
Abstract
Ecological speciation is an important factor in the diversification of plants. The distribution of the woody species Rhododendron indicum, which grows along rivers and is able to withstand water flow when rivers flood (i.e. it is a rheophyte), is disjunct, in contrast to the widespread distribution of its relative, Rhododendron kaempferi. This study aimed to elucidate the phylogenetic relationships between R. indicum and R. kaempferi and the evolutionary processes that gave rise to them. The sequences of three non-coding chloroplast DNA regions (total length 1977 bp) were obtained from 21 populations covering the ranges of the two species. In addition, genome-wide SNPs were genotyped from 20 populations using a genotyping by sequencing method. Leaf morphologies were measured for eight representative populations. Two chloroplast DNA haplotypes, which were detected in R. indicum, were shared between the two species. Genome-wide SNPs identified two lineages in R. indicum and these lineages did not constitute a monophyletic group. Each of these two lineages was related to geographically close populations of R. kaempferi. Leaf morphology, which is a characteristic feature in rheophytes, was not differentiated between the two lineages in R. indicum. The morphological similarity between the two heterogeneous lineages may be a result of parallel evolution from R. kaempferi or of introgressive hybridization between the species due to strong selective pressure imposed by flooding.
Similar content being viewed by others


Introduction
Adaptive evolution to diverse habitats is one of the fundamental mechanisms that shape biodiversity, and studies of evolution in response to novel environments can provide many insights facilitating an understanding of how biodiversity has been formed and maintained (Rundle and Nosil 2005; Hendry et al. 2007; Schluter 2009). Because the adaptive process increases the fitness of populations in particular environments and since natural selection can favor the evolution of similar forms and functions in distant taxa, if a selective pressure that is significant at the population level operates in geographically discrete regions, similar phenotypes, or ecotypes may originate independently in such locations (Streisfeld and Rausher 2009; Arnold et al. 2016). The adaptive parallel evolution of an ecotype within a species or among species has been observed in varied environments and taxa (Foster et al. 2007; Turner et al. 2010; Ostevik et al. 2012; Renaut et al. 2014; Trucchi et al. 2017). However, when investigating parallel evolution, it is difficult to distinguish between the effects of natural selection, hybridization or incomplete lineage sorting due to ancestral polymorphisms, though polyphyletic topology for a specific phenotype indicates evidence of parallel evolution (Twyford and Ennos 2012; Roda et al. 2013). Although there is no doubt that natural selection affects adaptive evolution, the evolutionary histories of adaptive species are still poorly understood in most taxa.
Accumulating lines of evidence suggest that riparian environments have had a significant influence on the adaptive evolution of riparian plants. Plant populations growing along rivers are subject to disturbance due to flooding after heavy rains, and this can have a significant effect on plant survival. The narrow and thick leaves of riparian plants, referred to as rheophytes, growing along rivers are considered to be a consequence of adaptation enabling them to tolerate spates of water (van Steenis 1981, 1987; Tsukaya 2002; Mitsui et al. 2011; Ueda et al. 2012). The plants grow at or above water level, and experience stress due to water flow when it rains heavily and the water level rises. Rheophytes have advantages in terms of inhabiting the riparian environment because of their specialized phenotype; however, they cannot survive outside this environment (Mitsui et al. 2011). Rheophytes are recognized in various taxa including angiosperms as well as pteridophytes and bryophytes (van Steenis 1981; Kato 2003). Most relatives of rheophytes grow in regions close to, but outside, riparian environments. This suggests that rheophytes have speciated within certain regions under the strong natural selection pressure imposed by riparian environments (Mitsui et al. 2011; Mitsui and Setoguchi 2012). For instance, over 23 taxa of rheophytes are recognized in the Japanese archipelago; in addition, some of them have close relatives, which are not rheophytes, in forests on the islands (Tsukaya 2002; Kato 2003; Yatabe et al. 2009; Mitsui and Setoguchi 2012).
Rhododendron is one of the world’s most diversified woody taxa, comprising over 1000 species; its range extends from arctic-alpine to tropic environments, thus the diversity of the genus has arisen following radiation to various habitats (Goetsch et al. 2005). In the Japanese archipelago, a riparian azalea, Rhododendron indicum (L.) Sweet, which belongs to the subgenus Tsutsusi, is known; it grows on exposed rocks along rivers, and the species has a relative, Rhododendron kaempferi Planch., in forests on the islands (Yamazaki 1996; Kato 2003). Rhododendron indicum and R. kaempferi are distinguished by the narrow leaves of the former, a characteristic of rheophytes. This morphology results from a decrease in the number of cells across the width of the leaf in both the adaxial epidermis and palisade tissues (Setoguchi and Kajimaru 2004). In addition, the two species have different flowering seasons: from late May to early July for R. indicum and from early April to May for R. kaempferi (Yamazaki 1996). Rhododendron indicum is horticulturally important; its narrow leaves, small mature size and late flowering season have long been favored by breeders (Ito 1692). In this study, we aimed to elucidate the evolutionary process of a rheophyte with a disjunct distribution by using sequences from three non-coding chloroplast DNA regions and a genotyping by sequencing method, multiplexed inter-simple sequence repeat (ISSR) genotyping by sequencing (MIG-seq; Suyama and Matsuki 2015), together with evidence from plant morphology. The findings of the present study may provide insights into the significance of selective pressure in the evolution of a rheophyte from a forest relative.
Materials and methods
Study species
Rhododendron indicum and its relative, R. kaempferi are deciduous or semi-evergreen species endemic in Japan. Rhododendron indicum is distributed in the central part of Honshu and in Yakushima, which is distant (over 500 km) from Honshu, whereas the related forest species, R. kaempferi, is widely distributed in Hokkaido, Honshu, Shikoku, and Kyushu, but is not found in Yakushima (Fig. 1). Rhododendron indicum occurs on rocky sites along rivers from 0 to 1000 m above sea level; this riparian species has narrow leaves (0.4–1 cm wide and 1–3 cm long) and mature individuals are 0.5–1 m tall (Yamazaki 1996). Rhododendron kaempferi occurs in forests or at forest edges from 0 to 1500 m above sea level; it has wide oblong leaves (1.5–4 cm wide, 2–7 cm long) and mature individuals are 1–5 m tall. Species of Rhododendron are pollinated by bumblebees and the seeds are dispersed by wind; in addition, seeds of R. indicum are dispersed by flowing water, as is also the case for another riparian species, R. ripense (Kondo et al. 2009). Other features, such as flower and fruit morphologies, are quite similar in the two species and they are therefore considered to be close relatives. Because R. kaempferi is widely distributed whereas R. indicum has a characteristic habitat, it has been considered that the riparian species speciated from the more widespread relative. The distributions of the two species overlap in Honshu, although their microhabitats are different.

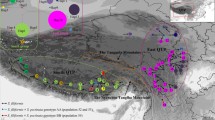
a Distributions of species in this study and chloroplast DNA haplotypes. Dots and triangles indicate locations of the populations analyzed for Rhododendron indicum and R. kaempferi respectively. Lowercase, non-bold letters indicate population codes corresponding to those in Table 1. Dashed and solid pie charts indicate frequencies of chloroplast DNA haplotypes for R. indicum and R. kaempferi respectively. Gray areas show the range of distribution of Rhododendron indicum and lines show major rivers on the islands. A haplotype network estimated by the parsimony method is superimposed on the map. It shows the relationships among the haplotypes detected in R. indicum and R. kaempferi using eight outgroup species. b, c Individual-based genetic structure elucidated by Bayesian clustering analysis. b Geographical distributions of ancestry for the two Rhododendron species when K = 3, a value which is supported by ΔK statistics, and c distributions of ancestry when the number of clusters ranged from K = 2 to 5
Plant sampling and DNA extraction
Leaf samples for molecular analysis were collected from 10 populations of Rhododendron indicum and from 11 populations of the closely related species, R. kaempferi (Table S1, Fig. 1), across their ranges. In addition, samples of R. macrosepalum and R. ripense were collected for use as outgroups. Leaves for DNA analysis were immediately dried using silica gel. Leaves for morphological analysis were collected from eight representative populations for each of the two species. Leaves to be used for analyzing leaf morphologies were stored in a refrigerator.
Genomic DNA was extracted using a modified CTAB (cetyltrimethylammonium bromide) method (Murray and Thompson 1980) or a DNeasy Plant Mini kit (Qiagen, Hilden, Germany) after clean-up treatment using sorbitol buffer to remove polysaccharides (Wagner et al. 1987).
Chloroplast DNA sequence analysis
Three non-coding regions of chloroplast DNA (trnL intron (Taberlet et al. 1991), trnG intron (Shaw et al. 2005), and rpl32-trnL (Shaw et al. 2007)) were sequenced from 84 samples (an average of 4.0 samples per population). PCRs were carried out in 5.0-μL volumes, each containing 10 ng of template DNA, 2.5 μL of PCR AmpliTaq Gold Master Mix (Applied Biosystems, Foster City, CA, USA) and 0.2 μm of each primer pair. PCR was performed with an initial denaturation for 4 min at 94 °C, followed by 35 cycles of denaturation for 1 min at 94 °C, annealing for 60 s at 60 °C and extension for 1 min at 72 °C, with a final extension for 7 min at 72 °C. After precipitation of PCR products using polyethylene glycol 8000 (Hartley and Bowen 1996), sequencing of PCR products was performed directly using a BIGDYE Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems, Foster City, CA, USA) and the sequencing products were separated by electrophoresis on an ABI 3130xl Genetic Analyzer (Applied Biosystems). Chloroplast DNA sequences were edited and assembled using DNA Baser 4 (Heracle BioSoft, Pitești, Romania) and aligned using MUSCLE implemented in MEGA 5 (Edgar 2004; Tamura et al. 2011). A haplotype network was constructed using TCS 1.06 (Clement et al. 2000).
MIG-seq experiment
Multiplexed ISSR genotyping by sequencing (MIG-seq) was used for genome-wide SNP detection; in this technique, loci between two ISSRs are amplified by PCR and sequence analysis is carried out using a next-generation sequencer (Suyama and Matsuki 2015). Two MIG-seq libraries were prepared following the protocol outlined in Suyama and Matsuki (2015). Repeat motifs and anchor sequences used for the ISSR amplification were as follows: (ACT)4TG, (CTA)4TG, (TTG)4AC, (GTT)4CC, (GTT)4TC, (GTG)4AC, (GT)6TC, (TG)6AC. The first round of PCR was conducted using these ISSR primers with tail sequences. The products of these first PCR reactions were diluted and used for the second round of PCR. This was conducted using primer pairs including tail sequences, adapter sequences for Illumina sequencing and six-base barcode sequences to identify each individual sample. The products of the second-round PCR for each individual were multiplexed in the size range of 300–800 bp and sequenced on an Illumina MiSeq platform (Illumina, San Diego, CA, USA) using an MiSeq Reagent Kit v3 (Illumina).
SNP detection
The MIG-seq data were de-multiplexed and low-quality reads were removed from raw reads using FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit). Reads excluding ISSR sequences (the remaining 80 bp) were used for de novo assembly. Prior to de novo assembly, samples with fewer than 105 reads were removed, and 158 samples (an average of 7.9 samples per population) were used for SNP detection. SNPs were called using Stacks 1.29 (Catchen et al. 2013). The minimum depth option for creating a stack was set to 10 (-m 10), and default settings for other options were used. After creating stacks, we extracted SNPs for population analysis. SNPs with a minor allele frequency (MAF) of at least 0.01 (-min_maf 0.01), with a genotyping rate of at least 20% of individuals within populations (-r 0.2) and present in half of the populations (-p 10) were extracted using Populations in Stacks. In addition, the FIS value for each SNP within each population was calculated using Populations, and SNPs whose FIS value was less than −0.75 or more than 0.75 were removed in order to exclude SNPs that were located on organelle genome and/or rich in null alleles. Pairwise R2 values for each SNP pair were calculated with PLINK 1.90b (Purcell et al. 2007), and if a value was higher than 0.5, an SNP locus exhibiting a low genotyping rate was removed so as to exclude linkages between SNPs. The extraction of these SNPs was conducted using whitelist in Populations.
In addition, we extracted another SNP set for demographic analysis. The objective of producing the SNP set was to force the coalescence of individuals within each lineage by regrouping samples in each population according to geographical group, and to obtain as many SNPs as possible by defining groups including many individuals. In the data set, we defined four groups: R. indicum in Honshu (in1–in8), R. indicum in Yakushima (in9 and in10), the eastern lineage of R. kaempferi (ka1–ka4 and ka6) and the western lineage of R. kaempferi (ka7–ka11), based on phylogenetic relationships among populations and genetic structure (see Results). SNPs that were observed in the four groups were extracted. Other settings in Populations were the same as in the methods used for the population analysis. Pairwise FST values between regions were calculated using GENEPOP 4.6 (Weir and Cockerham 1984; Raymond and Rousset 1995).
Population analysis
The number of polymorphic SNPs, observed and expected heterozygosity and fixation index for each population were calculated using Populations in Stacks. Phylogenetic relationships among populations were evaluated by constructing a neighbor-net based on DA distances between populations (Nei et al. 1983) using SplitTree 4 (Bryant and Moulton 2004). Significances for nodes were evaluated from bootstrap probabilities based on 1000 replicates of the neighbor-joining method using POPTREE2 (Takezaki et al. 2010). The individual-based genetic structure was estimated by model-based Bayesian clustering analysis using STRUCTURE (Pritchard et al. 2000). The F-model and admixture models were assumed (Falush et al. 2003). Ten independent MCMC simulations for each number of clusters (K = 1–10) were run with 80,000 iterations after a burn-in period of 40,000. The optimal number of clusters was determined by the log-likelihood of the data, Ln Pr(X|K) (Pritchard et al. 2000), and the ΔK statistic, which was calculated from the second-order rates of changes of Ln Pr(X|K) (Evanno et al. 2005).
Demographic analysis
The process of evolution of the two species, including the evolutionary origin of the two lineages within R. indicum, was inferred from SNPs using approximate Bayesian computation (ABC) implemented in DIYABC 2.1.0 (Bertorelle et al. 2010; Cornuet et al. 2014). DIYABC allows the testing of evolutionary scenarios with estimated divergence times, admixture and changes in population size among groups. We used SNPs that were called from four population groups, which were defined based on the genetic structure and distributions of the two species (see SNP detection). To infer the evolutionary pattern of the two groups in R. indicum, we tested five evolutionary scenarios using the same prior settings (Fig. 2a, Table S2). Scenario 1, a simple split model, assumed that all groups diverged at the same time t3. Scenario 2, a speciation model, was one in which the two species diverged at time t3 and two groups within each species (i.e. N1 and N2 for R. indicum and N3 and N4 for R. kaempferi) diverged at time t1 or t2. In scenario 3, a parallel evolution model, the western (N1 and N3) and eastern groups (N2 and N4), which were not concordant with species definitions, diverged at time t3, and each of the R. indicum groups (N1 and N2) diverged from geographically close R. kaempferi groups (N3 or N4) at time t1 or t2. Scenario 4, a speciation and subsequent admixture for R. indicum in Yakushima (N1), posited that the two species diverged at time t3, two groups within R. kaempferi (N3 and N4) diverged at time t2, and R. indicum in Yakushima (N1) was created by an admixture of R. indicum in Honshu (N2) and R. kaempferi in western region (N3). Scenario 5, a speciation and subsequent admixture for R. indicum in Honshu (N2), assumed that the two species diverged at time t3, two groups within R. kaempferi (N3 and N4) diverged at time t2, and R. indicum in Honshu (N2) was created by an admixture of R. indicum in Yakushima (N1) and R. kaempferi in eastern region (N4). In these scenarios, population size changes were allowed for each population, and in scenarios 4 and 5, admixture by migration was allowed. In total, five million simulations were run using the following summary statistics implemented in the software: proportion of zero values, mean of non-zero values and variance of non-zero values of the genetic diversity for each group (Nei 1987), pairwise Nei’s genetic distances between groups (Nei 1972) and the admixture summary statistics among groups (Choisy et al. 2004). These summary statistics showed a low level of discrepancy between observed and simulated data. The simulations generated a reference table containing about one million simulations per scenario. Scenarios were compared by directly counting the frequencies of the various scenarios in the 1% of simulated data sets closest to the observed data (direct approach) and by performing logistic regression of each scenario probability fores the closest simulated data sets on the differences between simulated and observed summary statistics (logistic approach; Cornuet et al. 2008). In addition, we evaluated the degree of confidence in scenario choice by estimating type I and type II errors; this was estimated by simulating 1000 pseudo-observed data sets drawn from a parameter prior distribution under each scenario. The 1% of simulated data sets closest to the observed data were used to estimate the posterior probabilities for the most likely scenario by means of local linear regression. Scaled divergence time (T) was estimated by multiplying generation time by divergence time (t). Estimating generation time is difficult for woody species because of their long maturation times, which are also variable among individuals; we used 10 years as the generation time for the species based on seeding experiments carried out in controlled environments (Morimoto et al. 2003; Yoichi personal observation).

a Demographic models examined for four populations of Rhododendron indicum and R. kaempferi, estimated by approximate Bayesian computation and b performance of the five models evaluated by direct and logistic regression estimates
Leaf morphological analysis
Leaf morphologies were measured in order to evaluate morphological variation between riparian and non-riparian species. Leaf area, width and length were measured on 79 individuals from eight populations. Because species of Rhododendron have different types of shoots and leaves during spring and summer (the latter is known as lammas growth), 15 spring leaves per individual were used for these measurements. Leaves were scanned using a digital scanner and digital images were analyzed by SHAPE 1.3 to calculate leaf area (Iwata and Ukai 2002). Leaf width and length were measured to the nearest 0.5 mm. Morphological differences between riparian and non-riparian species were evaluated by PCA using the prcomp function implemented in R 3.2.3 (R Development Core Team 2015).
Results
Distribution of chloroplast DNA haplotypes for the two species
The sequences of three cpDNA loci were obtained from 40 individuals of Rhododendron indicum and 44 individuals of R. kaempferi. The lengths of the aligned sequences for the three loci were 481 bp of the trnL intron, 545 bp of the trnG intron, and 951 bp for the rpl32-trnL. These sequences identified five haplotypes across the two species. The phylogenetic analysis revealed that the five haplotypes formed a group and the haplotypes were distinguished by one or two steps of substitution from each other (Fig. 1a). Rhododendron indicum has two haplotypes (H1 and H2) and R. kaempferi has all of the haplotypes identified in this study; thus H1 and H2 are shared by the two species. One of the shared haplotypes (H1) was recognized in four populations of R. indicum and half of the R. kaempferi populations. Another shared haplotype (H2) was recognized in populations of R. indicum and R. kaempferi in the central part of Honshu.
Phylogeographic patterns of SNPs for the two species
The total and average number of reads obtained from 158 individuals were 28,743,212 and 179,646 respectively. The de novo assembly yielded 675 loci, including 144 variable loci, for 20 populations after applying the filtering setting. The average read depth for individuals was 46.0. The data set produced 168 SNPs, with a 72.4% of genotyping rate for individuals. The number of polymorphic SNPs (S) within populations ranged from 9 to 47 for R. indicum and from 19 to 41 in R. kaempferi. The expected heterozygosity (HE) ranged from 0.018 to 0.079 for R. indicum and from 0.033 to 0.071 for R. kaempferi. The geographical distribution of values for these estimates tended to be low in northern populations and high in southern populations of R. indicum (Table 1). The fixation index (FIS) had a negative value for almost all populations.
The neighbor net based on the DA distance between populations across the two species did not show R. indicum to be monophyletic (Fig. 3). Populations of R. indicum were grouped into two phylogenetic groups, corresponding to the populations in Honshu and those in Yakushima. The populations in Yakushima (in9 and in10) and the other populations were distinguished by the highest possible bootstrap probability (100%). Phylogenetically, populations that were closest to the populations in Yakushima were populations of R. kaempferi in Kyushu (ka8−ka11). Populations of R. indicum in Honshu and the other populations were distinguished by a high bootstrap probability (96%). The genetic structure estimated by Bayesian clustering analysis supported K = 3 as the optimal number of clusters on the basis of ΔK statistics. In the case of K = 3, populations of R. indicum in Honshu and Yakushima were grouped into different clusters; in addition, populations of R. indicum in Yakushima and populations of R. kaempferi in the western region were grouped into the same cluster (Fig. 1b, c). On the other hand, when it was assumed K = 2, populations of R. indicum in Honshu and populations of R. kaempferi in the eastern region were grouped into the same cluster. When it was assumed that K = 4, populations of R. indicum in Honshu and Yakushima and those of R. kaempferi in the western and eastern regions were clearly distinguished, forming four clusters (Fig. 1c).

Phylogenetic relationships among populations constructed based on DA distance using the neighbor-net method. Bootstrap probabilities that exceeded 80% based on 1000 replicates are shown above nodes
Demographic analysis
The data set that SNPs were called from four population groups based on genetic structure and phylogenetic relationships produced 477 SNPs by de novo assembly. The pairwise genetic differentiation estimates (FST) between the four regions identified, i.e. R. indicum in Honshu (in1–in8); R. indicum in Yakushima (in9 and in10); the eastern lineage of R. kaempferi (ka1–ka4 and ka6); and the western lineage of R. kaempferi (ka7–ka11), ranged from 0.155 to 0.301 (Table S3). In the ABC analysis, scenario 3, which indicated that R. indicum in Yakushima and in Honshu diverged from R. kaempferi in the western and the eastern regions respectively, was supported by both direct (0.4636, 95% CI, 0.4198–0.5073) and logistic estimates (0.9999, 95% CI, 0.9998–1.0000, Fig. 2b). Sixty summary statistics showed few differences between observed and simulated data based on the posterior distributions. The type I error for scenario 3 (probability of erroneous rejection even if it was the true scenario) was 0.072 for the direct approach and 0.007 for the logistic regression approach, and the type II error for scenario 3 (probability of its being erroneously selected even if it was not the true scenario) was 0.010 for the direct approach and 0.002 for the logistic regression approach. The posterior mode and 95% highest posterior probability density (HPD) for the scaled divergence time between R. indicum in Yakushima (N1) and R. kaempferi in the western region (N3) was 179 ka (86.6-438) and that between R. indicum in Honshu (N2) and R. kaempferi in the eastern region (N4) was 205 ka (89.7–704) (Table 2, Supplementary Fig. 1). The posterior probability for the effective population size of R. indicum in Yakushima (N1) was lower than those for the other populations.
Morphological divergence between the two species
Leaf morphologies were measured on 585 leaves from 39 individuals of R. indicum and 585 leaves from 39 individuals of R. kaempferi. Leaf areas were 1.411 cm2 (SD, 0.540 cm2) and 5.159 cm2 (2.203 cm2) for R. indicum and R. kaempferi respectively. Leaf lengths were 2.736 cm (0.553 cm) and 4.349 cm (0.917 cm) for R. indicum and R. kaempferi respectively. Leaf widths were 0.877 cm (0.208 cm) and 2.192 cm (0.487 cm) for R. indicum and R. kaempferi respectively. The two species could be distinguished by principal component analysis of these data, whereas the two groups within R. indicum, which were distinguished by genetic analyses, were not separated on either of the two axes (Fig. 4).

Leaf morphology variation in R. kaemferi and R. indicum detected by principal component analysis. Open circles, black filled circles, and gray filled circles indicate average values of principal component scores for each individual of R. kaempferi, R. indicum on Honshu and R. indicum on Yakushima, respectively
Discussion
The large genetic divergence between the two lineages in R. indicum
The genetic evidence obtained in this study did not reflect the traditional taxonomic relationships between the species, since Rhododendron indicum consisted of two genetically distinct lineages; the species appears to have evolved independently from R. kaempferi in the distant regions, Honshu and Yakushima. When evolution occurs rapidly, few polymorphisms accumulate within and among species. In this study, we identified few chloroplast DNA variations so that only five haplotypes were identified within the two species and two haplotypes shared between them; these polymorphisms resulted from screening using many universal primers (Kress et al. 2005; Shaw et al. 2007). In addition, pairwise FST also showed low values between nearby populations of the two species rather than that between far populations within R. indicum. These results indicate that the two species are closely related.
The genetic structure, which was elucidated by Bayesian clustering analysis, was clearly different among populations across the two species; the pattern of genetic structure corresponded not to species difference but rather to the geographical distribution of populations within the two species. In the case of K = 2, populations of R. indicum in Yakushima and populations of R. kaempferi in the western region were assigned to the same cluster, and populations of R. indicum in Honshu and populations of R. kaempferi in the eastern region were assigned to the other cluster. As the number of clusters increased, populations of R. indicum in Yakushima and Honshu were clearly distinguished, failing into different clusters from those of the nearest R. kaempferi populations. In particular, when K = 4 was assumed, the four clusters corresponded largely to the four groups of populations. Whatever the number of clusters, clusters seldom contained admixtures of individuals and populations, indicating that there was clear genetic differentiation or genetic structure among the four genetic groups (i.e. populations of R. indicum in Yakushima and Honshu and populations of R. kaempferi in the western and eastern regions) and implying that there had been few migrations among groups (Pritchard et al. 2000). The analysis indicated that the two lineages of R. indicum are heterogeneous. In addition, the two groups of R. indicum did not form a monophyletic group, and were distinguished from R. kaempferi populations with high bootstrap probability (100 and 96%) according to the neighbor-joining method. The networks in the neighbor-net representing the possibility of migration and hybridization, which are not adequately modeled by a single tree, showed simple relationships between the species, and thus again indicates little migration between the species (Bryant and Moulton 2004). These results suggest the possibility of two origins for R. indicum.
The possibility of parallel evolution in R. indicum
The demographic analysis suggests that the two groups of R. indicum have evolved independently (scenario 3); the two scenarios in which secondary contact took place after the speciation event (scenarios 4 and 5) were rejected. In the scenario, age estimates obtained by demographic analysis imply that the two independent evolutionary processes have occurred over short evolutionary timescales (ca. 86–704 ka). Rheophytes are recognized in various taxa, including angiosperms as well as pteridophytes and bryophytes. The phenotypic differences in R. indicum compared to R. kaempferi are its narrow leaves, small mature height, and late flowering season; in contrast, the flower morphology of the two species is very similar. Despite significant polyphyly in R. indicum, leaf morphologies cannot be distinguished between the two lineages of the species; they are both characteristic of rheophytes. Similar morphologies between the two lineages, compared to the large phenotypic variation observed within R. kaempferi, are likely to have resulted from natural selection due to major flood disturbance and evolutionary constraint acting on R. indicum (Cronk 1998; Mitsui and Setoguchi 2012). For instance, Yakushima is an island that has a high rate of annual precipitation, up to 7500 mm per annum, due to the monsoon. The frequently flooding environment of the island is essential for sustaining rheophyte populations and many rheophytes, some of them endemic to Yakushima, are distributed here (Takahara and Matsumoto 2002; Kato 2003; Mitsui and Setoguchi 2012). The results may suggest that the lineage of R. indicum evolved, and has since been maintained, in this region. Although a high level of genetic diversity (HE) was observed within populations in the island, the effective population size on the island was lower than those for the other groups as estimated by demographic analysis. This may be a result of natural selection or an effect of the restricted habitat available for the species on the island (Lande, 1976). The demographic analysis also suggests that the evolution of two groups in R. indicum has not been influenced by any admixture between the two species, despite the proximity of their geographical distributions to those of R. kaempferi. The isolation between the species may be explained through differences in flowering time, geographical isolation (e.g. R. kaempferi is not distributed in Yakushima), or the existence of an ecological barrier that prevents hybridization between species because the low fitness level of hybrid descendants imposes strong natural selection (Nosil et al. 2005; Mitsui et al. 2011). These are known to be important processes in parallel evolution (Rundle and Schluter 2004; Roda et al. 2013; Renaut et al. 2014).
Although the possibility of parallel evolution in R. indicum should be noted, it should be treated with caution even though the data fit the model, because the evolutionary pattern may have been confounded by the small number of SNPs produced in this study. An insufficient degree of polymorphism in samples analyzed due to restricted genealogies causes incomplete lineage sorting; it is then difficult to distinguish among parallel evolution, ancestral polymorphisms, and migration after divergence, especially for closely related species (Nagl et al. 1998; Meyer et al. 2016). Although the 477 SNPs used in the demographic analysis carried out in the present study are fewer than those employed in the next-generation sequencing techniques (thousands to tens of thousands of SNPs for RAD-seq), the polymorphisms identified are expected to be similarly, or more, informative in comparison with previous studies, which used dozens of microsatellites (Haasl and Payseur 2011; Andrews et al. 2016; Kolář et al. 2016). In addition, the type II error for scenario 3 showed a low probability. The number of SNPs is therefore considered to be sufficiently informative for the analysis. However, evidence for hybridization in the genome may be unclear when a hybridization event has been introgressive (Seehausen 2004; Minder et al. 2007). In addition, because the scenarios in this study did not model secondary contacts following divergence or admixture, we were not able to detect such events in the genome by using only a small number of SNPs (Liu et al. 2014). For these reasons, it will be necessary to investigate the evolutionary process further by other methods. Even if we cannot reject the scenario that either of the R. indicum groups has evolved through introgressive hybridization, it is noteworthy that the two groups of R. indicum are heterogeneous despite their morphological similarity. If processes such as those referred to above have influenced either of the lineages, the similarity of the leaf morphology in the two lineages could have evolved and be maintained in response to strong selective pressure imposed by flooding (Mitsui et al. 2011; Roda et al. 2013; Sakaguchi et al. 2017).
Data archiving
The sequences of chloroplast DNA haplotypes reported in this study were deposited in GeneBank: accession numbers LC363560−LC363583. Genotype data were deposited in Dryad: doi:10.5061/dryad.tk563.
References
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92
Arnold BJ, Lahner B, DaCosta JM, Weisman CM, Hollister JD, Salt DE et al. (2016) Borrowed alleles and convergence in serpentine adaptation. Proc Natl Acad Sci USA 113:8320–8325
Bertorelle G, Benazzo A, Mona S (2010) ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Mol Ecol 19:2609–2625
Bryant D, Moulton V (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21:255–265
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Choisy M, Franck P, Cornuet JM (2004) Estimating admixture proportions with microsatellites: comparison of methods based on simulated data. Mol Ecol 13:955–968
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R et al. (2014) DIYABCv2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinforma (Oxf, Engl) 30:1187–1189
Cornuet J-M, Santos F, Beaumont MA, Robert CP, Marin J-M, Balding DJ et al. (2008) Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinforma (Oxf, Engl) 24:2713–2719
Cronk QCB (1998) The ochlospecies concept. In: Huxley CR, Lock JM, Cutler DF (eds) Chronology, taxonomy and ecology of the floras of Africa and Madagascar. Royal Botanic Gardens, Kew, pp 155–170
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Foster SA, McKinnon GE, Steane DA, Potts BM, Vaillancourt RE (2007) Parallel evolution of dwarf ecotypes in the forest tree Eucalyptus globulus. New Phytol 175:370–380
Goetsch L, Eckert AJ, Hall BD (2005) The molecular systematics of Rhododendron (Ericaceae): a phylogeny based upon RPB2 gene sequences. Syst Bot 30:616–626
Haasl RJ, Payseur BA (2011) Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity 106:158–171
Hartley JL, Bowen H (1996) PEG precipitation for selective removal of small DNA fragments. Focus (San Fr, Calif) 18:27
Hendry AP, Nosil P, Rieseberg LH (2007) The speed of ecological speciation. Funct Ecol 21:455–464
Ito I (1692) Kinsyu-makura. Matsue-Sanshiro, Edo
Iwata H, Ukai Y (2002) SHAPE: a computer program package for quantitative evaluation of biological shapes based on elliptic Fourier descriptors. J Hered 93:384–385
Kato M (2003) Evolution and adaptation in the rheophytes. Bunrui 3:107–122
Kolář F, Fuxová G, Záveská E, Nagano AJ, Hyklová L, Lučanová M et al. (2016) Northern glacial refugia and altitudinal niche divergence shape genome-wide differentiation in the emerging plant model Arabidopsis arenosa. Mol Ecol 25:3929–3949
Kondo T, Nakagoshi N, Isagi Y (2009) Shaping of genetic structure along Pleistocene and modern river systems in the hydrochorous riparian azalea, Rhododendron ripense (Ericaceae). Am J Bot 96:1532–1543
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci USA 102:8369–8374
Lande R. (1976) Natural selection and random genetic drift in phenotypic evolution. Evolution 30:314–334
Liu S, Lorenzen ED, Fumagalli M, Li B, Harris K, Xiong Z et al. (2014) Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157:785–794
Meyer BS, Matschiner M, Salzburger W (2016) Disentangling incomplete lineage sorting and introgression to refine species-tree estimates for lake Tanganyika cichlid fishes Syst Biol 66:531–550
Minder AM, Rothenbuehler C, Widmer A (2007) Genetic structure of hybrid zones between Silene latifolia and Silene dioica (Caryophyllaceae): evidence for introgressive hybridization. Mol Ecol 16:2504–2516
Mitsui Y, Nomura N, Isagi Y, Tobe H, Setoguchi H (2011) Ecological barriers to gene flow between riparian and forest species of Ainsliaea (Asteraceae). Evol; Int J Org Evol 65:335–349
Mitsui Y, Setoguchi H (2012) Demographic histories of adaptively diverged riparian and non-riparian species of Ainsliaea (Asteraceae) inferred from coalescent analyses using multiple nuclear loci. BMC Evolut Biol 12:254
Morimoto J, Shibata S, Hasegawa S (2003) Habitat requrement of Rhododendron reticulatum and R. macrosepalum in germination and seedling stages—field expriment for restoration of native Rhododendrons by seeding. J Jpn Soc Reveg Technol 29:135–140. (in Japanese)
Murray M, Thompson W (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326
Nagl S, Tichy H, Mayer WE, Takahata N, Klein J (1998) Persistence of neutral polymorphisms in Lake Victoria cichlid fish. Proc Natl Acad Sci USA 95:14238–14243
Nei M (1987) Molecular evolutionary genetics.. Columbia University Press, New York
Nei M (1972) Genetic distance between populations. Am Nat 106:283–291
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19:153–170
Nosil P, Vines TH, Funk DJ (2005) Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evol; Int J Org Evol 59:705–719
Ostevik KL, Moyers BT, Owens GL, Rieseberg LH (2012) Parallel ecological speciation in plants? Int J Ecol 2012:939862
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Human Genet 81:559–575
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Raymond M, Rousset F (1995) GENEPOP(version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Renaut S, Owens GL, Riseberg LH (2014) Shared selective pressure and local genomic landscape lead to repeatable patterns of genomic divergence in sunflowers. Mol Ecol 23:311–324
Roda F, Ambrose L, Walter GM, Liu HL, Schaul A, Lowe A et al. (2013) Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Mol Ecol 22:2941–2952
Rundle HD, Nosil P (2005) Ecological speciation. Ecol Lett 8:336–352
Rundle HD, Schluter D (2004) Natural selection and ecological speciation in sticklebacks. In: Dieckmann U, Doebeli M, Metz J, Tautz D (eds) Adaptive speciation. Cambridge University Press, Cambridge, pp 192–209
Sakaguchi S, Horie K, Ishikawa N, Nagano AJ, Yasugi M, Kudoh H et al. (2017) Simultaneous evaluation of the effects of geographic, environmental and temporal isolation in ecotypic populations of Solidago virgaurea. New Phytol 216:1268–1280
Schluter D (2009) Evidence for ecological speciation and its alternative. Science (New York, NY) 323:737–741
Seehausen O (2004) Hybridization and adaptive radiation. Trends Ecol Evol (Personal Ed) 19:198–207
Setoguchi H, Kajimaru G (2004) Leaf morphologyof the rheophyte, Rhododendeon indicum f. otakumi (Ericaceae). Geobot Acta Phytotaxa 55:45–54
Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J et al. (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot 92:142–166
Shaw J, Lickey EB, Schilling EE, Small RL (2007) Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot 94:275–288
Streisfeld MA, Rausher MD (2009) Genetic changes contributing to the parallel evolution of red floral pigmentation among Ipomoea species. New Phytol 183:751–763
Suyama Y, Matsuki Y (2015) MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci Rep 5:16963
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Takahara H, Matsumoto J (2002) Climatological study of precipitation distribution in Yaku-shima Island, Southern Japan. J Geogr 111:726–746
Takezaki N, Nei M, Tamura K (2010) POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–752
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Trucchi E, Frajman B, Haverkamp THA, Schönswetter P, Paun O (2017) Genomic analyses suggest parallel ecological divergence in Heliosperma pusillum (Caryophyllaceae). New Phytol 216:267–278
Tsukaya H (2002) Leaf anatomy of a rheophyte, Dendranthema yoshinaganthum (Asteraceae), and of hybrids between D. yoshinaganthum and a closely related non-rheophyte, D. indicum. J Plant Res 115:329–333
Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet 42:260–263
Twyford AD, Ennos RA (2012) Next-generation hybridization and introgression. Heredity 108:179–189
Ueda R, Minamiya Y, Hirata A, Hayakawa H, Muramatsu Y, Saito M et al. (2012) Morphological and anatomical analyses of rheophytic Rhododendron ripense Makino (Ericaceae). Plant Species Biol 27:233–240
van Steenis C (1981) Rheophytes of the world. Sijithoff & Noordhoff, Alphen aan den Rijin
van Steenis C (1987) Rheophytes of the world: supplement allertonia. Pacific Tropical Botanical Garden, Lawai
Wagner CE, Keller I, Wittwer S, Selz OM, Mwaiko S, Greuter L et al. (2013) Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol Ecol 22:787–798
Wagner DB, Furnier GR, Saghai-Maroof MA, Williams SM, Dancik BP, Allard RW (1987) Chloroplast DNA polymorphisms in lodgepole and jack pines and their hybrids. Proc Natl Acad Sci USA 84:2097–2100
Weir B, Cockerham C (1984) Estimating F-statics for the analysis of population structure. Evol; Int J Org Evol 38:1358–1370
Yamazaki T (1996) A revision of the genus Rhododendron in Japan, Taiwan, Korea and Sakhalin. Tsumura Laboratory, Tokyo
Yatabe Y, Tsutsumi C, Hirayama Y, Mori K, Murakami N, Kato M (2009) Genetic population structure of Osmunda japonica, rheophilous Osmunda lancea and their hybrids. J Plant Res 122:585–595
Acknowledgements
We are grateful to I. Honda, S. Fukumoto, S. Tagane, S. Sakaguchi, K. Higuchi, K. Yukitoshi, T. Morishige for their help in collecting plant material, and I. Tamaki for discussing and providing helpful comments on an early version of the manuscript.
Funding
The study was supported by a Grant-Aid for Scientific Research (16K18714) for the Japan Society for the Promotion of Science and JSPS Core-to-Core Program, A. Advanced Research Networks.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Yoichi, W., Kawamata, I., Matsuki, Y. et al. Phylogeographic analysis suggests two origins for the riparian azalea Rhododendron indicum (L.) Sweet. Heredity 121, 594–604 (2018). https://doi.org/10.1038/s41437-018-0064-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0064-3
- Springer Nature Switzerland AG
This article is cited by
-
The domestication and breeding history of Castanea crenata Siebold et Zucc. estimated by direction of gene flow and approximate Bayesian computation
Tree Genetics & Genomes (2023)
-
Population genetic structure of wild Angelica acutiloba, A. acutiloba var. iwatensis, and their hybrids by atpF–atpA intergenic spacer in chloroplast DNA and genome-wide SNP analysis using MIG-seq
Journal of Natural Medicines (2023)
-
From East Asia to Beringia: reconstructed range dynamics of Geranium erianthum (Geraniaceae) during the last glacial period in the northern Pacific region
Plant Systematics and Evolution (2022)
-
Phylogeographic and demographic modeling analyses of the multiple origins of the rheophytic goldenrod Solidago yokusaiana Makino
Heredity (2021)
-
Evidence of local adaptation despite strong drift in a Neotropical patchily distributed bromeliad
Heredity (2021)