
Abstract
The evolutionary history of species is a dynamic process as they modify, expand, and contract their spatial distributions over time. Range expansions (REs) occur through a series of founder events that are followed by migration among neighboring demes. The process usually results in structured metapopulations and leaves a distinct signature in the genetic variability of species. Explicitly modeling the consequences of complex demographic events such as REs is computationally very intensive. Here we propose an an alternative approach that requires less computational effort than a comprehensive RE model, but that can recover the demography of species undergoing a RE, by combining spatially explicit modelling with simplified but realistic metapopulation models. We examine the demographic and colonization history of Carcharhinus melanopterus, an abundant reef-associated shark, as a test case. We first used a population genomics approach to statistically confirm the occurrence of a RE in C. melanopterus, and identify its origin in the Indo-Australian Archipelago. Spatial genetic modelling identified two waves of stepping-stone colonization: an eastward wave moving through the Pacific and a westward one moving through the Indian Ocean. We show that metapopulation models best describe the demographic history of this species and that not accounting for this may lead to incorrectly interpreting the observed genetic variation as signals of widespread population bottlenecks. Our study highlights insights that can be gained about demography by coupling metapopulation models with spatial modeling and underscores the need for cautious interpretation of population genetic data when advancing conservation priorities.
Similar content being viewed by others
Introduction
Range expansions (REs) have likely occurred several times throughout the evolutionary history of many species, both as a consequence of environmental changes and as instances of invasion processes. While there is a growing interest in the genetic consequences of REs (Excoffier et al. 2009; Mona 2017; Mona et al. 2014), few empirical studies have explored REs quantitatively (but see Barbujani et al. 1995; Francois et al. 2008; Gaggiotti et al. 2009; Hamilton et al. 2005; Neuenschwander et al. 2008; Potter et al. 2016; Ray et al. 2005; Schneider et al. 2010). Rather, empirical population geneticists frequently employ simplistic population models, without concern that they may yield misleading inferences. For example, contemporary population genetics studies often use patterns of genetic variation to identify populations that are deemed to be in need of conservation intervention because they are at risk of extinction. But patterns of genetic variation that might superficially look as though they are the consequence of a population bottleneck in a panmictic population can be indistinguishable from those seen in demes that are part of a demographically stable structured metapopulation (Chikhi et al. 2018; Mazet et al. 2015; 2016). Unstructured panmictic models are widely used in empirical population genetics simply because they are computationally straightforward to implement and do not require detailed knowledge about the ecology and population structure of the study organism. However, they often lead to inaccurate inferences that can be misleading. Explicitly modeling the consequences of a RE is possible, but it requires detecting the timing and location of its origin(s) and estimating associated demographic parameters of each of the constituent demes, as well as migration rates among them (m). While considerable effort has been devoted to developing methods to identify the origin of a RE (He et al. 2017; Peter and Slatkin 2013; Ramachandran et al. 2005), estimating demographic parameters in the constituent demes remains challenging due to a lack of available analytical procedures. Comprehensive simulations coupled with approximate Bayesian computation can offer solutions (Mona 2017; Neuenschwander et al. 2008), but they still require detailed knowledge of the ecology and the distribution of the target species, which is usually unknown.
In this paper, we suggest an alternative approach that requires less computational effort than a comprehensive RE model, but that can recover the demography of species undergoing a RE, by combining spatially explicit modelling with statistical comparisons of realistic metapopulation models. Importantly, the method can be used to identify and correct some of the biases that arise as a consequence of neglecting the occurrence of a RE when inferring the historical demography of a species under models that assume panmixia. We apply the method in an empirical setting to reconstruct the pattern of colonization and demographic history of blacktip reef sharks, Carcharhinus melanopterus (Quoy and Gaimard 1824), a species that is associated with coral reefs throughout the Indian and Pacific Oceans.
Carcharhinus melanopterus is considered “Near Threatened” according to the International Union for the Conservation of Nature (IUCN) Red List of threatened species criteria. Population bottlenecks have been reported in some regions (Vignaud et al. 2014), but the species is known to be locally abundant in other parts of its range. Blacktip reef sharks have small home ranges. They exhibit strong site fidelity and restricted movements that appear to be closely tied to the distribution of available coral reef habitat (Papastamatiou et al. 2009; 2010; Stevens 1984). Their patterns of movement are consistent with the high levels of genetic population structure that have been reported previously (Maisano Delser et al. 2016; Vignaud et al. 2014) and indicate that long distance movements over oceanic expanses are uncommon. Accordingly, their demography is consistent with a metapopulation model that incorporates sub-population structure with occasional exchange of migrants among demes. A RE has previously been proposed to have occurred in this species, but has not yet been formally tested (Maisano Delser et al. 2016).
We combined a worldwide sampling design (partially overlapping with that of Vignaud et al. 2014 (Fig. 1) with the target gene capture and NGS approach described in Maisano Delser et al. 2016 to assemble a single nucleotide polymorphism (SNP) dataset with geographic representation across the distribution of C. melanopterus. We first tested for the signatures of a RE and then characterized it by estimating its center of origin using a spatially explicit framework (Peter and Slatkin 2013; Ramachandran et al. 2005). We used an Approximate Bayesian Computation (ABC; Beaumont et al. 2002) approach that compares unstructured versus computationally tractable, realistic metapopulation models. Here metapopulation is used in a broad sense, to signify a network of habitat patches in which species occur as discrete local populations connected by migration (Hanski 1998). Two simplifications are adopted here: (i) RE is approximated by analyzing the metapopulation at a smaller geographical scale (i.e., without considering the whole distribution of C. melanopterus in a single model); (ii) colonization of the array of demes is considered instantaneous (similar to Hamilton et al. 2005). The first simplification allows for regional differences: the pattern of connectivity does not have to be the same for all geographic areas where populations have been sampled. A full RE model fitting the whole dataset would certainly be the best option to take such differences into account, but it would come at the expense of computational tractability and the risk of over parameterization.

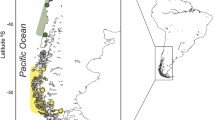
Distribution range of C. melanopterus. Distribution range of C. melanopterus is shown in light gray, sampling locations are in black. Population acronyms are reported in Table S1
We first estimated variation in the effective population size through time assuming an unstructured panmictic population model using an ABC-skyline approach (Maisano Delser et al. 2016). We then compared the posterior probability of this model against two structured metapopulation models: a finite island model and a non-equilibrium stepping-stone model as a proxy of a RE. We demonstrate that the structured metapopulation models were more consistent with the data, suggesting that reductions in population size detected using the unstructured model were the consequence of inadequately accounting for population structure. We also found that the demography inferred in the sampled population of C. melanopterus reflects the pattern of habitat availability, which, in turn, influences migration patterns, rather than local change in effective population size. We rejected previous claims of strong and recent population bottlenecks in some C. melanopterus population from French Polynesia (Vignaud et al. 2014) and instead show that the signatures obtained were due to locally restricted genetic exchanges between populations.
Materials and methods
Samples
A total of 140 samples of C. melanopterus were examined. These were collected from 11 different locations (Fig. 1): the Red Sea (N = 14), the Seychelles (N = 14), Western Australia (N = 14), the Great Barrier Reef (Australia, N = 9), Chesterfield (N = 11), Noumea (New Caledonia, N = 10), Kiribati (N = 9), Moorea (N = 15), Tetiaroa (N = 15), Fakahina (N = 15), and Vahanga (N = 14). In addition, previously collected data (Maisano Delser et al. 2016) from samples from Queensland (N = 5) and the Northern Territory (N = 6), Australia, were added to the dataset, bringing the total number of samples to 151 specimens from 13 locations (Table S1).
Bioinformatics pipeline
Briefly, sequence read data from several individuals was used to build a haploid reference sequence for the 1077 target exons and associated introns. Variant calling and filtering was conducted following (Corrigan et al. 2017; Maisano Delser et al. 2016) and details are reported in the SI.
Characterizing a range expansion in C. melanopterus
Heterozygosity, nucleotide diversity, pairwise, and global Hudson’s FST (Hudson et al. 1992) were calculated in arlsumstat (Excoffier and Lischer 2010), vcftools v 0.1.13 (Danecek et al. 2011) and using custom R scripts with the library “PopGenome” (Pfeifer et al. 2014), respectively. Principal Component Analysis (PCA) was performed with the function “prcomp” in the R environment (RCoreTeam 2014). A Mantel test was performed to determine the correlation between geographic and genetic distance. First, we computed simple geodesic distances between the sampled populations and correlated them with the Hudson’s FST pairwise matrix. Geodesic distances do not take barriers to dispersal into account and do not model local environmental features, such as the presence or absence of favorable habitat and suitable pathways for dispersal. Carcharhinus melanopterus has been shown to exhibit strong patterns of site fidelity. Its average daily activity space was estimated to be ~10 km2 (Mourier et al. 2012). Individuals spend ~70% of their time within an area 0.3 km2 over the course of a year and migrations are usually around an island or between neighboring islands (Mourier and Planes 2013). Open ocean environment therefore represents a significant barrier to dispersal. To take the reef-associated habit of C. melanopterus into account, we superimposed a raster on the distribution of C. melanopterus as estimated according to the Chondrichthyan Tree of Life Project (www.sharksrays.org). We excluded the Mediterranean Sea from its distribution as the occurrence of C. melanopterus in the Mediterranean is anecdotal and there are no known established populations there. The raster consists of 168,480 cells, each representing an area of approximately 30 km2. These values were chosen to be consistent with the known dispersal range of C. melanopterus, such that each cell roughly defines a deme. This resulted in 15,253 cells occupied by C. melanopterus, 114,497 empty open sea cells (unsuitable habitat) and the remaining cells representing land. We computed both geodesic and least-cost path distances in the R environment using the library gdistance. We assumed that cells have resistance values that are relative to the capacity for C. melanopterus movement through them, with empty cells presenting higher resistance. We tested several ratios between suitable and unsuitable cell resistance values. A value of 1 roughly corresponds to geodesic distance (because sharks cannot move over land).
Range expansions leave characteristic footprints in patterns of genetic diversity within species such that theoretical predictions can be used to both test for the occurrence of a RE and to estimate its center of origin. The first is based on the expected pattern of decay of genetic diversity with increasing geographic distance from the center of origin (Ramachandran et al. 2005). We used two indices of genetic diversity: the heterozygosity and nucleotide diversity for each population. We calculated the correlation coefficient r between within deme diversity and geographic distance to a lattice point, where each lattice point is considered a potential center of origin for the RE. Areas showing the lowest (i.e., most negative) correlation are candidates for the center of origin (i.e., areas showing high genetic diversity at low geographic distances). The second method is based on the directionality index (Ψ) proposed by Peter and Slatkin 2013. Shared derived alleles are expected to be at low frequency near the center of origin but to reach higher frequencies with increasing geographic distance from the origin due to serial founder effects. The directionality index, Ψ, is the average difference in the shared derived allele frequency between two populations (computed only on alleles not fixed in either of the two populations), and is expected to be around 0 in an equilibrium stepping-stone model but significantly different from 0 in a RE model. Alleles were polarized through comparison with an outgroup in order to detect the ancestral variant. We then computed the matrix of the pairwise Ψ and tested for significance using a permutation approach (i.e., whether Ψ is significantly different from 0). Finally, the origin of the expansion was identified using the Time Difference of Arrival (TDOA) algorithm (Gustafsson and Gunnarsson 2003) as implemented in the rangeExpansion library (Peter and Slatkin 2013) in the R environment. An incorrect polarization of the alleles may cause a bias in the computation of Ψ and consequently, in the localization of the center of origin of the RE. To minimize such bias, we performed these analyses three times by polarizing the alleles using three outgroups, Carcharhinus obscurus, Carcharhinus limbatus, and Carcharhinus fitzroyensis. Results were consistent, suggesting that errors in the polarization of the alleles were negligible.
Demographic inferences
We used an ABC (Beaumont et al. 2002) framework to estimate parameters and compare demographic models. The folded site frequency spectrum (SFS), the total number of SNPs and nucleotide diversity (π) were used as summary statistics to avoid phasing issues. We built our simulations such that each simulated dataset had the same configuration (number of loci, sequence lengths and sample sizes) as the observed data (Table S2). We let mutation and recombination rates vary across loci by setting a normal hyperprior distribution on both. Mutation rate was previously estimated in (Maisano Delser et al. 2016) and we considered the generation time for C. melanopterus to be seven years (Smith et al. 1998). The mean of the hyperprior distribution of the mutation rates was modeled as uniform, bounded between 8.05 and 8.54 × 10−9 per site per generation, following the calibration for C. galapagensis Maisano Delser et al. 2016. Having no prior information on the recombination rate of species closely related to C. melanopterus, a uniform distribution between 0 and 10-8 was chosen for the mean of the hyperprior distribution on the recombination rate. A uniform distribution between 10−11 and 10−10 was applied for the standard deviation of the hyperprior distributions on mutation and recombination rate. Such hyperprior distributions on mutation and recombination rates allowed us to account for their variation across the genome. Moreover, by modeling intra-locus recombination, we could use multiple SNPs that were derived from the same region.
We generated simulated data using three demographic models (100,000 simulations per model) using fastsimcoal2 v2.5.1 (Excoffier et al. 2013): (i) model CHG1 represents a single instantaneous demographic change from an ancestral population size, Nanc to a modern population size, Nmod occurring at the time Tc (Fig. 2a); (ii) model FIM represents a non-equilibrium finite island model with 100 demes (N1…N100) originating at the time Ti and exchanging Nm migrants following a symmetric migration matrix (Fig. 2b) and (iii) model SST is analogous to model FIM but is defined by a stepping-stone migration matrix (Fig. 2c). Nm is the product of the effective population size of a deme, N, and the migration rate per generation, m. In backward thinking, this value corresponds to the total number of migrants leaving a deme to go to any other demes within the metapopulation for model FIM and to a subset of neighboring demes defined by the stepping-stone model for model SST. Note that the three models were run independently for each of the 13 populations. We sampled either a random deme or the central deme of the array for FIM and SST respectively. We additionally modified model FIM and SST to allow one change of connectivity through time (model FIM2 and SST2 respectively, see SI). Prior distributions and parameter estimates under the most supported model for each population are reported in Tables S3 and S6. Model posterior probabilities were calculated by a weighted multinomial logistic regression (Beaumont 2008) for which we retained the best 25,000 simulations. The demographic parameters within each model (Nmod, Nanc, and Tc for CHG1; Nanc, Ti, and Nm for models FIM and SST) were estimated from the 5000 simulations deemed closest to the observed dataset using the neuralnet algorithm (Csillery et al. 2012). Analyses were performed in the R environment (R Core Team 2014) with the library abc (Csillery et al. 2012).

Demographic models. a Model CHG1 representing an unstructured population with one possible change in effective population size; b model FIM representing a non-equilibrium finite island model characterized by a symmetric migration matrix; c model SST representing a non-equilibrium finite island model characterized by a stepping-stone migration matrix
We performed cross-validation for both model selection and parameter estimation by randomly generating pseudo-observed datasets (pods) from the prior distributions of each model. For each cross-validation experiment we generated 1000 pods and we applied the same inferential procedure as for the observed data. We chose the datasets with the highest and lowest number of loci, represented by Kiribati and Noumea respectively, for the cross-validation of model selection experiment. We simulated 1000 pods under each model and then we checked how many pods were correctly assigned to the true model with several thresholds of probability (from 0.95 to 0.50, see Table S4).
The same procedure was used for the cross-validation of the parameter estimation, using the Kiribati dataset as an example. The coverage 95%, the scaled mean error (SME) and scaled root mean square error (SRMSE), calculated as in Walther and Moore (2005) were computed for each parameter (Table S4). SME and SRMSE were calculated on both the median and the mode of each estimated parameter.
Model CHG1 was also used to graphically reconstruct the variation of effective population size through time. For each combination of parameters retained by the ABC algorithm, we recorded the effective size at specific time points. The mean and median value of the posterior distribution of the effective size at each time point was calculated and plotted against time to obtain an ABC-skyline reconstruction following Maisano Delser et al. 2016 (Figure S1). Twenty-one time points were defined as described in Boitard et al. 2016 with an upper bound fixed at 300,000 generations ago. Each ABC-skyline was then reconstructed up to the estimated time of the most recent common ancestor (TMRCA). Analyses were performed in the R environment (R Core Team 2014) with the library abc (Csillery et al. 2012).
Results
Genetic diversity and data summary
After applying strict filters (see SI), we obtained a dataset comprising 144 samples sequenced for 431,257 bp spanning 879 independent loci. Overall, 1788 high quality SNPs were identified. A PCA was performed to assess the level of population structure within the dataset (Fig. 3). The first two components explain ~55% of the variance, showing a clear geographical pattern. PC1 identifies three clusters: the Red Sea and Seychelles, the Australian populations and the Pacific Ocean populations. Within the Australian cluster, samples from the Northern Territory and Western Australia group together while there is a gradient of diversity from Queensland to the Great Barrier Reef. The clear picture of geographical structure emerging from the PCA is consistent with the high value of global FST (FST = 0.53, p-value < 0.0001), as well as the pairwise FST matrix (Table S5). The Red Sea and Seychelles show the highest values of pairwise FST compared with all the other populations while the lowest values are observed among the three Australian populations (Western Australia, Northern Territory and Queensland) and between the samples of the Society archipelago (Tetiaroa and Moorea). Measures of genetic variability, computed both as heterozygosity and nucleotide diversity, appear also to be geographically structured. Indeed, northern and eastern Australia showed the highest genetic diversity (Table 1), which declines both eastward and westward towards French Polynesia and the Red Sea, respectively. Overall, these data suggest a strong geographical cline in diversity with a high level of population structure.

Principal Component Analysis of the complete dataset. Population acronyms are reported in Table S1
Range expansion
We used a Mantel test to explore the relationship between geographic and genetic distances, computing geodesic and least-cost distances (McRae and Beier 2007) in order to model the dispersal patterns of C. melanopterus. The highest correlation was between genetic and geodesic distances (Mantel test: Pearson’s r = 0.801, p-value < 0.001). Similar values were obtained with the alternative method by progressively increasing the capacity of C. melanopterus to cross open ocean habitat (i.e., reducing the resistance of unsuitable habitat cells).
We used two complementary approaches to identify the spatial origin of the range expansion. The first is based on the expectation that genetic diversity decays with increasing geographic distance from the center of origin of the expansion, resulting in a negative correlation between geographic distance from the origin and measures of genetic diversity (Ramachandran et al. 2005). Areas showing the lowest (i.e., most negative) correlation are candidates for the center of origin of the RE. The Indo-Australian Archipelago (IAA) was consistently identified as the most probable area for the origin of C. melanopterus when considering both measures of genetic diversity (i.e. nucleotide diversity and heterozygosity). Correlation values ranged between −0.6 and −0.7 (Fig. 4 and S2). The complementary approach is based on the concept that the serial founder effects that characterize REs create a pattern of neutral shared derived alleles that increase in frequency as one progresses away from the center of origin (Peter and Slatkin 2013). We rejected an equilibrium isolation by distance model, in favor of a range expansion model, based on the matrix of pairwise Ψ (p-value < 0.0001). Peripheral populations such as the Red Sea and French Polynesia displayed the highest frequency of shared derived alleles, while the northern Australian populations had the lowest incidence (Fig. 4 and S3). The TDOA algorithm identifies the South China Sea, located within the IAA, as the most likely origin of the expansion (Fig. 4 and S3), consistent with the results of our analysis based on the method of (Ramachandran et al. 2005). This result was robust to the choice of the outgroup that was chosen to polarize alleles.

Correlation map between genetic diversity and geographic distances. Each cell in the map shows the correlation coefficient between genetic diversity (measured as nucleotide diversity) and geographic distance from the putative origin of expansion. The red area represents the area of most negative correlation coefficients (i.e., the most likely area of the origin of C. melanopterus). The white cross indicates the location of origin of the range expansion using the directionality index by Peter and Slatkin (2013). Population acronyms are reported in Table S1
Demographic inferences
We first investigated the demographic history of each population with an unstructured demographic model, CHG1, that assumes a fully isolated population (Fig. 2a). We applied ABC to estimate the three parameters and to reconstruct the variation of the effective population size through time. For all populations in the Pacific Ocean, we identified a reduction of Ne, while a constant population size was observed for the four Australian populations. The Red Sea was the only location that showed a recent expansion. The Seychelles showed a decrease in Ne similar to that observed in the Pacific (Figure S1). We then separately applied FIM and SST non-equilibrium metapopulation (i.e., structured) models to the data. Results from the ABC model selection, for each population, are reported in Table 2. The metapopulation model SST shows the highest probability (between 0.69 and 0.94) for all populations in the Pacific Ocean, while the metapopulation model FIM is best supported for the four Australian populations (between 0.79 and 0.93). In the Seychelles, the metapopulation models FIM and SST show probabilities of 0.61 and 0.38, respectively, while the unstructured model CHG1 is most strongly supported (0.8) for the Red Sea. We report the estimated level of connectivity (Nm) obtained from the best fitting model for each population (Table 2, Table S3 and Fig. 5). Among the populations from the Pacific Ocean, Nm (mode) ranges between 8.7 and 23.2. This pattern is consistent with a geographical cline from French Polynesia toward Australia with progressively increasing values of Nm. The Australian populations show higher levels of connectivity with Nm (mode) ranging between 33.8 and 48.1 and an average of 43. Samples from the Seychelles are characterized by an Nm (mode) of ~10.2, highlighting a lower level of connectivity in the southwestern Indian Ocean and in the island systems (i.e., Polynesia, Seychelles). We also investigated a possible change in connectivity through time using model FIM2 and SST2. First, the simpler models received equal or higher support compared to the more complex model (Table S6). Parameter estimation of the ancestral level of connectivity (Nm2) and the time of the change in connectivity (Tcm) for both FIM2 and SST2 are uninformative and simply recover the prior distributions (Table S6).

Level of connectivity. Arrows indicate the demographic signal recovered with the ABC-skyline plot for each population based on model CHG1 (Fig. S1). The mode of the estimated Nm under the most supported metapopulation model (either model FIM or SST) is reported in parentheses. Up-arrow: demographic expansion; down-arrow: demographic reduction; right-arrow: constant population size. Population acronyms are reported in Table S1
Discussion
Range expansions have occurred frequently and recurrently (Excoffier et al. 2009) in nature, leaving unique signatures on the genetic diversity of species and sub-populations (Mona et al. 2014; Ray et al. 2003). As such, this special class of metapopulation model can provide a more realistic description of the evolution of a species than classic equilibrium models of population structure. Given that REs are widespread and likely account for a significant component of observed population structure, their consequences should be carefully examined and quantitatively tested when investigating the demographic history of a species. Unfortunately, this is rarely done, even in well-studied organisms such as humans, with relatively few exceptions (but see Chikhi et al. 2018; Currat and Excoffier 2005; Eriksson and Manica 2014; Mona et al. 2013). Rather, unstructured equilibrium models are commonly applied in empirical population genetics, with little examination of how inferences may be biased as a consequence of not accounting for metapopulation structure. Recognizing computational limitations associated with fully modeling range expansions, we argue that it is good practice in empirical studies of demographic history to begin by testing for a RE in the species under examination and to subsequently test and compare simplified metapopulation models with unstructured equilibrium models in order to make appropriate choices for inferring demographic parameters. In this paper we present a computationally tractable ABC approach for so doing, and apply it to study the demographic and colonization history of the blacktip reef shark C. melanopterus, as its ecology, behavior (Mourier and Planes 2013; Papastamatiou et al. 2010) and genetics (Maisano Delser et al. 2016; Vignaud et al. 2014) suggest that this species is highly structured and has likely experienced a RE (Maisano Delser et al. 2016). To achieve tractability we: (i) treated each population independently by simulating a reduced number of demes (i.e., 100) interacting with our focal deme; (ii) simulated an instantaneous colonization of the array of demes rather than a wave of advance typical of a RE (similar to the approach of Hamilton et al. 2005 and Stadler et al. 2009).
Results from the Mantel Test supported isolation by distance, which is compatible with both an equilibrium stepping-stone and a RE model. We confirmed a scenario of RE by examining patterns of decay in genetic diversity. The map of correlation coefficients estimated for two diversity indices (heterozygosity and nucleotide diversity) versus geographic distance to candidate centers of origin, highlighted an area of high negative correlation (with a maximum of −0.7) around the IAA, away from which genetic diversity decreased across geographical space (Fig. 4). This pattern is a typical signature of a RE and contrasts with an equilibrium model in which all populations are expected to show similar values of genetic diversity, if Nm is homogeneous across the lattice. This is not the case in C. melanopterus, with the Australian region showing the highest Nm values (Fig. 5, Table S3). A local decrease of Nm in both the Pacific and the Indian Ocean (compared to the IAA) could produce the same observed pattern. For this reason, we also exploited the directionality index proposed by Peter and Slatkin 2013 and consistently found a significant signature of RE (p-value < 0.0001) with the likely origin within the same region (Fig. 4 and S3). The Australian populations show the lowest frequencies of shared derived alleles, consistent with the expectation for locations in closest proximity to the center of the expansion. Finally, we also note that Carcharhinus cautus and Carcharhinus fitzroyensis, the two extant species most closely related to C. melanopterus, have a distribution restricted to an area ranging from Queensland to Western Australia (with some occurrence in southern Papua New Guinea) (Lyle 1987). This is consistent with an origin of the entire clade in the IAA, a known marine biodiversity hotspot that has previously been proposed as an “evolutionary pump” and a center of origin for tropical diversity (Bowen et al. 2013; Budd and Pandolfi 2010; Connolly et al. 2003; Hobbs et al. 2009). It is also possible that the observed patterns are the consequence of a contact zone centered at the IAA (Center of Overlap Hypothesis; Cowman et al. 2017). However, two patterns argue against this: first, given the high FST values observed between populations from the Indian and Pacific Oceans, demes in the contact zone would show a strong bottleneck signature—which they do not. Indeed, admixture between two divergent populations would be expected to yield a gene genealogy with longer internal relative to external branches, which would suggest a population decline when analyzed with an unstructured model (Tajima 1989). Secondly, if the IAA were indeed a contact zone we would have identified two range expansions converging at the IAA, with a higher frequency of shared derived alleles in the overlap region than in the periphery—which is also not observed. To our knowledge, this is the first time that a quantitative population genomics approach has been used to identify the historical origin of a marine species.
On the basis of indirect evidence (genetic diversity and biogeography of the sister species) and spatially explicit statistical tests (the decay of genetic diversity and the directionality index) we demonstrated that a RE likely occurred in C. melanopterus, suggesting that it would be inappropriate to adopt unstructured models to further infer demographic parameters. On the other hand, fully modeling a spatially explicit RE is time consuming, especially for a widely distributed species such as C. melanopterus (Figs. 1 and 4). In this study we adopted a compromise strategy in which each of sampled populations were treated independently and tested with three demographic models: an unstructured model (CHG1), and two non-equilibrium metapopulation models that are simplifications but that recreate the outcome of a range expansion (FIM and SST). Although inappropriate given that we detected metapopulation structure as a consequence of a range expansion, we chose to test the unstructured model CHG1 for two reasons: (i) to understand what would have been concluded had we disregarded the RE; (ii) to check whether it is possible to statistically reject conclusions from an unstructured model. The results of analyses under the unstructured model suggest that C. melanopterus has experienced local bottlenecks in both the Pacific Ocean (as already suggested for Moorea by Vignaud et al. 2014) and the Seychelles, while demographic stability is inferred around Australia and expansion in the Red Sea (Fig. 5). This interpretation suggests that there have been recent regional declines, raising questions about the potential impact of human activity on populations of C. melanopterus. The variation of Ne through time is often the focus of works on the demography of species and populations. Had we stopped here, we would have concluded that several populations in the IAA are healthy but conservation plans are needed in large areas of the Pacific and the Indian Ocean. However, our ABC model selection procedure revealed another story, indicating that structured metapopulation models (SST and FIM) have a better fit to the data for all but the Red Sea population (Table 2). This result implies that the bottlenecks that were inferred in the Pacific Ocean and the Seychelles under the unstructured model, CHG1, are likely artifacts of non-modeled population structure rather than a reflection of actual variation in Ne through time (Mazet et al. 2015). In the same vein, we suspect that the bottleneck in Moorea reported by Vignaud et al. 2014 might be better explained by a low Nm and associated metapopulation structure. It is likely that “population bottleneck” scenarios are reported more frequently in the literature than is warranted, because there will always be a metapopulation model that will produce similar patterns of genetic variability (Mazet et al. 2015). For this reason, we suggest that a statistical comparison of structured versus unstructured models be routinely explored when pursuing population genetic studies of demography, especially when applying the outcomes of such studies to infer impacts of anthropogenic activities and to set conservation priorities for vulnerable groups of animals.
The Nm estimated in Australia is much higher (~40) than in other regions, consistent with a constant effective size in this region. In the Red Sea, the unstructured model CHG1 is preferred, suggesting a strong signature of population expansion. These findings, in conjunction with the patterns observed for the Pacific Ocean populations, are better understood within the context of the distribution of C. melanopterus (Fig. 1). In the Pacific (from New Caledonia to French Polynesia), C. melanopterus has a patchy distribution, associated with the island system and discontinuous nature of suitable coral reef habitat. Carcharhinus melanopterus is unlikely to traverse large oceanic expanses, which, in turn, restricts migration across this part of the species range. The low Nm values and high statistical support for the SST metapopulation model is consistent with this finding (Table 2, Table S3). A similar scenario applies to the Seychelles, even though the FIM metapopulation model was found to be best supported (Table 2). In contrast to these scenarios, the IAA has more continuous habitat availability along continental shelfs that could conceivably support multiple colonies with higher levels of migration between them. This is consistent with the larger Nm values estimated across this area, as well as the fact that metapopulation model FIM better fit the data while a constant size signal was recovered under the unstructured model CHG1 (Table 2). We did not detect any change of connectivity through time for any of our populations (except the Red Sea) when tested with model FIM2 or SST2. This result suggests a similar migratory pattern across time and the absence of any environmental or human-mediated event that could have drastically changed the level of connectivity of C. melanopterus. The Red Sea population is the only location to return a result that is at odds with these expectations: this area is similar to the IAA in terms of habitat availability, but CHG1 was the preferred model. Clearly, the most recently colonized region appears to be the Red Sea. This likely explains why a signature of the metapopulation is not yet detectable in the data from this region. Interestingly, there is evidence that this colonization process is ongoing, with the recent detection of C. melanopterus along the coast of Costa Rica (López-Garro et al. 2012). After the initial range expansion, local patterns of connectivity likely formed, due to differential habitat availability. Populations inhabiting areas with high habitat availability could migrate more easily and show higher Nm (and a FIM model), while populations occupying areas with more isolated patches of habitat would show lower Nm values (and a SST model).
Conclusions
Here we used population genomics approaches to characterize the historical demography and colonization history of C. melanopterus throughout its range. We show that the demography of C. melanopterus is best described by metapopulation models. We were able to statistically reject an equilibrium metapopulation model suggesting instead that this species has experienced a range expansion. Spatial genetic modeling indicated that two waves of stepping-stone colonization originated in the Indo-Australian Archipelago, proceeding eastward through the Pacific and westward through the Indian Ocean, to give rise to the modern distribution range of the species. Signatures of population size changes in C. melanopterus previously described by Vignaud et al. 2014 are shown herein to be the consequence of metapopulation structure rather than local episodes affecting single demes.
Although the ecological characteristics of C. melanopterus make it a clear example of a species working as a metapopulation, our findings are more generally relevant to researchers wishing to study the demographic history of any species. Most empirical population genetics studies adopt computationally tractable models that assume that populations are fully isolated (Boitard et al. 2016; Li and Durbin 2011; Schiffels and Durbin 2014). In reality, most species belong to a network of sub-populations (demes) that exchange migrants to varying degrees. Quantifying and correcting the bias that can originate from adopting unstructured models to study demes sampled from metapopulations is therefore of crucial importance in molecular ecology. Appropriate model choice will become increasingly important as more empirical studies begin working with next-generation sequence data because the effects of model mis-specification are generally amplified when applied to large genomic datasets, leading to strong confidence in mislead inferences.
Data archiving
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.553cm8g.
References
Barbujani G, Sokal RR, Oden NL (1995) Indo-European origins: a computer-simulation test of five hypotheses. Am J Phys Anthropol 96:109–132
Beaumont MA (2008) Joint determination of topology, divergence time, and immigration in population trees. In: Matsumura, S., Forster, P. and Renfrew, C. (eds.) Simulation, genetics, and human prehistory. McDonald Institute for Archaeological Research, Cambridge, pp 135–154.
Beaumont MA, Zhang W, Balding DJ (2002) Approximate Bayesian computation in population genetics. Genetics 162(4):2025–2035
Boitard S, Rodriguez W, Jay F, Mona S, Austerlitz F (2016) Inferring population size history from large samples of genome-wide molecular data—an approximate Bayesian computation approach. PLoS Genet 12:e1005877
Bowen BW, Rocha LA, Toonen RJ, Karl SA (2013) The origins of tropical marine biodiversity. Trends Ecol Evol 28:359–366
Budd AF, Pandolfi JM (2010) Evolutionary novelty is concentrated at the edge of coral species distributions. Science 328:1558–1561
Chikhi L, Rodriguez W, Grusea S, Santos P, Boitard S, Mazet O (2018) The IICR (inverse instantaneous coalescence rate) as a summary of genomic diversity: insights into demographic inference and model choice. Heredity 120:13–24
Connolly SR, DR B, TP H (2003) Indo-Pacific biodiversity of coral reefs: deviations from a mid-domain model. Ecology 84:2178–2190
Corrigan S, Maisano Delser P, Eddy C, Duffy C, Yang L, Li C et al. (2017) Historical introgression drives pervasive mitochondrial admixture between two species of pelagic sharks. Mol Phylogenet Evol 110:122–126
Cowman P, Parravicini V, Kulbicki M, Floeter S (2017) The biogeography of tropical reef fishes: endemism and provinciality through time Biol Rev Camb Philos Soc 92:2112–2130
Csillery K, Francois O, Blum MGB (2012) abc: an R package for approximate Bayesian computation (ABC). Methods Ecol Evol 3:475–479
Currat M, Excoffier L (2005) The effect of the Neolithic expansion on European molecular diversity. Proc Biol Sci 272(1564):679–688
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Eriksson A, Manica A (2014) The doubly conditioned frequency spectrum does not distinguish between ancient population structure and hybridization. Mol Biol Evol 31:1618–1621
Excoffier L, Dupanloup I, Huerta-Sanchez E, Sousa VC, Foll M (2013) Robust demographic inference from genomic and SNP data. PLoS Genet 9:e1003905
Excoffier L, Foll M, Petit R (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481. 501
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Francois O, Blum MG, Jakobsson M, Rosenberg NA (2008) Demographic history of european populations of Arabidopsis thaliana. PLoS Genet 4:e1000075
Gaggiotti OE, Bekkevold D, Jorgensen HB, Foll M, Carvalho GR, Andre C et al. (2009) Disentangling the effects of evolutionary, demographic, and environmental factors influencing genetic structure of natural populations: Atlantic herring as a case study. Evolution 63:2939–2951
Gustafsson F, Gunnarsson F (2003) Positioning using time-difference of arrival measurements. In: 2003 IEEE International Conference on Acoustics, Speech, and Signal Processing, vol VI, Proceedings. pp 553–556
Hamilton G, Currat M, Ray N, Heckel G, Beaumont M, Excoffier L (2005) Bayesian estimation of recent migration rates after a spatial expansion. Genetics 170:409–417
Hanski I (1998) Metapopulation dynamics. Nature 396:41–49
He Q, Prado JR, Knowles LL` (2017) Inferring the geographic origin of a range expansion: Latitudinal and longitudinal coordinates inferred from genomic data in an ABC framework with the program x-origin Mol Ecol 26:6908–6920
Hobbs JPA, Frisch AJ, Allen GR, Van Herwerden L (2009) Marine hybrid hotspot at Indo-Pacific biogeographic border. Biol Lett 5:258–261
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132(2):583–589
Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475:493–496
López-Garro A, Zanella I, Golfín-Duarte G, Pérez-Montero M (2012) First record of the blacktip reef shark Carcharhinus melanopterus (Carcharhiniformes: Carcharhinidae) from the Tropical Eastern Pacific Rev Biol Trop 6:275–278
Lyle J (1987) Observations on the biology of Carcharhinus cautus (Whitley), C. melanopterus (Quoy & Gaimard) and C. fitzroyensis (Whitley) from Northern Australia. Aust J Mar Freshw Res 38:701–710
Maisano Delser P, Corrigan S, Hale M, Li C, Veuille M, Planes S et al. (2016) Population genomics of C. melanopterus using target gene capture data: demographic inferences and conservation perspectives. Sci Rep 6:33753
Mazet O, Rodriguez W, Chikhi L (2015) Demographic inference using genetic data from a single individual: Separating population size variation from population structure. Theor Popul Biol 104:46–58
Mazet O, Rodriguez W, Grusea S, Boitard S, Chikhi L (2016) On the importance of being structured: instantaneous coalescence rates and human evolution—lessons for ancestral population size inference? Heredity 116:362–371
McRae BH, Beier P (2007) Circuit theory predicts gene flow in plant and animal populations. Proc Natl Acad Sci USA 104:19885–19890
Mona S (2017) On the role played by the carrying capacity and the ancestral population size during a range expansion. Heredity 118:143–153
Mona S, Mordret E, Veuille M, Tommaseo-Ponzetta M (2013) Investigating sex-specific dynamics using uniparental markers: West New Guinea as a case study. Ecol Evol 3(8):2647–2660
Mona S, Ray N, Arenas M, Excoffier L (2014) Genetic consequences of habitat fragmentation during a range expansion. Heredity 112:291–299
Mourier J, Planes S (2013) Direct genetic evidence for reproductive philopatry and associated fine-scale migrations in female blacktip reef sharks (Carcharhinus melanopterus) in French Polynesia. Mol Ecol 22:201–214
Mourier J, Vercelloni J, Planes S (2012) Evidence of social communities in a spatially structured network of a free-ranging shark species. Anim Behav 83:389–401
Neuenschwander S, Largiader CR, Ray N, Currat M, Vonlanthen P, Excoffier L (2008) Colonization history of the Swiss Rhine basin by the bullhead (Cottus gobio): inference under a Bayesian spatially explicit framework. Mol Ecol 17:757–772
Papastamatiou Y, Friedlander A, Caselle J, Lowe C (2010) Long-term movement patterns and trophic ecology of blacktip reef sharks (Carcharhinus melanopterus) at Palmyra Atoll. J Exp Mar Biol Ecol 386:94–102
Papastamatiou Y, Lowe C, Caselle J, Friedlander A (2009) Scale-dependent effects of habitat on movements and path structure of reef sharks at a predator-dominated atoll. Ecology 90:996–1008
Peter BM, Slatkin M (2013) Detecting range expansions from genetic data. Evolution 67:3274–3289
Pfeifer B, Wittelsburger U, Ramos-Onsins SE, Lercher MJ (2014) PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol Biol Evol 31:1929–1936
Potter S, Bragg JG, Peter BM, Bi K, Moritz C (2016) Phylogenomics at the tips: inferring lineages and their demographic history in a tropical lizard, Carlia amax. Mol Ecol 25:1367–1380
Quoy JRC, Gaimard JP (1824) Quoy, J. R. C. & J. P. Gaimard, pp 185–232.
Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL (2005) Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA 102:15942–15947
Ray N, Currat M, Berthier P, Excoffier L (2005) Recovering the geographic origin of early modern humans by realistic and spatially explicit simulations. Genome Res 15:1161–1167
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20:76–86
R CoreTeam (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Schiffels S, Durbin R (2014) Inferring human population size and separation history from multiple genome sequences. Nat Genet 46:919–925
Schneider N, Chikhi L, Currat M, Radespiel U (2010) Signals of recent spatial expansions in the grey mouse lemur (Microcebus murinus). BMC Evol Biol 10:105
Smith S, Au D, Show C (1998) Intrinsic rebound potentials of 26 species of Pacific sharks. Mar Freshw Res 49:663–678
Stadler T, Haubold B, Merino C, Stephan W, Pfaffelhuber P (2009) The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics 182:205–216
Stevens J (1984) Life history and ecology of sharks at Aldabra Atoll, Indian Ocean Proc R Soc Lond B Biol Sci 222:79–106
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Vignaud TM, Mourier J, Maynard JA, Leblois R, Spaet J, Clua E et al. (2014) Blacktip reef sharks, Carcharhinus melanopterus, have high genetic structure and varying demographic histories in their Indo-Pacific range. Mol Ecol 23:5193–5207
Walther BA, Moore JL (2005) The concepts of bias, precision and accuracy, and their use in testing the performance of species richness estimators, with a literature review of estimator performance. Ecography 28:815–829
Acknowledgements
We are grateful to the genotoul bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo Genotoul) for providing computing resources (www.bioinfo.genotoul.fr). This work was funded by the Agence Nationale de la Recherche Demochips ANR-12-BSV7-0012, a LABEX CORAIL grant to SM and MV (CORALSHARK) and NSF Award DEB-01132229 to GN (Collaborative Research: Jaws and Backbone: Chondrichthyan Phylogeny and a Spine for the Vertebrate Tree of Life). We thank Ornella Weideli and Save Our Seas foundation for providing samples from the Seychelles. We also thank Andrew Chin, Jennifer Ovenden, Mark Meekan and Conrad Speed, Mael Imirizaldu, David Lecchini, Patrick Plantard, Jonathan Werry, Thomas Vignaud, Julia Spät and several students for providing samples or for assistance with blacktip reef shark population genetics sampling. We thank Johann Mourier, Andrea Manica, Anders Eriksson, Robert Beyer and Dave Swofford for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Maisano Delser, P., Corrigan, S., Duckett, D. et al. Demographic inferences after a range expansion can be biased: the test case of the blacktip reef shark (Carcharhinus melanopterus). Heredity 122, 759–769 (2019). https://doi.org/10.1038/s41437-018-0164-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0164-0
- Springer Nature Switzerland AG
This article is cited by
-
What Darwin could not see: island formation and historical sea levels shape genetic divergence and island biogeography in a coastal marine species
Heredity (2023)
-
Ecological and biogeographic features shaped the complex evolutionary history of an iconic apex predator (Galeocerdo cuvier)
BMC Ecology and Evolution (2022)
-
Genomic insights into the historical and contemporary demographics of the grey reef shark
Heredity (2022)