
Abstract
Environmental changes typically cause rapid gene expression responses in the exposed organisms, including changes in the representation of gene isoforms with different functions or properties. Identifying the genes that respond to environmental change, including in genotype-specific ways, is an important step in treating the undesirable physiological effects of stress, such as exposure to toxins or ethanol. Ethanol is a unique environmental stress in that chronic exposure results in permanent physiological changes and the development of alcohol use disorders. Drosophila is a classic model for deciphering the mechanisms of the response to alcohol exposure, as it meets the criteria for the development of alcohol use disorders, and has similar physiological underpinnings with vertebrates. Because many studies on the response to ethanol have relied on a priori candidate genes, broad surveys of gene expression and splicing are required and have been investigated here. Further, we expose Drosophila to ethanol in an environment that is genetically, socially, and ecologically relevant. Both expression and splicing differences, inasmuch as they can be decomposed, contribute to the response to ethanol in Drosophila melanogaster. However, we find that while D. melanogaster responds to ethanol, there is very little genetic variation in how it responds to ethanol. In addition, the response to alcohol over time is dynamic, suggesting that incorporating time into studies on the response to the environment is important.
Similar content being viewed by others

Introduction
Variation in gene expression, and variation in how gene expression responds to environmental changes, are important components of cellular and organismal phenotypes. For example, in the past gene expression in particular epithelial cells was assayed and found to differ in the glucocorticoid pathway between individuals with and without asthma, leading to the development of glucocorticoids to treat asthma (Christodoulopoulos et al. 2000; Poon et al. 2012; Bossé 2013; Chang et al. 2015; Li et al. 2015; Nieuwenhuis et al. 2016; Mukherjee et al. 2017). Alcohol use disorders are one of the leading causes of preventable death, affecting millions of people world-wide, and individuals differ in their susceptibility to alcohol use disorders. Chronic alcohol abuse results in changes in gene expression and chromosome organization, which likely contributes to abuse and dependence (Hyman and Malenka 2001; Pandey et al. 2008; Corl et al. 2009; Sasabe and Ishiura 2010; Aroor et al. 2010; Zhou et al. 2011; Farris and Miles 2012; Edenberg and Foroud 2013; Engel et al. 2016; Cervera-Juanes et al. 2017). Understanding the factors that contribute to alcohol use disorders is critical for the development of therapies for their prevention and treatment.
Drosophila melanogaster is one of the model organisms used to study alcohol use disorders, as the behavioral response to acute ethanol exposure is similar in Drosophila and mammals. At lower doses or upon initial exposure it is a stimulant, while at higher doses it acts as a depressant (Rodan and Rothenfluh 2010). Drosophila also develops tolerance after repeated exposures, and indeed has been shown to meet the DSM-IV criteria for addiction including relapse behavior (Scholz et al. 2000; Devineni and Heberlein 2009; Kaun et al. 2011; Devineni et al. 2011). Many of the genes predicted to be involved in ethanol response are conserved between Drosophila and humans (Heberlein et al. 2004), and a number of genes have been shown to affect alcohol-related phenotypes in both Drosophila and humans (Treistman and Martin 2009; MacKay et al. 2011; McClure et al. 2011; Ojelade et al. 2015). Ecologically ethanol is a common component of the fermenting fruit that makes up the primary habitat of D. melanogaster (Dorado and Barbancho 1984; Gibson and Wilks 1988; Milan et al. 2012). Higher ethanol concentrations are exploited by D. melanogaster for a number of reasons, including increased resistance to parasitism and caloric benefits (McClure et al. 2011; Milan et al. 2012; Pohl et al. 2012).
Expression differences in response to acute ethanol exposure or tolerance have been extensively documented, though variation in the methods used, including exposure time, amount, particular assay, and measurement method introduce significance variance in the comparability of results (Morozova et al. 2006, 2007). In addition, many studies on the genes involved in differences in ethanol tolerance or acute exposure rely on mutations, and given the systemic effects of ethanol it is likely that the potential number of genes that could affect ethanol response is larger than the actual number contributing to functional variation in the population. This is supported by the observation of Morozova et al. (2015), in that there is little overlap in genome-wide association studies and single gene studies, potentially because large effect genes are under strong selective pressure and will not vary in natural populations. Response to ethanol is also an extremely complex phenotype, and the links between short and long-term changes in gene expression and ethanol-related phenotypes (resistance, tolerance, and self-administration) are not entirely clear.
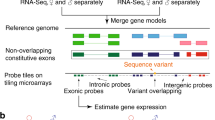
Alternative splicing of mRNA also has important effects on phenotypes, however, only rarely have the functional implications of different isoform usage been investigated. This is in part due to difficulty in confidently detecting different isoforms, given that many exons are shared between different isoforms or overlap exons from different isoforms (Fig. 1a). However, recent work has suggested that differential isoform usage is important for Alzheimer’s disease and several forms of cancer (Goehe et al. 2010; Lai et al. 2014; Caswell et al., 2015; Paronetto et al. 2016; Kędzierska et al. 2016). Totally, 15% of human diseases are thought to be attributable to aberrations in splice site selection (Naftelberg et al. 2015). Alternative splicing can affect mRNA stability, localization, translation, and/or produce different protein isoforms with different functions and/or localizations (Baralle and Giudice 2017). Previous work on gene expression changes in response to ethanol did not take alternative splicing into account, and given that gene expression and splicing cannot be disentangled with short-read RNA-seq or microarrays the results are likely confounded. Overall, the intersection between splicing and gene expression in response to ethanol has not been investigated, though in multiple animal models ethanol has been shown to affect alternative splicing (which could be mediated by preferential degradation or changes in splicing), epigenetic marks, and chromatin architecture (Oomizu et al. 2003; Morozova et al. 2006; Pandey et al. 2008; Sasabe and Ishiura 2010; Troutwine et al. 2016; Cervera-Juanes et al. 2017). Dynamic changes in alternative splicing, epigenetic marks, and chromatin architecture in response to environmental stimulus have been documented previously in Drosophila (Zraly and Dingwall 2012; Zaharieva et al. 2012).

a A representation of the classification scheme for exons, exonic regions, and exon fragments. In higher eukaryotes exons from different isoforms of a gene may overlap, and portions may be shared between some or all isoforms. In effect, this makes it essentially impossible to disentangle expression and splicing, and hence we classify exons as either being entire exons, which do not overlap other exons in different isoforms (exons), or fused regions consisting of a set of overlapping exons (exonic regions). Exonic regions can be decomposed into exon fragments, depending upon their overlap between different isoforms. Exons may be unique (found in a single isoform), common (found in some isoforms but not all), or constitutive (present in all isoforms), and if they are unique to a single isoform they may also be unique and constitutive if that is the only transcript annotated for that particular gene. As exonic regions require overlap between exons they may not be unique, but they may be common or constitutive. Exon fragments can be unique to a single isoform, common to several isoforms, or constitutive. Exon fragments may be unique and constitutive only in the situation of a multi-gene exonic region where one gene has a single isoform. b Some exons, exonic regions, and exon fragments could not be compared for expression levels for treatment because they were only unambiguously detected in one environment. Shown here is the number of exons, exonic regions, and exon fragments detected without ethanol, with ethanol, and in both environments. c The proportion of exons, exonic regions, and exon fragments that were unique, unique/constitutive, common, or constitutive and were detected either only without ethanol, only with ethanol, or in both environments. As expected constitutive exons and exonic regions are more commonly detected in both environments, which is suggestive of alternative exon usage in response to ethanol
We take an approach here that is sensitive to both expression differences within isoforms and between isoforms, in so far as the two can be decomposed (Fig. 1a) (Dalton et al. 2013; Newell et al. 2016; Fear et al. 2016). The accurate identification of a particular isoform with short-read RNA-seq requires that at least one exon or splicing event be unique to that isoform, however, this is usually not the case. More often, all or a portion of individual exons may be shared between some or all isoforms of a gene (Fig. 1a). Furthermore, even when there is a unique event, reads cannot be confidently assigned to other shared portions of a given transcript. Here we use an event based approach to quantify differences in the representation of portions of transcripts based on three broad categories—exons, exonic regions, and exon fragments—and quantify differences in their representation in response to genotype, treatment (ethanol), and time. Exons simply do not overlap any other exons, while exonic regions are a total fused set of overlapping exons (Fig. 1a). Exon fragments are decomposed from exonic regions (Fig. 1a). We also perform a rank test of exonic abundance within genes, which is additionally suggestive of a change in splicing.
We measure changes in the transcriptome due to ethanol during a single exposure, a phenotype referred to as ethanol resistance. Initial resistance to ethanol is well established as being a predictor of alcohol use disorders later in life (Schuckit and Smith 2011). This suggests that changes in gene expression during initial exposures to ethanol may be predictive of the metabolic and behavioral systems that induce long-term changes in response to chronic exposure. The vast majority of work on ethanol-related phenotypes has focused on major effect mutants, inbred homozygous lines, and experimental setups lacking in relevant social or ecological contexts. We address this drawback here by using heterozygous flies, inbred lines all crossed to a common tester, to more closely emulate wild flies. Given that the number of large effect mutations that could potentially effect ethanol response is much larger than the number that will actually contribute to alcohol-related phenotypes in wild flies, this represents a significant improvement. We also assayed gene expression in Drosophila heads only, to avoid detection of metabolic genes that are likely not involved in the development of long-term addiction behaviors. In addition, flies were exposed to ethanol within a chamber containing two peers—another male and a female—with the ethanol contained within the food substrate (see Fig. 2 for experimental setup). The mechanism of exposure is inhalation within the chamber as it evaporates from the food. Furthermore, by standardizing parental and F1 age, rearing density, and circadian rhythms, we significantly reduce the risk that the variation we detect is due to differences in offspring quality, maternal effects, or disrupted circadian entrainment. These modifications address the lack of repeatability between mutant- and wild-type assays of ethanol-response genes, by more closely recapitulating the natural environment within which flies will encounter ethanol while retaining the ability to replicate the experiments (Morozova et al. 2007; Berger et al., 2008; Morozova et al.2009; Awofala 2010; Fochler et al. 2017).

a This is an illustration of the social environment that each Drosophila male was exposed to during the course of ethanol exposure. Each chamber contained one female and two males, the female of which was not collected for analysis. Each plate contained 12 chambers, and experiments were conducted in groups of 12. b An illustration of experimental design. Chambers of 12 Drosophila were collected and flash frozen at either 10, 20, or 30 min. After flash freezing the males were collected from each chamber and the heads were isolated for RNA-seq. c A comparison of average relative expression between environments for genes significant in the main dataset (left) and the a priori dataset (right) for exons and exonic regions. d A comparison of relative expression between environments for genes that were significantly different at 10, 20, and 30 min for the main dataset (left) and the a priori dataset (right) for exons and exonic regions. e The proportion of exons and exonic regions (top) and exon fragments (bottom) that were significant for all components of variance. The left-hand row is from the main dataset, the right-hand row is from the a priori dataset
Materials and methods
Fly lines
Male flies came from six natural genotypes collected from an orchard in Winters, California in 1998 and were made isogenic by at least 40 generations of full sibling inbreeding (Yang and Nuzhdin 2003; Campo et al. 2013). Flies in natural conditions are heterozygous, so each inbred genotype was crossed to a reference strain (w1118, Bloomington stock number 3605) to create the F1 flies used in the RNA-seq. With this design, we have the ability to replicate observations of gene expression because the flies resemble wild flies genetically but are identical twins. Flies were reared on a standard medium at 25 °C with a 12-h light/12-h dark cycle. To standardize offspring quality all F1 flies were produced from females of the same age and held at the same density (ten individuals of each sex per vial). Male F1 flies used for the assays were collected as virgins and reared in single sex vials at a density of 24–30 individuals per vial.
Within each chamber a female fly was included as stimulus, but not as a part of the collections for RNA-seq (Signor et al 2017a, 2017b). The female genotype was an inbred laboratory strain y1w1 (Bloomington stock number 1495). Females were produced and reared in the same manner as described for males. Both males and females were aged 3–4 days prior to collection.
Experiment setup
Each chamber contained 12 isolated, circular arenas with a diameter of 2.54 cm (VWR cat. no. 89093-496, Fig. 2a). The flies were sedated through exposure at 4 °C for 10 min, and placed in the chambers with a paintbrush (two males and one female per arena) (Fig. 2a). The flies were also allowed to recover for 10 min within the chambers prior to the start of timing the assay, as these experiments were also intended for behavioral analysis and this is a standard recovery time (Signor et al. 2017a). The chambers contained either grapefruit medium or medium in which 15% of the water has been replaced with ethanol. The ethanol is contained within the food, however, the mechanism of administration is ethanol vapor within the chamber. The flies were recorded using VideoGrabber (http://code.google.com/p/video-grabber/), and set‐up of the assays was facilitated with FlyCapture (PointGrey, Canada). To standardize the transcriptional response, the videos of each assay were used to determine if flies mated during the assay or were damaged during setup, and flies in those chambers were not collected.
Flies were assayed for 10, 20, or 30 min for three replicates of each of the two conditions (Fig. 2b). The assays were conducted within a 2-h window after dawn, to standardize for the effect of circadian rhythms. Replicates were conducted randomly under standardized conditions (25 °C, 70% humidity). At the end of the 10, 20, or 30-min period flies were flash frozen and collected for RNA-seq. Please note that a behavioral analysis of this exposure to ethanol has been published and shows evidence of intoxication as well as genotype-specific differences in the behavioral response to ethanol (Signor et al. 2017a).
Sample preparation and RNA sequencing
mRNA purification
Flash-frozen flies were carefully vortexed followed by freeze drying. Ten to twelve heads were placed into individual mini tubes of a 96-tube plate formate (Axygen MTS-11-C-R). mRNA purification, cDNA synthesis and library preparation were carried out by RAPiD GENOMICS (http://rapid-genomics.com) in 96-well plate format using a robot. In brief, mRNA was purified using Dynbeads mRNA DIRECT Micro kit (Invitrogen # 61021) with slight modifications. Heads were homogenized in 200 µL Lysis Binding Buffer using a GenoGrinder for 2 min. A 0.5 µL of a 1/20 dilution of ERCC spike (Invitrogen # 4456739) was added to the lysate prior to combining with the magnetic beads. Samples were incubated at 65 °C for 5 min, dropped to 20 °C then incubated at RT for 10 min with rocking. mRNA-beads were collected using a magnetic stand for 2 min and supernatants discarded. mRNA-beads were washed six times with 200 µL Wash Buffer A by gentle pipetting and collection of the mRNA-beads on a magnetic stand for 2 min followed by discarding supernatants. mRNA-beads were washed an additional six times with 200 µL low salt buffer (20 mM Tris-HCl ph 7.5, 150 mM NaCl, 1 mM EDTA). mRNA was eluted from the beads in 10 µL of 10 mM Tris-HCl ph 7.5 at 80 °C for 2 min followed by dropping to 20 °C. mRNA was rebound to the beads by addition of 200 µL lysis binding buffer and incubating 10 min at room temperature with rocking. mRNA-beads were washed with 200 µL wash buffer A, 200 µL wash buffer B, and 200 µL low salt buffer by gentle pipetting and collecting the mRNA-beads on the magnetic stand.
mRNA fragmentation
mRNA-beads were resuspended in 10 µL 2× first strand buffer (Invitrogen # 18064-014), incubated at 80 °C for 2 min and placed on ice. Supernatant containing fragmented mRNA was transferred to a new tube after 5 min on magnetic stand.
First strand cDNA synthesis
Random hexamers (1 µL of 3 µg/uL, Invitrogen #48190-011) and Superase N (1 µL of 20 U/µL, Ambion #AM2694) was added to fragmented mRNA and incubated at 65 °C for 5 min then placed on ice. DTT (2 µL of 0.1 M), dNTP mix (1.25 µL of 10 mM), Superscript II (1 µL of Invitrogen #18640-014) and water (4.75 µL) were then added to yield mRNA in 1× first strand buffer. After reverse transcription (25 °C 10 min, 42 °C for 50 min, 70 °C for 15 min, 10 °C hold), second strand synthesis was carried out immediately.
Second strand synthesis
Second strand synthesis reagents were added to the first strand reaction (4 µL 10× NEB2 buffer, 2 µL 10 mM dNTP mix, 2 µL RNaseH, 2 µL DNA Pol I, 1 µL 100 mM DTT, 10 µL water) and incubated at 16 °C for 2.5 h. cDNA was purified with 1.8 volume of AMPure XP following manufactures instructions (Beckman Coulter A63880).
Library preparation
Illumina RNAseq libraries were prepared by Rapid Genomics (http://rapid-genomics.com/home/) using dual barcodes.
Sequencing was performed using the Illumina HiSeq 2500 as either 2 × 150 bp or 2 × 50 bp reads. The second run was intended to provide extra coverage, and all replicates were sequenced in both runs. There were between 8.2 and 16.3 million reads per sample in total (~11.5 million reads on average and 1065 million reads in total).
Gene expression analysis
In higher eukaryotes exons often overlap with one another, and/or are shared across multiple isoforms (Fig. 1a). This complicates assessments of isoform abundance and gene expression when using short-read RNA-seq data. In the past, an exonic region was quantified without regard to differences in donor/acceptor sites (Dalton et al. 2013; Graze et al. 2014; Newell et al. 2016; Fear et al. 2016) to avoid double counting reads, and in regions where differences are small (less than 10 bp) there is no meaningful loss of information in this approach. However, the overlap is often much larger than this, so to address this issue we use a classification scheme where reads may be assigned to exons, exonic regions, or exon fragments. The primary difference is the separation of exonic regions and exon fragments, the latter of which is a subset of the former belonging to portions of individual exons that do or do not overlap (Fig. 1a). Exon boundaries were determined using the D. melanogaster FlyBase 6.17 genome features file. Reads are assigned to the exon category when the exon does not overlap any other exons, and may be unique to a single isoform, common to several isoforms, or constitutive to all isoforms (Fig. 1a). If an exon is both unique and constitutive, it indicates that the gene has one transcript. If the exon overlaps other exons, we grouped overlapping exons from the D. melanogaster FlyBase 6.17 genome features file into exonic regions. Exonic regions may be common or constitutive, however, as they by necessity of definition require overlap between exons from different isoforms they are never unique. Where exonic regions are comprised of overlapping exons that differ measurably, the 5′ and 3′ positions of exons within the exonic region are used to separate the region into exon fragments. Each exon fragment within an exonic region is annotated to indicate whether it is exclusive to a single exon (unique) or is shared among sets of exons (common or constitutive) (Fig. 1a). Note that in the case where a fusion contains exons from two genes, one of which is a gene with only a single transcript, once these are split into fragments the fragment that belongs to the single transcript can be both unique and constitutive (Fig. 1a). Distinct paired reads were aligned to the genome with BWA-MEM version 0.7.15 and BED files for exonic regions were used to count reads in the region and the length adjusted read count (reads in region divided by the length of region), the average per nucleotide (APN) (Li 2015).
As there was a mixture of read lengths for each sample (2 × 150 bp and 2 × 50 bp), the APN was summed for technical replicates of the same read length, then averaged between different read lengths. An exonic region was considered detected if the APN was greater than zero in at least half of all samples per condition. Several approaches were considered to the normalize the coverage counts, and upper-quartile normalization with log-transformation and median centering within time × treatment × genotype were selected due to better performance of the residuals (Bullard et al. 2010; Dillies et al. 2013).
To test main effects, the log APN for each exonic region was modeled as
for each genotype (g), treatment (t), and time point (m). The six genotypes are referred to using subscript i (gi), and the two treaments as j (tj; j = ethanol or no ethanol). Lastly the time points are differentiated using the subscript k (mk; k = 10, 20, 30 min), and lth replicate (three replicates per genotype per treatment per time point). For contrasts between treatments and time points, and the interaction between treatment and time point, the log APN for each exonic region was modeled as
for the ith condition (time × treatment) and jth replicate. Contrasts to compare treatments within time point (ethanol versus no ethanol, for 10, 20, and 30 min) were conducted. Residuals were evaluated for conformation with normality assumptions, and assumptions were met in excess of 95% of the models.
To evaluate whether there was evidence for splicing differences among times or treatments, exonic regions for each gene and for each sample were ranked and the most expressed region ranked as one, the least expressed exonic region as three and all others as two. Exon ranks for each gene were modeled as
where \({\mathrm{\Upsilon }}_{ijk}\) is the exon rank (1–3) of the ith exonic region of the gene, jth condition (time × treatment), and the kth replicate; ri is the exonic region of the gene; tj is condition; and \((rt)_{ij}\) is the interaction between exonic region and condition. Due to a lack of normality in the distribution of model residuals, a more traditional general linear model test could not be used to look for changes in exon or exonic region representation between exons of a gene, which is why we have chosen to use a rank test to summarize changes in exon representation (Supplemental File 4). F-tests for the significance of the mean square attributed to the effect tested versus the mean square attributed to error, or the appropriate interaction term, were used.
Correction for multiple testing
The false discovery rate (FDR) was controlled using the Benjamini–Hochberg procedure, with a significance cutoff of α = 0.05 (Benjamini and Hochberg 1995). Three gene lists were exempted from correction due to an a priori expectation that they would be involved in the response to ethanol (Supplemental File 1). This included (1) All genes documented as being involved in the response to alcohol in Drosophila or humans (with an ortholog in Drosophila), (2) immune and defense genes, given the well documented involvement of the immune system in response to ethanol (Troutwine et al. 2016), and (3) splicing factors and genes that are sex-specifically spliced. These a priori gene lists were filtered according to two criteria, first for all tests other than genotype only genes with an average fold change of at least ½ were retained. For differences at 10, 20, and 30 min an additional filter was applied, wherein the difference between relative abundance at ten and 30 min needed to be at least 0.1. This cutoff was determined empirically by visualizing the data, and serves largely to remove gene expression differences that occurred in the nonethanol environment rather than due to ethanol.
Results and discussion
Six heterozygous genotypes of D. melanogaster were used to identify changes in gene expression related to immediate (10 min), moderate (20 min), and severe (30 min) responses to ethanol exposure (Fig. 2a). Control flies were exposed to the same experimental setup, but the food was made entirely with water rather than 15% ethanol, as described in (Signor et al. 2017a, 2017b). In general, more than three replicates of each experiment were needed to produce enough material for an RNA library, and these were combined randomly to create three biological replicates for each genotype and six treatment/control types.
Gene expression and splicing cannot be fundamentally disentangled using short-read RNA-seq, therefore we approach quantification of differences in transcript representation by quantifying exons which do not overlap with other exons, exonic regions which represent a fused region of overlapping exons, and fragments, the decomposition of exonic regions (Fig. 1a). We will describe exons, exonic regions, and exonic fragments that were only detected in one environment and therefore could not have their expression level formally compared. We will summarize changes in the representation of exons, exonic regions, and fragments for each component of variance for the FDR corrected and a priori gene list (Supplemental File 1–3). Lastly, we will summarize the results of a rank test for changes in abundance of exons and exonic regions within a gene, which provides additional evidence for alternative splicing.
To determine the appropriateness of the genes selected for the a priori gene lists (defense, ethanol, and splicing) we wanted to test for an enrichment of significant genes in these categories compared to the main dataset. We chose to focus on one component of variance as the goal is only to demonstrate the appropriateness of the chosen gene lists (treatment). Using the same criteria for significance (p < 0.05, fold change in expression of 0.5) we determined the number of genes that would be identified in the entire dataset for the effect of ethanol. Using a χ2 test in R with simulated p values we found that every category was enriched for significant genes (defense, p = 0.0005, ethanol p = 0.0005, and splicing p = 0.01).
Events detected in a single environment
In addition to detecting changes in the representation of exons, exonic regions, and fragments, the presence and absence of a given exon in different environments is suggestive of differential exon usage. This cannot be formally approached as in the following sections, but they can be qualitatively understood—for example “not detected” refers to a variety of ambiguous situations such as detection in a single replicate or random minority of replicates, but without enough replicability to have confidence in its presence. A total of 644 exons and exonic regions were only detected with confidence in nonethanol environments, and 717 exons and exonic regions were only detected with confidence in ethanol environments (Fig. 1b). This includes six exons from Dscam1, which has 75 annotated transcripts and is important for synaptic targeting (Hattori et al. 2007). Totally, 465 exon fragments were only detected in nonethanol environments, while 418 exon fragments were only observed in the presence of ethanol (Fig. 1b). Among exons and exonic regions detected in both environments, they were far more likely to be constitutive than those detected in one or another environment, which is suggestive of alternative isoform usage (Fig. 1c). The same was true among exon fragments (to a lesser degree), which is expected given that a constitutive exon fragment is alternatively spliced (Fig. 1c).
GO enrichment of events detected only with ethanol
Exons and exonic regions present only in the ethanol environment were enriched for biological process gene ontology (GO) terms detection of stimulus involved in sensory perception and cilium assembly. This list was also enriched for molecular functions transmembrane signaling receptor activity. GO terms for cellular components were enriched for BBSome (a protein complex which traffics proteins to cilia) (Klink et al. 2017), intraciliary transport particles, neuron projection membrane, dendrite, transmembrane transporter complex, integral component of plasma membrane, and cytoskeletal part. In general these GO terms relate to known important components of the response to ethanol, suggesting that the presence of these exons and exonic regions in the ethanol treated flies may be functional. For example, alcohol-induced ciliary dysfunction is a known consequence of alcohol exposure (Yang et al. 2015; Price et al. 2017). As exon fragments are primarily a subset of the above list they were not tested independently for GO enrichment.
Genotype
A total of 1444 exons, 631 exonic regions, and 1135 exon fragments changed their expression level in response to genotype (Table 1). We are not interested in gene expression differences between genotypes per se, but this serves to demonstrate that we have the power to detect differences in thousands of genes, or potentially that gene expression differences due to genotype are large. Among the a priori gene lists regardless of treatment, 257 exons, 99 exonic regions, and 177 exon fragments from defense genes were different among genotypes. Totally, 132 exons, 59 exonic regions, and 28 exon fragments from ethanol-related genes varied among genotypes, as well as 133 exons, 55 exonic regions, and 123 exon fragments from splicing factors. The average expression difference between environments for genes implicated in genotype-specific differences is 0.0013, while for genes implicated in differences between treatments at 30 min it is 0.19, which suggests that detecting more genes in response to genotype is not due to larger differences in expression. Again, we are not inherently interested in differences between genotypes, but it serves to demonstrate our power to detect many differences.
Treatment
In all, 15 exons, 13 exonic regions, and 21 exon fragments changed their representation in response to ethanol, including many genes previously predicted to be involved in ethanol metabolism and response (Pinocchio, sugarbabe, cabut, Drat, CG32512) (Fig. 2c) (Morozova et al. 2006; Kong et al. 2010; Awofala et al. 2012). For example, in response to treatment an exonic region from CG32425 was significantly different, however among exonic fragments only one fragment from this multiexon region significantly changed its representation, suggesting that a particular isoform is changing its representation. Three exons implicated in the response to treatment were also unique but not constitutive, which suggests a change in isoform representation (CG32103, CG32512, fatty acid synthase 1). Representation of these exons and exonic regions was higher in ethanol, and expression was significantly correlated between environments (Fig. 1c, Spearman’s rho = 0.98, p < 2.2 × 10−16, 88%). Cabut is a transcription factor that has been observed previously as being upregulated early in response to ethanol, and potentially responsible for later changes in ethanol-related genes (Awofala et al. 2012). In general, cabut is highly regulated by stress and metabolic conditions (Havula and Hietakangas 2012; Bartok et al. 2015). Interestingly, many of the implicated genes are thought to belong to related pathways with essential roles in systemic metabolic regulation (Mattila et al. 2015). For example, dawdle (significant for treatment × time) and sugarbabe are thought to be direct targets of the Mondo-Mlx sugar sensing pathway, and sugarbabe expression was found to be dependent on dawdle (Mattila et al. 2015). While cabut is not directly regulating dawdle or sugarbabe it is also downstream of the Mondo-Mlx sugar sensing pathway, and it represses Phosphoenolpyruvate carboxykinase and circadian cycling of metabolic genes (Havula and Hietakangas 2012; Bartok et al. 2015). There is evidence linking the Mondo-Mlx pathway with human disease phenotypes such as severe obesity, high circulating triglycerides, and tumorigenesis (Kathiresan et al. 2008; Kooner et al. 2008; Herman et al. 2012; Carroll et al. 2015).
Among the a priori gene lists 24 exons, one exonic region, and 9 exon fragments from defense genes were significantly different between treatments, including the most frequent with five significant exons, Hemolectin, similar to the human von Willebrand factor (Goto et al. 2001), which in humans who drink heavily is associated with changes in the frequency of coagulating factors and with heart disease (Fig. 2c; Mukamal et al. 2001). Among ethanol-related genes 15 exons, one exonic region, and 12 exon fragments showed significantly different expression patterns due to treatment. Two of these exons are unique but not constitutive, suggesting a change in expression of a particular isoform. Three exons, two exonic regions, and eight exon fragments are different between treatments for splicing-related genes, including P-element somatic inhibitor (Fig. 2c). P-element somatic inhibitor controls the alternative splicing patterns of more than 1200 genes in the Drosophila brain, including those important for male courtship behavior such as fruitless (Wang et al. 2016). Expression of these genes is correlated between environments, though less so than in the main data set, and only a slim majority were more highly expressed in ethanol (Fig. 2c) (Spearman’s rho = 0.73, p = 8.525 × 10−9, 56%).
Genotype × treatment
For the interaction between genotype and treatment three exons, no exonic regions, and no exonic fragments were significant. Exons from Molecule interaction with CasL, wings apart like, and a noncoding RNA CR44660 altered their representation in response to the interaction between genotype and ethanol. CR44660 has only one exon and is not alternatively spliced, and the other two exons are common, thus no inferences about changes in isoform representation can be made. Note that for categories such as this with only three significantly different exons it is not reasonable to investigate correlations between environments. Furthermore, while response to treatment is an indication of plasticity, and a number of genes were implicated, three exons being implicated in genotype by treatment suggests that there is very little genetic variation for plasticity in these D. melanogaster genotypes. That is, all genotypes respond to ethanol in similar ways.
Four exons, no exonic regions, and four exon fragments from defense genes were significant, with the largest fold change in TGF-β activated kinase 1. The mammalian ortholog of TGF-β activated kinase 1 has been associated with increases in alcohol consumption and the development of liver disease (Harris and Blednov 2012). Four exons, one exonic region, and two exon fragments from ethanol-related genes are significant for the interaction between genotype and treatment, including Tao. Tao interacts with par-1 to control microtubule dynamics and behavioral responses to ethanol (King et al. 2011). Two exons, no exonic regions, and two exon fragments from splicing-related genes were significant, including P-element somatic inhibitor. Expression level of these exon fusions was significantly correlated between environments, and the majority were not more highly expressed in ethanol (Spearman’s rho = 0.75, p = 0.0085, 45%).
Time x treatment
There were 22 exons, 8 exonic regions, and 12 fragments with a significant interaction between treatment and time, including some aforementioned genes such as Drat and sugarbabe (Fig. 2). This includes several noncoding RNAs (CR44603 and CR44350), genes implicated in neurodegenerative disease (telomere fusion (Petersen et al. 2013)), and sugar-response (dawdle (Mattila et al. 2015)). None of the exons are unique and not constitutive, however, for example an exonic region containing four exons from the gene cabut is significant, but only one exon fragment from cabut is significantly different, suggesting differences in transcript representation. As this is the interaction between time and treatment we examined expression at 30 min, and expression of these genes is highly correlated between environments and expression was higher in ethanol (Spearman’s rho = 0.98, p < 2.2 × 10−16, 86%).
Four exons, no exonic regions, and four exon fragments from defense genes respond to treatment × time, including icarus (involved in ethanol-induced sedation (Ojelade et al. 2015)). icarus has also been shown to be linked to a network of genes implicated in ethanol self-administration, downstream of estimated glomerular filtration rate (EGFR) (Fochler et al. 2017). Two exons, one exonic region, and three exon fragments from ethanol-related genes are significant, where both exons are from NMDA receptor 1 and 2. NMDA receptors are thought to be the most sensitive of all glutamatergic receptors to the effects of ethanol, and the usage of different subunits within these receptors is seen in mice exposed to chronic ethanol (Rodan and Rothenfluh 2010; Most et al. 2014). Two exons, one exonic region, and four exon fragments have a significant interaction between treatment and time. Expression of these exons and exonic regions is not correlated between environments (Spearman’s rho = 0.37, p = 0.26), nor where they significantly correlated at any specific time point. In all, 54% of differences in expression were higher in ethanol environments.
Genotype × treatment × time
No exons, two exonic regions, and three exon fragments were significant for the interaction between treatment, genotype, and time. This suggests again that while there is plasticity for the response to ethanol (treatment and treatment × time), there is very little genetic variation for plasticity in D. melanogaster. Seven exons, no exonic regions, and six exon fragments from defense genes were significant for the interaction between all three main effects. Four exons, two exonic regions, and six exon fragments from ethanol-related genes had an interaction between genotype, treatment, and time, including Krasavietz which is known to affect ethanol preference (Devineni and Heberlein 2009, 2010). No exons, no exonic regions, and two fragments from splicing-related genes were significant. Relative average expression of exon fusions was weakly correlated overall but expression at individual time points was not, and a slim majority of expression differences were positive in ethanol (Spearman’s rho = 0.64, p = 0.02, 53%).
Differences between time points
While treatment by time interaction terms capture some of the effects of time since the exposure, here we follow with more biologically driven comparisons between three time points contrasting them between ethanol exposed and control flies (Figs. 2d and 3a). At 10 min, there is a significant differences in two exons, one exonic region, and three exon fragments in cabut, CG44004, and CR43803 in ethanol and nonethanol exposed flies (Fig. 2d; Fig. 3). All of the exon fragments are from cabut, and represent three of the four possible exon fragments that are a member of the exonic region, suggesting differences in the representation of particular transcripts. At 20 min this list includes one exon, four exonic regions, and five exonic fragments primarily from cabut, and Drat. Of the four exon fragments making up the exon fusion for cabut, only two change their expression, and in Drat one exonic region consisting of two fragments shows a change in expression of only one fragment, both suggesting differences in transcript usage (Fig. 3). At 30 min 46 exons, 24 exonic regions, and 20 exon fragments are different between ethanol exposed and unexposed flies (Figs. 2d and 3a). This includes the aforementioned Drat, dawdle, Phosphoenolpyruvate carboxykinase, cabut, sugarbabe, Pinocchio, and CG32512. Four exons are unique and not constitutive, suggesting differences in transcript representation. As another example, an exonic region from dawdle is composed of the overlap between four exons, however, only one of these fragments is significantly different at this time point, suggesting alternative transcript usage. The level of representation of these exons and exonic regions is highly correlated between environments, and largely higher in ethanol (Spearman’s rho = 0.98, p = 2.2 × 10−16, 94%). A gene involved in glycogen metabolism that was also found in a previous study was significantly different at 30 min (Glycogen binding subunit 70E) (Kong et al. 2010). Glycogen metabolism is an important mediator of the effects of long-term alcohol exposure, such as the development of fatty liver disease in humans (Gu et al. 2015). The fact that in the main dataset the vast majority of differences in representation were higher in the presence of ethanol suggests that we are not detecting preferential degradation of transcripts in the presence of ethanol. However, this has been shown to be one of the mechanisms for changes in the abundance of splice variants in Drosophila, for example there is preferential degradation of ethanol sensitive slowpoke isoforms (Cowmeadow et al. 2005, 2006; Pietrzykowski et al. 2008).

a Gene expression changes over time for genes that were significant in the main dataset at 10, 20, or 30 min (top row), and genes that were significant in the a priori dataset at 10, 20, or 30 min (bottom row). Each time point is shown for all genes, and the y axis is the relative difference in expression between environments. b Differences in exon representation in three genes that were frequently implicated—cabut, Drat, and sugarbabe. Intensity of color indicates level of expression of each exon (with the scale shown at the bottom of each gene). Each isoform is illustrated at the top of the image, with the representation of exons belonging to each isoform for each component of variation shown below
Overall, we observe that the number of significant effects increases with the time since exposure to ethanol, which may be for several reasons. Firstly, it is possible that early responding genes such as cabut require time to signal downstream genes. Second, genes that respond to ethanol exposure likely have different thresholds, and longer exposures may be required for the response of genes with higher thresholds. Lastly, the effect of ethanol may be mediated by its high nutritional value, as many genes which respond to ethanol also have metabolic roles. This latter interpretation is unlikely as we have not observed males engaged in extensive feeding in our behavioral observations of these experiments (Signor et al. 2017a, 2017b).
Defense genes exhibit the largest number of changes at 10 min, with 18 exons, 1 exonic region, and 6 fragments, compared to 20 and 30 min where 4 and 9 exons, 1 and 3 exonic regions, and 11 and 13 exon fragments exhibit significant differences (Figs. 2d and 3a). Ethanol-related genes show differences in 8 exons, 1 exonic region, and 4 fragments at 10 min, 8 exons, no exonic regions, and 4 exon fragments at 20 min, and 12 exons, 1 exonic region, and 5 fragments at 30 min (Figs. 2d and 3a). This includes many aforementioned genes such as lush (Ader et al. 2010), slowpoke, Ecdysone receptor, and 5-HT2A. Splicing-related genes are significantly different for 5 exons at 10 min, 4 exonic regions, and 15 exon fragments, 4 exons, 2 exonic regions, and 3 exon fragments at 20 min, and 5 exons, no exonic regions, and 9 exon fragments at 30 min (Figs. 2d and 3a). This includes fruitless and P-element somatic inhibitor. fruitless is a direct target of P-element somatic inhibitor (Wang et al. 2016). At 10, 20, and 30 min these exons and exonic regions were correlated between environments and more highly expressed under ethanol (Spearman’s rho = 0.67, p = 5.34 × 10−6, 73%; Spearman’s rho = 0.75, p = 0.0002, 70%; Spearman’s rho = 0.67, p = 4.25 × 10−5, 70%).
Shared and unique exons
For each component of variance described above the exons, exonic regions, and exon fragments may be unique, common, or constitutive (Fig. 2e). Exons and exonic regions were more likely to be unique and constitutive (meaning the gene has one transcript), when they were from the main FDR corrected dataset (Fig. 2e, top left). These genes will also not have differences in transcript usage, given that there is only one. From the a priori gene set they were more likely to be unique but not unique/constitutive, meaning they are exons that do not overlap other exons and are found in a single transcript among more than one possible transcript (Fig. 2e, top right). This is also one of the few cases where the change in expression can be attributed to a single transcript, as are changes in the representation of any unique exon fragment. Among exon fragments the main dataset was more likely to contain unique fragments (Fig. 2e, bottom left), meaning that a portion of the exon fusion belonged to a single transcript, indicative of differential transcript representation. The a priori gene set was far more likely to contain common fragments (Fig. 2e, bottom right), meaning that a portion of the exonic region belonged to several but not all transcripts of the gene. The presence of more unique exon fragments in the main dataset is suggestive of alternative exon usage, as the uniquely assigned fragment is changing its representation. An example of the complexity of the situation is given for several genes that were significant for many components of variance, cabut, Drat, and sugarbabe, in Fig. 3b. For example, cabut is composed of an exon fusion with four exon fragments. Only one of these fragments is unique to a particular isoform, and it is more frequently detected at 20 and 30 min than at 10 min, as well as more frequently than at any time point without ethanol.
Changes in rank abundance
Due to a lack of normality in the distribution of model residuals, we used a rank test to summarize changes in exon representation for the response to ethanol and ethanol by time. In response to treatment 71 genes altered the rank abundance of their constitutive exons and exonic regions. This includes many genes implicated previously in expression changes in response to ethanol, such as Acetyl Coenzyme A synthase, which is a known part of the alcohol dehydrogenase pathway (Montooth 2006; Kong et al. 2010; Yampolsky et al. 2012). Beadex was also significant and has been implicated in altered behavioral responses to alcohol and cocaine in Drosophila and mice (Lasek et al. 2011). Beadex was also inferred to be a part of a transcriptional response network to ethanol, as were par-1 and Darkener of apricot which also showed changes in rank abundance (Morozova et al. 2011). par-1 is involved in regulating ethanol-stimulated behavior through the mushroom body, though nothing about the potential role of different isoforms is documented (King et al. 2011). Immune-related genes with differences in rank abundance also include α-Tubulin at 84B, Pabp2, Relish (De Gregorio et al. 2002), Spatzle (Troutwine et al. 2016), and Serine protease 7 (Tang et al. 2006). Among the a priori gene lists 31 defense genes, 13 ethanol-related genes, and 18 splicing-related genes changed the rank abundance of their exons in response to ethanol. Of the ethanol-related genes, several genes, which are involved in ecdysone signaling or EGFR signaling were implicated. EGFR has been robustly linked to ethanol-related phenotypes, and it appears to contribute to the action of ethanol in the central nervous system. Inhibitors of EGFR increase the sensitivity of both Drosophila and mice to ethanol, (Corl et al. 2009; Forsyth et al. 2010). The interaction between ethanol treatment and time also induces 145 significant changes in rank abundance. This includes several genes implicated previously in the response to ethanol, including the aforementioned Acetyl Coenzyme A synthase, as well as homer (Urizar et al. 2007), IA-2 protein tyrosine phosphatase (Morozova et al. 2015), longitudinals lacking (Morozova et al. 2006, 2007), schizo (Morozova et al. 2006), FK506-binding protein 14 (Morozova et al. 2006), ligand-gated chloride channel homolog 3 (Morozova et al. 2007), and tipE homolog 2 (Derst et al. 2006; Ghezzi et al. 2013). Interestingly, a splice variant of longitudinals lacking is thought to regulate JIL-1 kinase, which is also significantly differently spliced in response to ethanol × time, and is important in the regulation of heterochromatization and gene expression (Zhang et al. 2003; Regnard et al. 2011; Silva-Sousa et al. 2012). 14-3-3ζ is thought to be an important component of cellular pathways involved in cancer (most prominently the Ras/MAPK cascade), it is known to be alternatively spliced, and it is significantly differently spliced (Ashton-Beaucage et al. 2014). It has also been reported as having different expression in the brains of alcoholic humans (Tiebe et al. 2015). Small ribonucleoprotein particle protein SmD1 modulates the RNA interference pathway as well as pre-mRNA splicing, suggesting that differences in the splicing of this gene could change the splicing of other genes as well as altering the inhibition of gene expression or translation of other genes (Xiong et al. 2013, 2015). Interestingly, we did not detect a change in splicing of slowpoke, which has previously been shown to have functional differences in isoform abundance in response to ethanol (Cowmeadow et al. 2005, 2006; Pietrzykowski et al. 2008). Among the a priori gene lists 38 exons or exonic regions from defense genes changed their rank abundance, 24 ethanol-related genes, and 27 splicing-related genes. cAMP signaling has long been recognized as important for the response to ethanol in Drosophila, and genes thought to respond to cAMP signaling such as rutabaga, protein kinase regulatory subunit 1, and dunce are represented here (Moore et al. 1998; Ron and Messing 2011; Ron and Barak 2016). Splicing-related genes included fruitless, dunce, glorund, and hu li tai shao. fruitless has been implicated previously in mediating ethanol sensitivity, however, this study did not control for baseline differences in male and female movement which are large (Devineni and Heberlein 2012).
GO enrichment analysis
Exons, exonic regions, and exon fragments collectively from all components of variance were not significantly enriched for any category of genes, which is not inherently surprising given the small number of exons and exonic regions implicated in the differences between components of variance (and that of these 15% are noncoding genes with no associated GO terms). However, genes that significantly changed the rank abundance of their exons in response to ethanol × time were enriched for cellular component actin cytoskeleton. The actin cytoskeleton is a well-recognized component of the response to ethanol, for example Rho genes which transduce extracellular signals to changes in the actin skeleton are alternatively spliced and implicated in the response to ethanol in Drosophila, mice, and humans (Rothenfluh et al. 2006; Iwata et al. 2011; Ojelade et al. 2015).
Comparison with other studies
While significant methodological differences exist between the current study and others that have been conducted with the same goal in mind, a comparison between gene lists may help to illuminate core moderators of the ethanol pathway. Unfortunately, we do not have access to enough raw data from these other studies to perform a test of the significance of overlap, but the results can be discussed qualitatively. Morozova et al. (2011) performed qPCR to model alcohol interaction networks, and two genes that were identified in this study overlap with our exons, exonic regions, or exon fragments: fatty acid synthase 1 and glycogen binding subunit 70E. In fact, many more genes were in common between our list of genes with significant changes in rank abundance, including kayak, outspread, brother of ihog, par-1, Beadex, Darkener of apricot, and longitudinals lacking. Morozova et al. (2011) states that they designed primers to encompass common exons, and perhaps it was in fact a confounding between isoform representation and expression that was detected. Kong et al. (2010) performed a microarray study of gene expression differences after a 30-min exposure, and during the recovery period. This identified many more genes than the Morozova et al. (2011) study, thus the expectation is that the overlap will be larger—and indeed fatty acid synthase 1, glycogen binding subunit 70E, Drat, and 9 other genes are shared between exons, exonic regions, and exon fragments and this study. Genes with exons who change their rank abundance also overlapped with the Kong et al. (2010) dataset, including the previously mentioned Acetyl Coenzyme A synthase, Jabba, Tetraspanin 42Eg and Ed, santa-maria, clot, Cyp4d8, CG10863, CG5326, and CG18302. Morozova et al. (2015) published another study on alcohol in D. melanogaster using GWA and extreme quantitative trait locus (QTL) mapping. This set of genes overlaps, again, more frequently with the rank abundance dataset where tweek, RNA binding protein 6, thioester-containing protein 4, CG5778, beat-ILLc, IA-2, and lingerer are found in both datasets. Among exons, exonic regions, exon fragments, and Morozova et al. (2015) only bunched and stonewall were shared, and between fragments forked is shared.
There are many differences between the methodology used in these studies—including the use of targeted qPCR (which requires candidate genes) (Morozova et al. 2011), microarrays which are notoriously unreliable (Kong et al. 2010), and differences in the assay type (inebriometer (Morozova et al. 2011, 2015), booz-o-mat (Kong et al. 2010)). There are also many differences in the amount of time that Drosophila were exposed to ethanol, and at what concentration, for example in most inebriometer studies the mean time to elution of the flies (when they fall through the contraption and can no longer right themselves) is 6 min. In our assays the flies were still active at 30 min, suggesting a lower dose of ethanol (Signor et al. 2017a, 2017b). As many of the targets of these studies will have been partial transcripts, it is possible that many of the detected changes included differences in splicing. Genes that are implicated in multiple studies may be especially good targets for future research, though we note that many genes that did not overlap with these lists have been validated in other systems or in single gene studies.
Summary
While many exons, exonic regions, and exon fragments, were detected in only one environment and ambiguously detected in the other, only a handful of genes that were detected in both environments showed a response to ethanol. In particular, while D. melanogaster showed a response to ethanol (treatment and treatment × time), there was little evidence for genetic variation in this response (genotype × treatment and genotype × treatment × time). As few as three to four exons, exonic regions, or exon fragments were implicated in genetic variation for the response to ethanol. While other work on the response to ethanol in D. melanogaster has found balanced polymorphisms due to heterogeneity in ethanol concentration, a lack of genetic variation for the response to the environment suggests that there has been selection on this reaction norm (Chakraborty and Fry 2016). Time was also an important component of the response to ethanol, with 23× as many exons and exonic regions responding at 30 min compared to 10 and 7× as many exon fragments. This reflects that the fact that the response to the environment has a temporal component and will be detected differently depending upon the time scale at which it is sampled, which may explain some of the discrepancies between other studies of environmental response (Saltz et al. 2018).
Furthermore, the response to ethanol has a much larger effect on rank abundance of exons than on the representation of particular exons or exonic regions that were detected in both environments. For example, only 28 exons and exonic regions were significantly different in response to treatment, and 31 in response to treatment and time. However, for rank abundance 71 genes were different in response to treatment and 146 genes were different in response to treatment over time. This is a markedly different picture in comparison with earlier papers in which hundreds or thousands of genes are differently expressed in response to ethanol treatment, even in response to much shorter exposures. This could be due to differences in methodology such as whole body versus head sequencing, or the use of RNA-seq as opposed to microarray. Rigorously controlling offspring quality and rearing environment likely also reduces variation amongst individuals. However, overlap between this and other studies lends extra confidence to the involvement of the identified genes.
There are some important patterns that differ between the main dataset and the a priori gene list, which bear mentioning. Firstly, the genes in the main dataset tend to be much more highly expressed than in the a priori gene list, and more highly correlated between environments. In addition, the vast majority of representation differences are positive in response to ethanol in the main dataset, while it is a small majority in the a priori dataset. While more highly represented genes are more easily detected in RNA-seq, the fact that they are more commonly positive and correlated between environments cannot be explained by coverage. Rapid responses to environmental changes are sometimes thought to be mediated more by preferential degradation of transcripts than de novo transcription, as the production of new transcripts and proteins will require a certain amount of time (López-Maury et al. 2008). However, the fact that the majority of representation differences in the main dataset were positive, and the time delay resulting in a larger number of gene expression differences suggests that this is not the primary mechanism in this case.
Changes in gene expression in response to ethanol have been quantified, however, past studies failed to incorporate the complexity of gene expression and splicing given the common overlap between exons and sharing of exons between isoforms. Changes in splicing in response to ethanol have been investigated in specific cases (namely slowpoke), but a broad survey that incorporates differences that can be attributable to both expression and splicing has not been performed. The fact that there are more changes in rank abundance in response to ethanol than changes in expression level of particular exons or exonic regions suggests that in D. melanogaster adaptation to ethanol may have been mediated by the regulation of splicing. In turn, splicing may be an important part of the response to ethanol. Understanding the functional differences between isoforms of a gene is rare, and it is a difficult question to answer given the limitations of our research methods. This will be an important area to consider for future research into alcohol use disorders (Table 2).
Data archiving
Sequence data have been submitted to GenBank: accession number PRJNA482662.
References
Ader L, Jones DNM, Lin H (2010) Alcohol binding to the odorant binding protein LUSH: multiple factors affecting binding affinities. Biochemistry 49:6136–6142
Aroor AR, James TT, Jackson DE, Shukla SD (2010) Differential changes in MAP kinases, histone modifications, and liver injury in rats acutely treated with ethanol. Alcohol Clin Exp Res 34:1543–1551
Ashton-Beaucage D, Udell CM, Gendron P, Sahmi M, Lefrançois M, Baril C et al. (2014) A functional screen reveals an extensive layer of transcriptional and splicing control underlying RAS/MAPK signaling in Drosophila. PLoS Biol 12:e1001809
Awofala AA (2010) Gene regulation in Drosophila melanogaster in response to an acute dose of ethanol. University of Sussex Ph.D. Diss
Awofala AA, Davies JA, Jones S (2012) Functional roles for redox genes in ethanol sensitivity in Drosophila. Funct Integr Genomics 12:305–315
Baralle FE, Giudice J (2017) Alternative splicing as a regulator of development and tissue identity. Nat Neurosci 18:437–451
Bartok O, Teesalu M, Ashwall-Fluss R, Pandey V, Hanan M, Rovenko BM et al. (2015) The transcription factor Cabut coordinates energy metabolism and the circadian clock in response to sugar sensing. EMBO J 34:1538–1553
Benjamini YB, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing J. R. Stat. Soc., B 57:289–300
Berger KH, Kong EC, Dubnau J, Tully T, Moore MS, Heberlein U (2008) Ethanol Sensitivity and Tolerancein in Long-Term Memory Mutants of Drosophila melanogaster. Alcohol Clin Exp R 32:895–908
Bossé Y (2013) Genome-wide expression quantitative trait loci analysis in asthma. Curr Opin Allergy Clin Immun 13:487–494
Bullard JH, Purdom E, Hansen KD, Dudoit S (2010) Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinforma 11:94–13
Campo D, Lehmann K, Fjeldsted C, Souaiaia T, Kao J, Nuzhdin SV (2013) Whole-genome sequencing of two North American Drosophila melanogaster populations reveals genetic differentiation and positive selection. Mol Ecol 22:5084–5097
Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J et al. (2015) Deregulated Myc requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 27:271–285
Caswell JL, Camarda R, Zhou AY, Huntsman S, Hu D, Brenner SE et al. (2015) Multiple breast cancer risk variants are associated with differential transcript isoform expression in tumors. Human Molecular Genetics 24:7421–7431
Cervera-Juanes R, Wilhelm LJ, Park B, Grant KA, Ferguson B (2017) Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Transl Psychiatry 7:e994
Chakraborty M, Fry JD (2016) Evidence that environmental heterogeneity maintains a detoxifying enzyme polymorphism in Drosophila melanogaster. Curr Biol 26:219–223
Chang P-J, Michaeloudes C, Zhu J, Shaikh N, Baker J, Chung KF et al. (2015) Impaired nuclear translocation of the Glucocorticoid receptor in corticosteroid-insensitive airway smooth muscle in severe asthma. Am J Respir Crit Care Med 191:54–62
Christodoulopoulos P, Leung DYM, Elliott MW, Hogg JC, Muro S, Toda M et al. (2000) Increased number of glucocorticoid receptor-β-expressing cells in the airways in fatal asthma. J Allergy Clin Imm 106:479–484
Corl AB, Berger KH, Ophir-Shohat G, Gesch J, Simms JA, Bartlett SE et al. (2009) Happyhour, a Ste20 family kinase, implicates EGFR signaling in ethanol-induced behaviors. Cell 137:949–960
Cowmeadow RB, Krishnan HR, Atkinson NS (2005) The slowpoke gene is necessary for rapid ethanol tolerance in Drosophila. Alcohol Clin Exp R 29:1777–1786
Cowmeadow RB, Krishnan HR, Ghezzi A, Al’Hasan YM, Wang YZ, Atkinson NS (2006) Ethanol tolerance caused by slowpoke induction in Drosophila. Alcohol Clin Exp R 30:745–753
Dalton JE, Fear JM, Knott S, Baker BS, McIntyre LM, Arbeitman MN (2013) Male-specific Fruitless isoforms have different regulatory roles conferred by distinct zinc finger DNA binding domains. BMC Genom 14:659
De Gregorio E, Spellman PT, Tzou P, Rubin GM, Lemaitre B (2002) The Toll and Imd pathways are the major regulators of the immune response in Drosophila. EMBO J 21:2568–2579
Derst C, Walther C, Veh RW, Wicher D, Heinemann SH (2006) Four novel sequences in Drosophila melanogaster homologous to the auxiliary Para sodium channel subunit TipE. Biochem Biophys Res Comm 339:939–948
Devineni AV, Heberlein U (2009) Preferential ethanol consumption in Drosophila models features of addiction. Curr Biol 19:2126–2132
Devineni AV, Heberlein U (2010) Addiction-like behavior in Drosophila. Commun Integr Biol 3:357–359
Devineni AV, Heberlein U (2012) Acute ethanol responses in Drosophila are sexually dimorphic. Proc Natl Acad Sci 109:21087–21092
Devineni AV, McClure K, Guarnieri D, Corl A, Wolf F, Eddison M et al. (2011) The genetic relationships between ethanol preference, acute ethanol sensitivity, and ethanol tolerance in Drosophila melanogaster. Fly 5:191–199
Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N et al. (2013) A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14:671–683
Dorado G, Barbancho M (1984) Differential responses in Drosophila melanogaster to environmental ethanol: modification of fitness components at the Adh locus. Heredity 53:309–320
Edenberg HJ, Foroud T (2013) Genetics and alcoholism. Nat Neurosci 10:487–494
Engel GL, Marella S, Kaun KR, Wu J, Adhikari P, Kong EC et al. (2016) Sir2/Sirt1 links acute inebriation to presynaptic changes and the development of alcohol tolerance, preference, and reward. J Neurosci 36:5241–5251
Farris SP, Miles MF (2012) Ethanol modulation of gene networks: implications for alcoholism. Neurobiol Dis 45:115–121
Fear JM, León-Novelo LG, Morse AM, Gerken AR, Van Lehmann K, Tower J et al. (2016) Buffering of genetic regulatory networks in Drosophila melanogaster. Genetics 203:1177–1190
Fochler S, Morozova TV, Davis MR, Gearhart AW, Huang W, Mackay TFC et al. (2017) Genetics of alcohol consumption in Drosophila melanogaster. Genes Brain Behav 39:W323–11
Forsyth CB, Tang Y, Shaikh M, Zhang L, Keshavarzian A (2010) Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial–mesenchymal transition in colon and breast cancer cells. Alcohol Clin Exp Res 34:19–31
Ghezzi A, Krishnan HR, Lew L, Prado FJ, Ong DS, Atkinson NS (2013) Alcohol-induced histone acetylation reveals a gene network involved in alcohol tolerance. PLoS Genet 9:e1003986
Gibson JB, Wilks AV (1988) The alcohol dehydrogenase polymorphism of Drosophila melanogaster in relation to environmental ethanol, ethanol tolerance and alcohol dehydrogenase activity. Heredity 60:403–414
Goehe RW, Shultz JC, Murudkar C, Usanovic S, Lamour NF, Massey DH et al. (2010) hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest 120:3923–3939
Goto A, Kumagai T, Kumagai C, Hirose J, Narita H, Mori H et al. (2001) A Drosophila haemocyte-specific protein, hemolectin, similar to human von Willebrand factor. Biochem J 359:99–108
Graze RM, McIntyre LM, Morse AM, Boyd BM, Nuzhdin SV, Wayne ML (2014) What the X has to do with it: differences in regulatory variability between the sexes in Drosophila simulans. Genome Biol Evol 6:818–829
Gu J, Zhang Y, Xu D, Zhao Z, Zhang Y, Pan Y et al. (2015) Ethanol-induced hepatic steatosis is modulated by glycogen level in the liver. J Lipid Res 56:1329–1339
Harris RA, Blednov YA (2012) Neuroimmune genes and alcohol drinking behavior. Cui, Changhai, Grandison, Lindsey, Noronha, Antonio (Eds.). Neural–immune interactions in brain function and alcohol-related disorders. Springer US, Boston, MA
Hattori D, Demir E, Kim HW, Viragh E, Zipursky SL, Dickson BJ (2007) Dscam diversity is essential for neuronal wiring and self-recognition. Nature 449:223–227
Havula E, Hietakangas V (2012) Glucose sensing by ChREBP/MondoA–Mlx transcription factors. Sem Cell Dev Biol 23:640–647
Heberlein U, Wolf FW, Rothenfluh A, Guarnieri DJ (2004) Molecular genetic analysis of ethanol intoxication in Drosophila melanogaster. Integr Comp Biol 44:269–274
Herman MA, Peroni OD, Villoria J, Schön MR, Abumrad NA, Blüher M et al. (2012) A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484:333–338
Hyman SE, Malenka RC (2001) Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2:695–703
Iwata T, Nozu F, Imawari M (2011) Ethanol impairs the assembly and disassembly of actin cytoskeleton and cell adhesion via the RhoA signaling pathway, catenin p120 and E-cadherin in CCK-stimulated pancreatic acini. Biochem Biophys Res Commun 405:558–563
Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ et al. (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40:189–197
Kaun KR, Azanchi R, Maung Z, Hirsh J, Heberlein U (2011) A Drosophila model for alcohol reward. Nat Neurosci 14:612–619
Kędzierska H, Popławski P, Hoser G, Rybicka B, Rodzik K, Sokół E et al. (2016) Decreased expression of SRSF2 splicing factor inhibits apoptotic pathways in renal cancer. IJMS 17:1598–16
King I, Tsai LTY, Pflanz R, Voigt A, Lee S, Jackle H et al. (2011) Drosophila tao controls mushroom body development and ethanol-stimulated behavior through par-1. J Neurosci 31:1139–1148
Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, Wittinghofer A (2017) A recombinant BBSome core complex and how it interacts with ciliary cargo. eLife 6:743
Kong EC, Allouche L, Chapot PA, Vranizan K, Moore MS, Heberlein U et al. (2010) Ethanol-regulated genes that contribute to ethanol sensitivity and rapid tolerance in Drosophila. Alcohol Clin Exp Res 34:302–316
Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, Warnes GR et al. (2008) Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet 40:149–151
Lai MKP, Esiri MM, Tan MGK (2014) Genome-wide profiling of alternative splicing in Alzheimer’s disease. GDATA 2:290–292
Lasek AW, Giorgetti F, Berger KH, Tayor S, Heberlein U (2011) Lmo genes regulate behavioral responses to ethanol in Drosophila melanogaster and the mouse. Alcohol Clin Exp R 35:1600–1606
Li H (2015). Aligning sequence reads, clone sequences and assemble contigs with BWA-MEM arXiv 1–3.
Li L, Lyu X, Hou C, Takenaka N, Nguyen HQ, Ong C-T et al. (2015) Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell 58:216–231
Li X, Hastie AT, Hawkins GA, Moore WC, Ampleford EJ, Milosevic J et al. (2015) eQTL of bronchial epithelial cells and bronchial alveolar lavage deciphers GWAS-identified asthma genes. Allergy 70:1309–1318
López-Maury L, Marguerat S, Bähler J (2008) Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet 9:583–593
MacKay RK, Colson NJ, Dodd PR, Lewohl JM (2011) Differential expression of 14-3-3 isoforms in human alcoholic brain. Alcohol Clin Exp Res 35:1041–1049
Mattila J, Havula E, Suominen E, Teesalu M, Surakka I, Hynynen R et al. (2015) Mondo-Mlx mediates organismal sugar sensing through the Gli-Similar transcription factor sugarbabe. Cell Rep 13:350–364
McClure KD, French RL, Heberlein U (2011) A Drosophila model for fetal alcohol syndrome disorders: role for the insulin pathway. Dis Model Mech 4:335–346
Milan NF, Kacsoh BZ, Schlenke TA (2012) Alcohol consumption as self-medication against blood-borne parasites in the fruit fly. Curr Biol 22:488–493
Montooth KL (2006) Membrane lipid physiology and toxin catabolism underlie ethanol and acetic acid tolerance in Drosophila melanogaster. J Exp Biol 209:3837–3850
Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U (1998) Ethanol intoxication in Drosophila: Genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell 93:997–1007
Morozova TV, Anholt RRH, Mackay TFC (2007) Phenotypic and transcriptional response to selection for alcohol sensitivity in Drosophila melanogaster. Genome Biol 8:R231
Morozova TV, Ayroles JF, Jordan KW, Duncan LH, Carbone MA, Lyman RF et al. (2009) Alcohol Sensitivityin in Drosophila: Translational Potential of Systems Genetics. Genetics 183:733–745
Morozova TV, Huang W, Pray VA, Whitham T, Anholt RRH, Mackay TFC (2015) Polymorphisms in early neurodevelopmental genes affect natural variation in alcohol sensitivity in adult Drosophila. BMC Genomics 16:865
Morozova TV, Mackay TFC, Anholt RRH (2011) Transcriptional networks for alcohol sensitivity in Drosophila melanogaster. Genetics. 187:1193–1205
Morozova T, Anholt R, Mackay TF (2006) Transcriptional response to alcohol exposure in Drosophila melanogaster. Genome Biol 7:R95
Most D, Ferguson L, Harris RA (2014) Molecular basis of alcoholism. Handb Clin Neurol 125:89–111
Mukamal KJ, Jadhav PP, D’Agostino RB, Massaro JM, Mittleman MA, Lipinska I et al. (2001) Alcohol consumption and hemostatic factors: analysis of the Framingham offspring cohort. Circulation 104:1367–1373
Mukherjee M, Svenningsen S, Nair P (2017) Glucocortiosteroid subsensitivity and asthma severity. Curr Opin Pulm Med 23:78–88
Naftelberg S, Schor IE, Ast G, Kornblihtt AR (2015) Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem 84:165–198
Newell NR, New FN, Dalton JE, Genes LMG Genomes, 2016 2016) Neurons that underlie Drosophila melanogaster reproductive behaviors: detection of a large male-bias in gene expression in fruitless-expressing neurons G3 6:2455–2465
Nieuwenhuis MA, Siedlinski M, van den Berge M, Granell R, Li X, Niens M et al. (2016) Combining genome-wide association study and lung eQTL analysis provides evidence for novel genes associated with asthma. Allergy 71:1712–1720
Ojelade SA, Jia T, Rodan AR, Chenyang T, Kadrmas JL, Cattrell A et al. (2015) Rsu1 regulates ethanol consumption in Drosophila and humans. Proc Natl Acad Sci USA 112:E4085–E4093
Oomizu S, Boyadjieva N, Sarkar DK (2003) Ethanol and Estradiol modulate alternative splicing of Dopamine D2 receptor messenger RNA and abolish the inhibitory action of bromocriptine on prolactin release from the pituitary gland. Alcohol Clin Exp Res 27:975–980
Pandey SC, Ugale R, Zhang H, Tang L, Prakash A (2008) Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28:3729–3737
Paronetto MP, Passacantilli I, Sette C (2016) Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ 23:1919–1929
Petersen AJ, Katzenberger RJ, Wassarman DA (2013) The innate immune response transcription factor relish is necessary for neurodegeneration in a Drosophila model of ataxia-telangiectasia. Genetics 194:133–142
Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM et al. (2008) Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron 59:274–287
Pohl JB, Baldwin BA, Dinh BL, Rahman P, Smerek D, Prado FJ et al. (2012) Ethanol preference in Drosophila melanogaster is driven by its caloric value. Alcohol Clin Exp Res 36:1903–1912
Poon AH, Eidelman DH, Martin JG, Laprise C, Hamid Q (2012) Pathogenesis of severe asthma. Clin Exp Allergy 42:625–637
Price M, Yang F, Sisson JH, Wirschell M (2017) Ciliary dynein dysfunction caused by chronic alcohol exposure. Steven M. King (Ed.) Dyneins. Second Edn Academic Press, San Diego
Regnard C, Straub T, Mitterweger A, Dahlsveen IK, Fabian V, Becker PB (2011) Global analysis of the relationship between JIL-1 Kinase and transcription. PLoS Genet 7:e1001327
Rodan AR, Rothenfluh A (2010) The genetics of behavioral alcohol responses in Drosophila. Int Rev Neurobiol 91:25–51
Ron D, Barak S (2016) Molecular mechanisms underlying alcohol-drinking behaviours. Nat Rev Neurosci 17:576–591
Ron D, Messing RO (2011) Signaling pathways mediating alcohol effects. Curr Top Behav Neurosci 13:87–126
Rothenfluh A, Threlkeld RJ, Bainton RJ, Tsai LTY, Lasek AW, Heberlein U (2006) Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell 127:199–211
Saltz JB, Bell AM, Flint J, Gomulkiewicz R, Hughes KA, Keagy J (2018) Why does the magnitude of genotype-by-environment interaction vary? Ecol Evol 8:6342–6353
Sasabe T, Ishiura S (2010) Alcoholism and alternative splicing of candidate genes. IJERPH 7:1448–1466
Scholz H, Ramond J, Singh CM, Heberlein U (2000) Functional ethanol tolerance in Drosophila. Neuron 28:261–271
Schuckit MA, Smith TL (2011) Onset and course of alcoholism over 25 years in middle class men. Drug Alcohol Depend 113:21–28
Signor SA, Abbasi M, Marjoram P, Nuzhdin SV (2017a) Social effects for locomotion vary between environments in Drosophila melanogaster females. Evolution 71:1765–1775
Signor SA, Abbasi M, Marjoram P, Nuzhdin SV (2017b) Conservation of social effects (Ψ) between two species of Drosophila despite reversal of sexual dimorphism. Ecol Evol 7:10031–10041
Silva-Sousa R, López-Panadès E, Piñeyro D, Casacuberta E (2012) The chromosomal proteins JIL-1 and Z4/Putzig regulate the telomeric chromatin in Drosophila melanogaster. PLoS Genet 8:e1003153
Tang H, Kambris Z, Lemaitre B, Hashimoto C (2006) Two proteases defining a melanization cascade in the immune system of Drosophila. J Biol Chem 281:28097–28104
Tiebe M, Lutz M, La Garza De A, Buechling T, Boutros M, Teleman AA (2015) REPTOR and REPTOR-BP regulate organismal metabolism and transcription downstream of TORC1. Dev Cell 33:272–284
Treistman SN, Martin GE (2009) BK channels: mediators and models for alcohol tolerance. Trends Neurosci 32:629–637
Troutwine BR, Ghezzi A, Pietrzykowski AZ, Atkinson NS (2016) Alcohol resistance in Drosophila is modulated by the Toll innate immune pathway. Genes Brain Behav 15:382–394
Urizar NL, Yang Z, Edenberg HJ, Davis RL (2007) Drosophila homer is required in a small set of neurons including the ellipsoid body for normal ethanol sensitivity and tolerance. J Neurosci 27:4541–4551
Wang Q, Taliaferro JM, Klibaite U, Hilgers V, Shaevitz JW, Rio DC (2016) The PSI-U1 snRNP interaction regulates male mating behavior in Drosophila. Proc Natl Acad Sci USA 113:5269–5274
Xiong X-P, Kurthkoti K, Chang K-Y, Lichinchi G, De N, Schneemann A et al. (2013) Core small nuclear ribonucleoprotein particle splicing factor SmD1 modulates RNA interference in Drosophila. Proc Natl Acad Sci USA 110:16520–16525
Xiong X-P, Vogler G, Kurthkoti K, Samsonova A, Zhou R (2015) SmD1 modulates the miRNA pathway independently of its pre-mRNA splicing function. PLoS Genet 11:e1005475
Yampolksy L, Glazko GV, Fry JD (2012) Evolution of gene expression and expression plasticity in long-term experimental populations of Drosophila melanogaster maintained under constant and variable ethanol stress. Mol Ecol 21:4287–4299
Yang H-P, Nuzhdin SV (2003) Fitness costs of Doc expression are insufficient to stabilize its copy number in Drosophila melanogaster. Mol Biol Evol 20:800–804
Yang F, Pavlik J, Fox L, Scarbrough C, Sale WS, Sisson JH et al. (2015) Alcohol-induced ciliary dysfunction targets the outer dynein arm. Am J Physiol Lung Cell Mol Physiol 308:L569–L576
Zaharieva E, Chipman JK, Soller M (2012) Alternative splicing interference by xenobiotics. Toxicology 296:1–12
Zhang W, Wang Y, Long J, Girton J, Johansen J, Johansen KM (2003) A developmentally regulated splice variant from the complex lola locus encoding multiple different zinc finger domain proteins interacts with the chromosomal kinase JIL-1. J Biol Chem 278:11696–11704
Zhou Z, Yuan Q, Mash DC, Goldman D (2011) Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci USA 108:6626–6631
Zraly CB, Dingwall AK (2012) The chromatin remodeling and mRNA splicing functions of the Brahma (SWI/SNF) complex are mediated by the SNR1/SNF5 regulatory subunit. Nucleic Acids Res 40:5975–5987
Acknowledgments
The authors would like to thank Jeremy Newman and Lauren McIntyre for contributions to the manuscript. The authors would like to thank J. Butler and T. Bobert for advice on this manuscript, and N. Shadman, V. Paterson, Z. Polonus, K. Cortez, L. Hassanzadeh, L. Cline, A. Khokhar, E. Lee, K.L. Yee, M. Ling, S. Sarva, O. Akintonwa, A. Gupta, R. Manson, P. Hassanzadeh, and K. Kavoussi for assistance in the production of this data.
Funding
This work was supported by grants GM102227 and MH09156.
Author contributions
S.S. performed the experiments, analyzed the dataset, and wrote the manuscript. S.V.N. conceived of the experiment and assisted in writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Please note: Supplemental Files 1–4 are currently available at: https://github.com/signor-molevol/melanogaster_alcohol.git
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Signor, S., Nuzhdin, S. Dynamic changes in gene expression and alternative splicing mediate the response to acute alcohol exposure in Drosophila melanogaster. Heredity 121, 342–360 (2018). https://doi.org/10.1038/s41437-018-0136-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0136-4
- Springer Nature Switzerland AG
This article is cited by
-
It is not just about transcription: involvement of brain RNA splicing in substance use disorders
Journal of Neural Transmission (2024)
-
Genome- and transcriptome-wide splicing associations with alcohol use disorder
Scientific Reports (2023)
-
Alternative splicing in seasonal plasticity and the potential for adaptation to environmental change
Nature Communications (2022)
-
Studying alcohol use disorder using Drosophila melanogaster in the era of ‘Big Data’
Behavioral and Brain Functions (2019)