoceptor expression exhibits a common regulatory pattern with GRK2 and GRK5 in human and animal models of cardiovascular diseases. J Cardiovasc Pharm. 2015;66:478–86.
Gros R, Chorazyczewski J, Meek MD, Benovic JL, Ferguson SS, Feldman RD. G-Protein-coupled receptor kinase activity in hypertension : increased vascular and lymphocyte G-protein receptor kinase-2 protein expression. Hypertension. 2000;35:38–42.
Oliver E, Flacco N, Arce C, Ivorra MD, D’Ocon MP, Noguera MA. Changes in adrenoceptors and G-protein-coupled receptor kinase 2 in L-NAME-induced hypertension compared to spontaneous hypertension in rats. J Vasc Res. 2014;51:209–20.
Gros R, Tan CM, Chorazyczewski J, Kelvin DJ, Benovic JL, Feldman RD. G-protein-coupled receptor kinase expression in hypertension. Clin Pharm Ther. 1999;65:545–51.
Izzo R, Cipolletta E, Ciccarelli M, Campanile A, Santulli G, Palumbo G, et al. Enhanced GRK2 expression and desensitization of beta AR vasodilatation in hypertensive patients. Clin Transl Sci. 2008;1:215–20.
Cohn HI, Xi Y, Pesant S, Harris DM, Hyslop T, Falkner B, et al. G protein-coupled receptor kinase 2 expression and activity are associated with blood pressure in Black Americans. Hypertension. 2009;54:71–76.
Li Y, Li N, Yao X, Heizati M, Zhang D, Zhu Q, et al. Association between polymorphisms of ADRBK1 gene and plasma renin activity in hypertensive patients: a case-control study. Med Sci Monit. 2016;22:2981–8.
Napolitano R, Campanile A, Sarno L, Anastasio A, Maruotti GM, Morlando M, et al. GRK2 levels in umbilical arteries of pregnancies complicated by gestational hypertension and preeclampsia. Am J Hypertens. 2012;25:366–71.
Lv Z, Xiong LL, Qin X, Zhang H, Luo X, Peng W, et al. Role of GRK2 in trophoblast necroptosis and spiral artery remodeling: implications for preeclampsia pathogenesis. Front Cell Dev Biol. 2021;9:694261.
Yi XP, Zhou J, Baker J, Wang X, Gerdes AM, Li F. Myocardial expression and redistribution of GRKs in hypertensive hypertrophy and failure. Anat Rec A Discov Mol Cell Evol Biol. 2005;282:13–23.
Oliver E, Rovira E, Montó F, Valldecabres C, Julve R, Muedra V, et al. beta-Adrenoceptor and GRK3 expression in human lymphocytes is related to blood pressure and urinary albumin excretion. J Hypertens. 2010;28:1281–9.
Vinge LE, von Lueder TG, Aasum E, Qvigstad E, Gravning JA, How OJ, et al. Cardiac-restricted expression of the carboxyl-terminal fragment of GRK3 uncovers distinct functions of GRK3 in regulation of cardiac contractility and growth: GRK3 controls cardiac alpha1-adrenergic receptor responsiveness. J Biol Chem. 2008;283:10601–10.
von Lueder TG, Gravning J, How OJ, Vinge LE, Ahmed MS, Krobert KA, et al. Cardiomyocyte-restricted inhibition of G protein-coupled receptor kinase-3 attenuates cardiac dysfunction after chronic pressure overload. Am J Physiol Heart Circ Physiol. 2012;303:H66–H74.
Sedaghat K, Tiberi M. Cytoplasmic tail of D1 dopaminergic receptor differentially regulates desensitization and phosphorylation by G protein-coupled receptor kinase 2 and 3. Cell Signal. 2011;23:180–92.
Maimari T, Krasel C, Bünemann M, Lorenz K. The N-termini of GRK2 and GRK3 simulate the stimulating effects of RKIP on β-adrenoceptors. Biochem Biophys Res Commun. 2019;520:327–32.
Chen K, Fu C, Chen C, Liu L, Ren H, Han Y, et al. Role of GRK4 in the regulation of arterial AT1 receptor in hypertension. Hypertension. 2014;63:289–96.
Wang Z, Zeng C, Villar VA, Chen SY, Konkalmatt P, Wang X, et al. Human GRK4γ142V variant promotes AT1R-mediated hypertension via renal HDAC1 inhibition. Hypertension. 2016;67:325–34.
Sanada H, Yatabe J, Midorikawa S, Katoh T, Hashimoto S, Watanabe T, et al. Amelioration of genetic hypertension by suppression of renal G protein-coupled receptor kinase type 4 expression. Hypertension. 2006;47:1131–9.
Yatabe J, Sanada H, Midorikawa S, Hashimoto S, Watanabe T, Andrews PM, et al. Effects of decreased renal cortical expression of G protein-coupled receptor kinase 4 and angiotensin type 1 receptors in rats. Hypertens Res. 2008;31:1455–64.
Huang H, Li X, Zheng S, Chen Y, Chen C, Wang J, et al. Downregulation of renal G protein-coupled receptor kinase type 4 expression via ultrasound-targeted microbubble destruction lowers blood pressure in spontaneously hypertensive rats. J Am Heart Assoc. 2016;5:e004028.
Yang Y, Li M, Zou X, Chen C, Zheng S, Fu C, et al. Role of GRK4 in the regulation of the renal ETB receptor in hypertension. FASEB J. 2020;34:11594–604.
Diao Z, Asico LD, Villar VAM, Zheng X, Cuevas S, Armando I, et al. Increased renal oxidative stress in salt-sensitive human GRK4γ486V transgenic mice. Free Radic Biol Med. 2017;106:80–90.
Allen SJ, Parthasarathy G, Darke PL, Diehl RE, Ford RE, Hall DL, et al. Structure and function of the hypertension variant A486V of G protein-coupled receptor kinase 4. J Biol Chem. 2015;290:20360–73.
Rankin ML, Marinec PS, Cabrera DM, Wang Z, Jose PA, Sibley DR. The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol Pharm. 2006;69:759–69.
Watanabe H, Xu J, Bengra C, Jose PA, Felder RA. Desensitization of human renal D1 dopamine receptors by G protein-coupled receptor kinase 4. Kidney Int. 2002;62:790–8.
Ye Z, Lu X, Deng Y, Wang X, Zheng S, Ren H, et al. In Utero exposure to fine particulate matter causes hypertension due to impaired renal dopamine D1 receptor in offspring. Cell Physiol Biochem. 2018;46:148–59.
Wang X, Luo H, Chen C, Chen K, Wang J, Cai Y, et al. Prenatal lipopolysaccharide exposure results in dysfunction of the renal dopamine D1 receptor in offspring. Free Radic Biol Med. 2014;76:242–50.
Sun D, Chen K, Wang J, Zhou L, Zeng C. In-utero cold stress causes elevation of blood pressure via impaired vascular dopamine D1 receptor in offspring. Clin Exp Hypertens. 2020;42:99–104.
Tao Y, Luo W, Chen Y, Chen C, Chen S, Li X, et al. Exercise ameliorates skeletal muscle insulin resistance by modulating GRK4-mediated D1R expression. Clin Sci. 2023;137:1391–407.
Villar VA, Jones JE, Armando I, Palmes-Saloma C, Yu P, Pascua AM, et al. G protein-coupled receptor kinase 4 (GRK4) regulates the phosphorylation and function of the dopamine D3 receptor. J Biol Chem. 2009;284:21425–34.
Hu C, Tao Y, Deng Y, Cai Q, Ren H, Yu C, et al. Paternal long-term PM2.5 exposure causes hypertension via increased renal AT1R expression and function in male offspring. Clin Sci (Lond). 2021;135:2575–88.
Zhang F, Lei L, Huang J, Wang W, Su Q, Yan H, et al. G-protein-coupled receptor kinase 4 causes renal angiotensin II type 2 receptor dysfunction by increasing its phosphorylation. Clin Sci. 2022;136:989–1003.
Lu X, Ye Z, Zheng S, Ren H, Zeng J, Wang X, et al. Long-term exposure of fine particulate matter causes hypertension by impaired renal D1 receptor-mediated sodium excretion via upregulation of G-protein-coupled receptor kinase type 4 expression in Sprague-Dawley rats. J Am Heart Assoc. 2018;7:e007185.
Chugh G, Lokhandwala MF, Asghar M. Altered functioning of both renal dopamine D1 and angiotensin II type 1 receptors causes hypertension in old rats. Hypertension. 2012;59:1029–36.
Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, et al. Renal dopaminergic defect in C57Bl/6J mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1660–R1669.
Jones ES, Spence JD, Mcintyre AD, Nondi J, Gogo K, Akintunde A, et al. High frequency of variants of candidate genes in Black Africans with low renin-resistant hypertension. Am J Hypertens. 2017;30:478–83.
Kimura L, Angeli CB, Auricchio MT, Fernandes GR, Pereira AC, Vicente JP, et al. Multilocus family-based association analysis of seven candidate polymorphisms with essential hypertension in an African-derived semi-isolated Brazilian population. Int J Hypertens. 2012;2012:859219.
Sanada H, Yatabe J, Midorikawa S, Hashimoto S, Watanabe T, Moore JH, et al. Single-nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin Chem. 2006;52:352–60.
Gu D, Su S, Ge D, Chen S, Huang J, Li B, et al. Association study with 33 single-nucleotide polymorphisms in 11 candidate genes for hypertension in Chinese. Hypertension. 2006;47:1147–54.
Speirs HJ, Katyk K, Kumar NN, Benjafield AV, Wang WY, Morris BJ. Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J Hypertens. 2004;22:931–6.
Bengra C, Mifflin TE, Khripin Y, Manunta P, Williams SM, Jose PA, et al. Genotyping of essential hypertension single-nucleotide polymorphisms by a homogeneous PCR method with universal energy transfer primers. Clin Chem. 2002;48:2131–40.
Jeong S, Kim JY, Cho Y, Koh SB, Kim N, Choi JR. Genetically, dietary sodium intake is causally associated with salt-sensitive hypertension risk in a community-based cohort study: a mendelian randomization approach. Curr Hypertens Rep. 2020;22:45.
Jiang W, Wang X, Li R, Wang P, Shan G, Jia X, et al. Targeted capture sequencing identifies genetic variations of GRK4 and RDH8 in Han Chinese with essential hypertension in Xinjiang. PLoS One. 2021;16:e0255311.
Du B, Jia X, Tian W, Yan X, Wang N, Cai D, et al. Associations of SUCNR1, GRK4, CAMK1D gene polymorphisms and the susceptibility of type 2 diabetes mellitus and essential hypertension in a northern Chinese Han population. J Diabetes Complic. 2021;35:107752.
Zhang H, Sun ZQ, Liu SS, Yang LN. Association between GRK4 and DRD1 gene polymorphisms and hypertension: a meta-analysis. Clin Int Aging. 2015;11:17–27.
Carey RM, Schoeffel CD, Gildea JJ, Jones JE, McGrath HE, Gordon LN, et al. Salt sensitivity of blood pressure is associated with polymorphisms in the sodium-bicarbonate cotransporter. Hypertension. 2012;60:1359–66.
Williams SM, Ritchie MD, Phillips JA 3rd, Dawson E, Prince M, Dzhura E, et al. Multilocus analysis of hypertension: a hierarchical approach. Hum Hered. 2004;57:28–38.
Liu C, Xi B. Pooled analyses of the associations of polymorphisms in the GRK4 and EMILIN1 genes with hypertension risk. Int J Med Sci. 2012;9:274–9.
Zilbermint M, Hannah-Shmouni F, Stratakis CA. Genetics of hypertension in African Americans and others of African descent. Int J Mol Sci. 2019;20:1081.
Bhatnagar V, O’Connor DT, Brophy VH, Schork NJ, Richard E, Salem RM, et al. G-protein-coupled receptor kinase 4 polymorphisms and blood pressure response to metoprolol among African Americans: sex-specificity and interactions. Am J Hypertens. 2009;22:332–8.
Martinez Cantarin MP, Ertel A, Deloach S, Fortina P, Scott K, Burns TL, et al. Variants in genes involved in functional pathways associated with hypertension in African Americans. Clin Transl Sci. 2010;3:279–86.
Lohmueller KE, Wong LJ, Mauney MM, Jiang L, Felder RA, Jose PA, et al. Patterns of genetic variation in the hypertension candidate gene GRK4: ethnic variation and haplotype structure. Ann Hum Genet. 2006;70:27–41.
Staessen JA, Kuznetsova T, Zhang H, Maillard M, Bochud M, Hasenkamp S, et al. Blood pressure and renal sodium handling in relation to genetic variation in the DRD1 promoter and GRK4. Hypertension. 2008;51:1643–50.
Cheng CF, Lin YJ, Lin MC, Liang WM, Chen CC, Chen CH, et al. Genetic risk score constructed from common genetic variants is associated with cardiovascular disease risk in type 2 diabetes mellitus. J Gene Med. 2021;23:e3305.
Lee M, Kim MK, Kim SM, Park H, Park CG, Park HK. Gender-based differences on the association between salt-sensitive genes and obesity in Korean children aged between 8 and 9 years. PLoS One. 2015;10:e0120111.
Zhu H, Lu Y, Wang X, Snieder H, Treiber FA, Harshfield GA, et al. The G protein-coupled receptor kinase 4 gene modulates stress-induced sodium excretion in Black normotensive adolescents. Pediatr Res. 2006;60:440–2.
Marzano F, Liccardo D, Elia A, Mucio I, de Lucia C, Lucchese AM, et al. Genetic catalytic inactivation of GRK5 impairs cardiac function in mice via dysregulated P53 levels. JACC Basic Transl Sci. 2022;7:366–80.
de Lucia C, Grisanti LA, Borghetti G, Piedepalumbo M, Ibetti J, Lucchese AM, et al. G protein-coupled receptor kinase 5 (GRK5) contributes to impaired cardiac function and immune cell recruitment in post-ischemic heart failure. Cardiovasc Res. 2022;118:169–83.
Xu B, Li M, Wang Y, Zhao M, Morotti S, Shi Q, et al. GRK5 controls SAP97-dependent cardiotoxic β1 adrenergic receptor-CaMKII signaling in heart failure. Circ Res. 2020;127:796–810.
Ishizaka N, Alexander RW, Laursen JB, Kai H, Fukui T, Oppermann M, et al. G protein-coupled receptor kinase 5 in cultured vascular smooth muscle cells and rat aorta. Regulation by angiotensin II and hypertension. J Biol Chem. 1997;272:32482–8.
Keys JR, Zhou RH, Harris DM, Druckman CA, Eckhart AD. Vascular smooth muscle overexpression of G protein-coupled receptor kinase 5 elevates blood pressure, which segregates with sex and is dependent on Gi-mediated signaling. Circulation. 2005;112:1145–53.
Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension. 2010;56:696–704.
Wang L, Shen M, Wang F, Ma L. GRK5 ablation contributes to insulin resistance. Biochem Biophys Res Commun. 2012;429:99–104.
da Silva AA, do Carmo JM, Li X, Wang Z, Mouton AJ, Hall JE. Role of hyperinsulinemia and insulin resistance in hypertension: metabolic syndrome revisited. Can J Cardiol. 2020;36:671–82.
Lutz SZ, Falcenberg M, Machicao F, Peter A, Kächele M, Randrianarisoa E, et al. Single nucleotide polymorphisms in the G-protein coupled receptor kinase 5 (GRK5) gene are associated with plasma LDL-cholesterol levels in humans. Sci Rep. 2018;8:7745.
Kang S, Hong X, Ruan CW, Yu P, Yu SS, Chen M, et al. Effects of GRK5 and ADRB1 polymorphisms influence on systolic heart failure. J Transl Med. 2015;13:44.
Cresci S, Kelly RJ, Cappola TP, Diwan A, Dries D, Kardia SL, et al. Clinical and genetic modifiers of long-term survival in heart failure. J Am Coll Cardiol. 2009;54:432–44.
Stegen M, Frey UH. The role of G protein-coupled receptor kinase 6 regulation in inflammation and pain. Int J Mol Sci. 2022;23:15880.
Fraga S, Luo Y, Jose P, Zandi-Nejad K, Mount DB, Soares-da-Silva P. Dopamine D1-like receptor-mediated inhibition of Cl/HCO3- exchanger activity in rat intestinal epithelial IEC-6 cells is regulated by G protein-coupled receptor kinase 6 (GRK 6). Cell Physiol Biochem. 2006;18:347–60.
Fraga S, Jose PA, Soares-da-Silva P. Involvement of G protein-coupled receptor kinase 4 and 6 in rapid desensitization of dopamine D1 receptor in rat IEC-6 intestinal epithelial cells. Am J Physiol Regul Integr Comp Physiol. 2004;287:R772–R779.
Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–9.
Wang L, Bao H, Wang KX, Zhang P, Yao QP, Chen XH, et al. Secreted miR-27a induced by cyclic stretch modulates the proliferation of endothelial cells in hypertension via GRK6. Sci Rep. 2017;7:41058.
Soliman RH, Pollock DM. Circadian control of sodium and blood pressure regulation. Am J Hypertens. 2021;34:1130–42.
Palaniappan B, Arthur S, Sundaram VL, Butts M, Sundaram S, Mani K, et al. Inhibition of intestinal villus cell Na/K-ATPase mediates altered glucose and NaCl absorption in obesity-associated diabetes and hypertension. FASEB J. 2019;33:9323–33.
Nwia SM, Li XC, Leite APO, Hassan R, Zhuo JL. The Na+/H+ Exchanger 3 in the intestines and the proximal tubule of the kidney: localization, physiological function, and key roles in angiotensin II-induced hypertension. Front Physiol. 2022;13:861659.
Sakr HF, Sirasanagandla SR, Das S, Bima AI, Elsamanoudy AZ. Insulin resistance and hypertension: mechanisms involved and modifying factors for effective glucose control. Biomedicines. 2023;11:2271.
Varney MJ, Steyaert W, Coucke PJ, Delanghe JR, Uehling DE, Joseph B, et al. G protein-coupled receptor kinase 6 (GRK6) regulates insulin processing and secretion via effects on proinsulin conversion to insulin. J Biol Chem. 2022;298:102421.
Guo J, Chen H, Ho J, Mancini J, Sontag T, Laporte SA, et al. TGFbeta-induced GRK2 expression attenuates Ang II-regulated vascular smooth muscle cell proliferation and migration. Cell Signal. 2009;21:899–905.
Gildea JJ, Tran HT, Van Sciver RE, Bigler Wang D, Carlson JM, Felder RA. A novel role for c-Myc in G protein-coupled receptor kinase 4 (GRK4) transcriptional regulation in human kidney proximal tubule cells. Hypertension. 2013;61:1021–7.
Yang M, Lin Y, Wang Y, Wang Y. High-glucose induces cardiac myocytes apoptosis through Foxo1/GRK2 signaling pathway. Biochem Biophys Res Commun. 2019;513:154–8.
Islam KN, Koch WJ. Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J Biol Chem. 2012;287:12771–8.
Stegen M, Engler A, Ochsenfarth C, Manthey I, Peters J, Siffert W, et al. Characterization of the G protein-coupled receptor kinase 6 promoter reveals a functional CREB binding site. PLoS One. 2021;16:e0247087.
Taguchi K, Kobayashi T, Matsumoto T, Kamata K. Dysfunction of endothelium-dependent relaxation to insulin via PKC-mediated GRK2/Akt activation in aortas of ob/ob mice. Am J Physiol Heart Circ Physiol. 2011;301:H571–H583.
Penela P, Ruiz-Gómez A, Castaño JG, Mayor F Jr. Degradation of the G protein-coupled receptor kinase 2 by the proteasome pathway. J Biol Chem. 1998;273:35238–44.
Zha Z, Han XR, Smith MD, Lei QY, Guan KL, Xiong Y. Hypertension-associated C825T polymorphism impairs the function of Gβ3 to target GRK2 ubiquitination. Cell Discov. 2016;2:16005.
Banday AA, Lokhandwala MF. Oxidative stress reduces renal dopamine D1 receptor-Gq/11alpha G protein-phospholipase C signaling involving G protein-coupled receptor kinase 2. Am J Physiol Ren Physiol. 2007;293:F306–F315.
Yu P, Asico LD, Luo Y, Andrews P, Eisner GM, Hopfer U, et al. D1 dopamine receptor hyperphosphorylation in renal proximal tubules in hypertension. Kidney Int. 2006;70:1072–9.
Villar VA, Armando I, Sanada H, Frazer LC, Russo CM, Notario PM, et al. Novel role of sorting nexin 5 in renal D1 dopamine receptor trafficking and function: implications for hypertension. FASEB J. 2013;27:1808–19.
Tiu AC, Yang J, Asico LD, Konkalmatt P, Zheng X, Cuevas S, et al. Lipid rafts are required for effective renal D1 dopamine receptor function. FASEB J. 2020;34:6999–7017.
Yang J, Villar VAM, Rozyyev S, Jose PA, Zeng C. The emerging role of sorting nexins in cardiovascular diseases. Clin Sci. 2019;133:723–37.
Okawa T, Aramaki Y, Yamamoto M, Kobayashi T, Fukumoto S, Toyoda Y, et al. Design, synthesis, and evaluation of the highly selective and potent G-protein-coupled receptor kinase 2 (GRK2) inhibitor for the potential treatment of heart failure. J Med Chem. 2017;60:6942–90.
Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, et al. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol. 2012;7:1830–9.
Sun X, Zhou M, Wen G, Huang Y, Wu J, Peng L, et al. Paroxetine attenuates cardiac hypertrophy via blocking GRK2 and ADRB1 interaction in hypertension. J Am Heart Assoc. 2021;10:e016364.
Carotenuto A, Cipolletta E, Gomez-Monterrey I, Sala M, Vernieri E, Limatola A, et al. Design, synthesis and efficacy of novel G protein-coupled receptor kinase 2 inhibitors. Eur J Med Chem. 2013;69:384–92.
Ciccarelli M, Sorriento D, Fiordelisi A, Gambardella J, Franco A, Del Giudice C, et al. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020;7:1571–84.
Rainbow RD, Brennan S, Jackson R, Beech AJ, Bengreed A, Waldschmidt HV, et al. Small-molecule G protein-coupled receptor kinase inhibitors attenuate G protein-coupled receptor kinase 2-mediated desensitization of vasoconstrictor-induced arterial contractions. Mol Pharm. 2018;94:1079–91.
Echeverría E, Velez Rueda AJ, Cabrera M, Juritz E, Burghi V, Fabián L, et al. Identification of inhibitors of the RGS homology domain of GRK2 by docking-based virtual screening. Life Sci. 2019;239:116872.
Echeverría E, Ripoll S, Fabián L, Shayo C, Monczor F, Fernández NC. Novel inhibitors of phosphorylation independent activity of GRK2 modulate cAMP signaling. Pharm Res Perspect. 2022;10:e00913.
Waldschmidt HV, Bouley R, Kirchhoff PD, Lee P, Tesmer JJG, Larsen SD. Utilizing a structure-based docking approach to develop potent G protein-coupled receptor kinase (GRK) 2 and 5 inhibitors. Bioorg Med Chem Lett. 2018;28:1507–15.
Wu Y, Wang S, Wang H, Hu B, Wang J. Selectivity mechanism of GRK2/5 inhibition through in silico investigation. Comput Biol Chem. 2022;101:107786.
Homan KT, Waldschmidt HV, Glukhova A, Cannavo A, Song J, Cheung JY, et al. Crystal structure of G protein-coupled receptor kinase 5 in complex with a rationally designed inhibitor. J Biol Chem. 2015;290:20649–59.
Duarte JD, Cavallari LH. Pharmacogenetics to guide cardiovascular drug therapy. Nat Rev Cardiol. 2021;18:649–65.
Vandell AG, Lobmeyer MT, Gawronski BE, Langaee TY, Gong Y, Gums JG, et al. G protein receptor kinase 4 (RK4) polymorphisms: β-blocker pharmacogenetics and treatment-related outcomes in hypertension. Hypertension. 2012;60:957–64.
Sanada H, Yoneda M, Yatabe J, Williams SM, Bartlett J, White MJ, et al. Common variants of the G protein-coupled receptor type 4 are associate ed with human essential hypertension and predict the blood pressure response to angiotensin receptor blockade. Pharmacogenom J. 2016;16:3–9.
Cao N, Tang H, Tian M, Gong X, Xu Z, Zhou B, et al. Genetic variants of GRK4 influence circadian rhythm of blood pressure and response to candesartan in hypertensive patients. Clin Exp Hypertens. 2021;43:597–603.
Wagner F, Malice MP, Wiegert E, McGrath HE, Gildea J, Mitta S, et al. A comparison of the natriuretic and kaliuretic effects of cicletanine and hydrochlorothiazide in prehypertensive and hypertensive humans. J Hypertens. 2012;30:819–27.
Muskalla AM, Suter PM, Saur M, Nowak A, Hersberger M, Krayenbuehl PA. G-protein receptor kinase 4 polymorphism and response to antihypertensive therapy. Clin Chem. 2014;60:1543–8.
Rayner B, Ramesar R, Steyn K, Levitt N, Lombard C, Charlton K. G-protein-coupled receptor kinase 4 polymorphisms predict blood pressure response to dietary modification in Black patients with mild-to-moderate hypertension. J Hum Hypertens. 2012;26:334–9.
Xia X, Zeng Y, Li Z, Luo H, Wang W, He Y, et al. Effect of GRK4 on renal gastrin receptor regulation in hypertension. Clin Exp Hypertens. 2023;45:2245580.
Wang Y, Li B, Zhao W, Liu P, Zhao Q, Chen S, et al. Association study of G protein-coupled receptor kinase 4 gene variants with essential hypertension in northern Han Chinese. Ann Hum Genet. 2006;70:778–83.
Montasser ME, Shimmin LC, Gu D, Chen J, Gu C, Kelly TN, et al. Variation in genes that regulate blood pressure are associated with glomerular filtration rate in Chinese. PLoS One. 2014;9:e92468.
Frey MK, Dao F, Olvera N, Konner JA, Dickler MN, Levine DA. Genetic predisposition to bevacizumab-induced hypertension. Gynecol Oncol. 2017;147:621–5.
Yaqub DA, Moore SC, Rozyyev S, Konkalmatt P, Asico LD, Hunt J, et al. G protein-coupled receptor kinase 4 (GRK4) variant 65L-mediated impairment of dopamine D1 receptor function causes hypertension. FASEB J. 2021;35:03664.
Rozyyev S, Konkalmatt P, Villar VA, Asico LD, Kumar M, Hunt J, et al. Molecular mechanisms underlying the GRK4 65L-mediated hypertension in mice. FASEB J. 2019;33:597.2.
Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, et al. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38:291–303.
Kavelaars A, Vroon A, Raatgever RP, Fong AM, Premont RT, Patel DD, et al. Increased acute inflammation, leukotriene B4-induced chemotaxis, and signaling in mice deficient for G protein-coupled receptor kinase 6. J Immunol. 2003;171:6128–34.
Funding
This work was supported in part by grants from National Natural Science Foundation of China (82070442, 82370435), Program of Chongqing Medical University for Youth Innovation in Future Medicine (W0085), Project of Chongqing Medical Talent Studio (2022), Natural Science Foundation Project of Chongqing (2023NSCQ-MSX1358), Key Program of The Third Affiliated Hospital of Chongqing Medical University (KY22037), and the National Institutes of Health (P01HL074940, DK039308, DK119652, and DK134574).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
PAJ, who is the Scientific Director of Hypogen, Inc., owns US Patent Number 6660474B1 for GRK4. The other authors report no conflicts.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, F., Armando, I., Jose, P.A. et al. G protein-coupled receptor kinases in hypertension: physiology, pathogenesis, and therapeutic targets. Hypertens Res (2024). https://doi.org/10.1038/s41440-024-01763-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41440-024-01763-y
- Springer Nature Singapore Pte Ltd.