
Abstract
The involvement of necroptosis in the control of influenza A virus (IAV) infection has been reported in multiple studies. Downstream of the nucleic acid sensor ZBP1, RIPK3 kinase activity is critically involved in the induction of necroptotic cell death by phosphorylating MLKL, while RIPK3 as a scaffold can induce apoptosis. Paradoxically, RIPK3-deficiency of mice may result in increased or decreased susceptibility to IAV infection. Here, we critically review the published reports on the involvement of RIPK3 in IAV infection susceptibility and try to identify differences in experimental settings that could explain seemingly conflicting outcomes. Analysis of the experimental reports revealed differences in the IAV challenge dose, the IAV inoculum preparation, IAV titer assessment, as well as the route of inoculation between studies. Furthermore, differences were noticed in the inclusion of littermate controls, which show high variance in viral sensitivity. Our evaluation argues for a standardized setup for IAV infection experiments including the preparation of the IAV virus, the use of different IAV infectious doses description and the proper experimental genetic controls of the mouse strains to increase inter-laboratory consistency in this field.

Workflow for IAV infection studies in vivo: Viral preparation and titer assessment should be as standardized as possible with the use of a universal repository (such as BEI resources). Infection studies in genetically modified mice and littermate controls should include dose-response experimentation, following a defined infection route and inoculation volume. Data are generated by consistent analysis methods.
Similar content being viewed by others

Implication of pattern recognizing receptors and cell death modalities during IAV infection
Influenza A virus (IAV) infection of epithelial cells of the upper and lower respiratory tract and lung parenchyma leads to cell death contributing to inflammation and barrier loss [1]. Besides apoptosis, necroptosis and pyroptosis are triggered both in epithelial and immune cells [2, 3] leading to an inflammatory response by releasing proinflammatory cytokines and danger-associated molecular patterns (DAMPs) [4]. IAV is recognized by the innate immune system by at least three distinct classes of pattern-recognition receptors (PRRs) including the Toll-like receptors TLR3, −7 and −8 (recognizing IAV nucleic acids), the retinoic acid-inducible gene I (RIG-I) (recognizing the 5′- triphosphate RNA) and the NOD-like receptor family member NOD-, LRR- and pyrin domain-containing 3 (NLRP3) (recognizing different stimuli) [5, 6]. The activation of TLR3 by IAV dsRNA activates the production of nuclear factor-κB (NF-κB)-dependent cytokines and of type I and III interferons (IFN) [6]. Viral RNA in the cytosol is recognized by RIG-I which activates the mitochondrial antiviral signaling protein (MAVS), inducing the production of pro-inflammatory cytokines and type I and III IFN [7]. The matrix 2 (M2) proton-selective ion channel of IAV at the Golgi apparatus initiates the assembly of the NLRP3 inflammasome which leads to the caspase-1-mediated activation of interleukin-1β (IL-1β) and IL-18 [8]. Caspase-1 also cleaves and activates gasdermin D (GSDMD) resulting in pyroptosis. Z-DNA binding protein 1 (ZBP1) is another innate immune sensor that recognizes Z-RNA generated during IAV-infection leading to RHIM-mediated recruitment and activation of receptor interacting kinase 3 (RIPK3) followed by apoptosis and necroptosis [9]. During IAV-infection ZBP1 can also engage pyroptosis by caspase-1 activation and gasdermin D (GSDMD) pore formation [10] (Supplementary Figure 1).
RIPK3 at crossroad between apoptosis and necroptosis during IAV infection
In mouse and human the RIPK1, RIPK3, ZBP1 and TIR-domain containing adaptor protein inducing interferon-β (TRIF) are the only four proteins containing a RHIM, allowing homotypic protein-protein interactions and initiating the assembly of the necrosome [11]. The necrosome contains the phosphorylated forms of RIPK3 and RIPK1, as well as FADD, caspase-8 and MLKL [12]. In IAV-infected mouse fibroblasts and alveolar epithelial cells, RIPK3 can mediate either necroptosis or apoptosis, pending on the availability of the necrosome constituting proteins [13]. The RHIM and kinase activity of RIPK3 is required to phosphorylate and activate downstream MLKL, while the RHIM, but not the kinase activity, is needed for apoptosis. In the context of apoptosis, RIPK3 recruits RIPK1 through RHIM-RHIM interactions and RIPK1 then acts as the scaffold downstream of ZBP1 to recruit FADD and caspase-8 [13]. Mouse fibroblasts and alveolar epithelial cells lacking RIPK3 cells are almost completely resistant to IAV-induced cell death compared to WT cells [13]. ZBP1 deletion in fibroblasts, respiratory epithelial cells and macrophages leads to complete cell death resistance, compared to RIPK3 deletion [13, 14, 15] suggesting that ZBP1 can also induce cell death independetly from RIPK3, most likely by directly engaging RIPK1-FADD-Caspase-8-mediated apoptosis [13].
In vivo IAV infection studies: variables that may affect the outcome
Role of different cell death modulators and modalities
Cell death plays a crucial role in limiting viral spread and immune activation but if uncontrolled it can contribute pathogenic inflammation and increased lung pathology [4]. In vivo, Ripk3−/− mice and Zbp1−/− mice were reported to be more susceptible to IAV PR8 (H1N1 strain) infection compared to wild type mice, exhibiting elevated pulmonary viral load and heightened morbidity and mortality [9, 15, 16]. Apart from inducing cell death, RIPK3 may regulate type I IFN signaling at both the transcriptional and post-transcriptional levels to inhibit viral replication and protect the host against influenza infection [17]. In contrast, Ripk3 deficiency promotes survival of mice infected with H7N9 influenza virus [18]. ZBP1/RIPK3/RIPK1/Caspase-8-mediated apoptosis limits IAV without the need for necroptosis, but in the absence of apoptosis by a mutant Casp8DA allele preventing caspase-8 autoprocessing, necroptosis may also function as an independent, “stand-alone” cell death mechanism in antiviral host defense [19]. We observed that the requirement of RIPK3 for protection against IAV PR8 (H1N1) infection in vivo is only apparent within a limited dose range of IAV challenge [17]. Moreover, this protective outcome is independent from RIPK3 kinase activity and from MLKL, demonstrating that the scaffolding function of RIPK3 rather than its kinase activity is required for protection. Altogether, this suggests that a RIPK3 function independent of necroptosis is implicated in protection against IAV-associated pathology [16] in line with the prominent role of apoptosis [19].
Apparently, the involvement of necroptosis in the protection against IAV in vivo is only observed in conditions where apoptosis is prevented by blocking caspase-8 activity or deleting of Fadd [13, 16, 19]. Since RIPK3 as a scaffold can be involved in apoptosis [20] while as a kinase it promotes necroptosis, it is important to also evaluate the contribution of MLKL as the final executioner of necroptosis [13, 19]. Interestingly, several studies showed that Mlkl−/− mice in conditions without deletion of caspase-8 are equally susceptible to IAV compared to Mlkl+/+ littermates, regardless of the IAV dose of mouse adapted A/Puerto Rico/8/34 (H1N1) (PR8) strain [13, 16, 19, 21], confirming that apoptosis is the primary modality to protect against IAV infection. However, two independent studies reported that MLKL deficiency increased the protection in mice infected with a lethal dose of PR8 [22] or with A/California/7/2009 virus strain [23]. In these two studies, however, the WT controls were different: either C57BL/6J mice (The Jackson Laboratory) or C57BL/6 N (#B6NTac) mice were used as controls. Another group reported that Mlkl−/− mice display similar survival and body weight loss as WT mice when challenged with PR8 virus [21]. A recent study showed that 70% of the Mlkl-/- mice recovered after lethal infection with IAV PR8 strain (H1N1) compared to WT controls, demonstrating that necroptosis contributes to detrimental inflammation during severe infections. Indeed, therapeutic treatment of mice with a newly developed RIPK3 kinase inhibitor improved lung function, decreased inflammation and prevented mortality [22]. However, it could noted from the same paper that also p38MAPK, JAK2 and JAK3 are targeted by the RIPK3 inhibitor at 0.5 µM, which suggests a combined contribution of the targeted kinases in the improved lung function following severe IAV infection.
Moreover, we here document that the kinase activity of RIPK3 is not required for protection against IAV PR8 in vivo, confirming that necroptosis is not the main cell death mechanism implicated [16]. In line with this observation, we also showed that Fadd−/− Ripk3−/− mice are even more susceptible than their WT Ripk3+/+ littermates regardless of the IAV inoculum doses that were used, suggesting an important role for FADD-mediated apoptosis independent of RIPK3 in the control of IAV-associated mortality [16]. It should be noted that Fadd−/− Ripk3−/− mice develop autoimmune lymphoproliferative syndrome due to a failure of mature T cells to undergo Fas-mediated apoptosis [24]. An alternitive explanation for the extreme susceptibility of these mice may be related to a disruption in T cell function. Moreover, Casp8DA Mlkl−/− Fadd−/− and Casp8DA Mlkl−/− mice were more susceptible to IAV-associated morbidity compared to WT controls, confirming that caspase-8-mediated apoptosis is crucial for protection against IAV [13, 19]. Finally, Cellular Inhibitors of ApoPtosis (cIAPs) are E3 ligases that keep RIPK1 in the survival mode of signaling [25]. Mice deficient in cIAP2 exhibit increased susceptibility and mortality to IAV infection, which can be rescued by pharmacological RIPK1 kinase inhibition or Rip3-deficiency [26], suggesting that sensitization to cell death death can also worsen the outcome.
Concerning the involvement of the NLRP3 inflammasome, there are also conflicting results in Nlrp3−/− mice infected with IAV. It was shown that Nlrp3 deficiency results in the susceptibility of the mice infected with PR8 [27], but Nlrp3−/− mice were more likely to survive than the WT controls when infected with influenza A/Shanghai/4664 T/2013 (H7N9) strain [28].
Virus strains
Looking in more detail at the in vivo IAV studies investigating the role of RIPK3 reveals that the different outcomes or involvement of particular cell death pathways partially could also be explained by differences in the IAV preparation, dose and route of inoculation of the mice (Table 1).
The most commonly used influenza A virus strain used to examine disease pathogenesis and the immune responses in vivo is the mouse adapted PR8 virus strain. This strain was adapted to mice by multiple serial sequential passages of A/Puerto Rico/8/34 in mouse lung. During this process, the strain acquired mutations allowing a high replication efficiency and evasion of the immune response, therefore becoming highly virulent for the mice [29]. For instance, contrary to other strains, such as H7N9, PR8 (H1N1) cannot infect macropahges [29].
Viral preparations are mostly generated by allantoic inoculation of embryonated eggs or by propagation in Madin-Darby canine kidney (MDCK) cells. Embryonated chicken eggs are the standard procedure for producing viral stocks, and has the advantage of producing high virus titers in a short period of time. The mammalian cell culture-based production of viral stocks for influenza vaccine use, such as using MDCKs, produces about 4-fold lower titers than the egg-based process, increasing manufacturing costs [30, 31]. Moreover, cell culture-based viruses have increased batch-to-batch variation and risk for mycoplasma contamination [31].
Viral titers are assessed by egg infectious dose (determine EID50) or by plaque froming unit assay (determine the pfu) [9, 14, 16, 19, 32]. However, some labs do not inform on the precise method of the viral preparation [17, 26]. Table 1 provides a systematic overview of the experimental settings and the different outcomes following IAV infection (Table 1). In the next paragraphs, we highlight how differences in the IAV inoculum dose, virus preparation and titration as well as the route of inoculation may impact the comparison and the conclusions.
Viral dose of infection
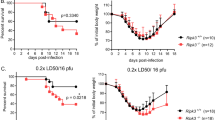
Most studies use the mouse-adapted H1N1 laboratory strain of influenza A, PR8 [33]. Despite the use of the same strain, the lethal outcome following IAV infection looks often very different. One study found that the IAV infection with a dose that is expected to be sublethal for most of the control mice (0.4x LD50; 12.9 hemagglutination units (HAU)/ mouse) did not reveal the presumed sensitized lethality in RIPK3-deficient mice compared to their controls [26]. In contrast, other studies found that RIPK3-deficient mice were more susceptible to IAV when challenged with a dose that was sublethal for the majority of the control mice [13]. Yet, another study reported by comparing two doses that a low IAV dose (50 pfu/mouse) displayed higher lethality, while this difference is not observable with a higher challenge dose (90 pfu/mouse), likely because of the lethal outcome for the majority of the control mice [17]. This finding is in accordance with our results (see below), i.e., that RIPK3 is required for protection against IAV infection in vivo in response to a limited range of challenging dose, while at high dose it is dispensable for protection. We demonstrate that Ripk3−/− mice were more susceptible than their Ripk3+/+ counterparts when infected with medium IAV doses (in this study corresponding to 0.2x LD50/16 pfu). However Ripk3−/− mice were not more susceptible compared to their littermate controls when infected with very low or low doses (0.05x LD50/4 pfu and 0.1x LD50/8 pfu) and high doses (0.5x LD50/40 pfu) of PR8 (see Fig. 1) [16]. These results underline that the Ripk3 phenotype is restricted to a range of infectious doses. Also, the involvement of ZBP1 in modulating lethality following IAV-infection shows various outcomes. When challenged with 1x LD50 of PR8, Zbp1−/− mice were shown to be either better protected from IAV infection compared to their WT controls [14, 15] or to be more susceptible compared to their Zbp1+/+ controls [15]. Another study revealed that the infection route could explain these results. Intranasal challenge revealed that Zbp1−/− mice were more susceptible to IAV compared to Zbp1+/− mice, while intratracheal virus inoculum administration resulted in reduced mortality with the same IAV dose (1x LD50; 50 pfu) [32]. Both Zbp1−/− and Ripk3−/− mice displayed increased mortality compared to C57BL/6 J controls when challenged with a sublethal as well as a lethal IAV dose [9]. All these experiments underline the importance of testing a range of viral doses to address the susceptibility of genetically modified mice. It also shows that at higher infectious doses both ZBP1 and RIPK3 may contribute to immunopathology resulting in increased lethality.

Pooled littermate controls (Ripk3+/+, Ripk3 KD-KI+/+, Mlkl+/+, and Ripk3+/+Fadd +/+ mice) compared with the Ripk3−/−, Ripk3 KD-KIK51A/K51A, Mlkl−/−, and Ripk3−/−Fadd −/−. A Mice were challenged with different IAV doses (here 0.2x LD50/16 pfu). Survival curves were plotted for indicated groups and evaluated statistically according to Kaplan–Meier (GraphPad Prism 8). ns, not significant; ****p < 0.0001 [16]. Survival of littermate controls (B−D): Ripk3+/+, Ripk3 KD-KI+/+, Mlkl+/+, and Ripk3+/+Fadd +/+ mice at different IAV challenge doses Mice were challenged with different IAV doses (0.1 × LD50/8 pfu; 0.2 × LD50/16 pfu; 0.5 × LD50/40 pfu). Survival curves were plotted for indicated groups [16]. Mice: Ripk3−/− were kindly provided by Dr. Vishva Dixit (Genentech, San Francisco) [44], Mlkl−/− by Dr. Alexander Warren and Dr. James Murphy [45] and Ripk3 KD-KIK51A/K51A mice by Dr. John Bertin by GlaxoSmithKline [20]. The Ripk3−/− animals were congenic to the C57BL/6 N background, while all other strains were of the C57BL/6 J background, and were therefore compared with the appropriate littermate controls. Ripk3−/− mice were housed in individually ventilated cages in a conventional animal house. The other mice were bred and housed in the SPF facility in individually ventilated cages. Three weeks prior to the experiment all mice were transferred to the conventional animal house and allowed to go through a quarantine and accommodation period of minimum 3 weeks before the infection experiment. Littermate controls of Ripk3−/−, Mlkl−/−, Ripk3−/−Fadd−/− and Ripk3 KD-KIK51A/K51A were used in each experiment. In all experiments, 10–15- week-old mice were used. All animal experiments were done under conditions specified by law (European Directive and Belgian Royal Decree of November 14, 1993) and approved by the Institutional Ethics Committee on Experimental Animals. Viral infection: Age-matched mice were anesthetized with a cocktail of 87,5 mg/kg ketamine and 12,5 mg/kg xylazine intraperitoneally and infected intranasally with 50 μl/20 g phosphate-buffered saline (PBS) containing different doses of influenza virus A/PR/8/3443, as described in the legends. The plaque-forming units (pfu) were determined by plaque assay on Madin-Darby Canine Kidney (MDCK) cells [31]. The LD50 of the viral batch was determined on BALB/c mice and 1x LD50 represented 80 pfu, as determined in the lab of Prof. Saelens. Although the LD50 is not referring to 50% of death in the mice that were used in this study, the nomenclature is used together with the pfu to have a supplementary information regarding the power of the virus in vivo. This terminology is often used in the papers cited here. Age- and sex-matched mice were challenged with 0.05x LD50 (4 pfu), 0.1x LD50 (8 pfu), 0.2x LD50 (16 pfu) or 0.5x LD50 (40 pfu) and monitored for survival and weight loss over a period of at least 18 days. We used the following 4 scores of clinical symptoms: 0 = no visible signs of disease; 1 = slight ruffling of fur; 2 = ruffled fur, reduced mobility; 3 = ruffled fur, reduced mobility, rapid breathing; 4 = ruffled fur, minimal mobility, huddled appearance, rapid and/or labored breathing indicative of pneumonia and body temperature below 32 °C. For the combination of body weight loss by 30% and a clinical score 4 the mice were considered moribund and euthanized by CO2 asphyxiation or cervical dislocation (EC2016–17). The lethality includes an ethical endpoint during which euthanasia was performed. This ethical endpoint is a combination of 30% weight loss and a clinical score of 4. Survival curves: All the survival data were plotted using the Kaplan–Meier survival analysis with the software Prism 10.1.0.
Besides the range of viral dose for challenge, it is important to note that the humane endpoints for each study can vary. The maximum weight loss that are used to determine the humane endpoint beyond which the mice are euthanized are very important in determining survival numbers and impact survival curves [34]. Some institutional protocols impose euthanasia of mice that have lost 25% of their body weight relative to the time of virus inoculation. In this particular IAV infection model, mice can lose more than 30% of their weight and still recover [34]. We noticed that these humane endpoints vary between labs: some apply ethical endpoint of 30% body weight loss [9], some more than 35% [19, 32], a combination of weight loss and other clinical symptoms [16], while for other studies we were not able to retrieve this information from the methodology [4, 17, 21, 23, 26, 28].
Interestingly, Mlkl-deficient mice were shown to be equally susceptible to IAV as their WT littermates [13], demonstrating that the necroptotic execution mechanism is not implicated in the protective effect. However, another independent study also showed that Mlkl−/− mice were equally susceptible to lethality as WT control in response to a moderate IAV dose (described as EID2500, approximately 1x LD20) but exhibited a better survival when challenged with a higher dose (EID6000), indicating that the presence of MLKL is associated with a worse outcome at high IAV challenge dose. In this context, MLKL was shown to drive a pathogenic recruitment of neutrophils and lethality during severe IAV disease [9]. We found, however, that different doses (0.1x LD50/8 pfu, 0.2x LD50/16 pfu, and 0.5x LD50/40 pfu,) of IAV did not reveal any role for MLKL neither protective nor worsening [16]. We showed that the RIPK3 kinase activity is not required for partial protection against IAV infection at different viral doses and neither is MLKL, suggesting that necroptosis is not involved in the protection against small, medium or high IAV doses in vivo [16]. However, the conclusions from these experiments might be difficult to draw, since it has been reported that RIPK3Ripk3 K51A kinase dead knock-in (Ripk3 KD-KIK51A/K51A) mice used here express very low levels of RIPK3 protein compared to wild type littermate [20] which may affect the scaffolding function of RIPK3. Therefore, differences in viral challenge due to non-standardized doses in titer and volume of application may explain the various outcome ranging from no [26], partial [16] to complete protection of Ripk3−/− mice [13, 17].
Viral preparation, titer assessment and route of infection
It is important that experiments performed in different laboratories can be compared in order to contribute to a better understanding of cell death induced upon IAV infection. Such a comparison relies on an exact description of the viral dose used in infection experiments. While performing these comparisons, we found that the description of the IAV challenge inoculum differs between labs (Table 1). For example, Downey and colleagues inoculated the mice with a low IAV dose defined as 50 pfu per mouse [17]. One group challenged the mice with a sublethal dose described as 0.4x LD50 corresponding to 12.9 hemagglutination units (HAU) [26]. Another group reported their viral titers as egg infectious dose 50 (EID50) [9, 13, 15]. We used the LD50 terminology as determined in BALB/c mice as well as the pfu determined by plaque assay [16].
Indeed, such different ways of determining the IAV doses may affect the conclusions. The infectious dose of a certain IAV preparation may be assessed by determining the 50% egg infectious dose (EID50), the plaque forming unit assay (pfu/mL) or the median tissue culture infectious dose (TCID50). We propose that viral dose for in vivo use should be standardized by cellular assays (pfu/mL) and the median lethal dose for 50% of wild type mice (LD50). It is also important to note that the virulence of PR8 IAV strains may vary between laboratories and the mouse strains used. A uniform source of IAV challenge strains, e.g., use of strains that can be procured from a repository such as BEI Resources, managed by the American Type Culture Collection, could also help in this process. Finally, other variables such as the volume used (30 µl, 50 µl) and the route of infection (intranasal, versus intratracheal) may affect the outcome of the experiment [32] and also requires standardization. Regarding the inoculum volume, a study has shown that mice challenged with a 25 µl inoculum volume rapidly recovered infection with a dose considered lethal when inoculated in 35 or 50 µl volumes [35]. This could be due to poor distribution and subsequent infection of the lung when using relatively small inoculum volumes (such as 20 µL) when compared to higher inoculum volumes [36].
A similar discussion regarding the roles of RIPK3, MLKL, and ZBP1 has been raised previously [37]. In this review, the authors indicated the possible outcomes of upper and lower respiratory tract administration of IAV. Human IAV strains primarily infect human epithelial cells of the upper respiratory tract and typically are associated with mild to moderate disease. Lower respiratory tract infections are associated with increased higher morbidity and mortality [37]. This is an important aspect that needs to be considered when analyzing tract-specific pathophysiology of IAV, respiratory epithelial damage, and inflammation.
Littermate controls and genetic backgrounds
From Table 1 it is obvious that an evenness in the use of littermate controls and comparison between appropriate genetic backgrounds in the experimental setup is also essential for reliable conclusions between genetically modified mouse strains [9, 38]. Littermates have comparable genetic background to the knock-out (KO) mice and are exposed to the same environment during development and birth. C57BL/6 N ES cells have been an important source for the construction of transgenic mice by the International Mouse Phenotyping Consortium (IMPC) [39] while some spontaneous mutations on the C57BL/6 J background are used in many biomedical studies [40]. It is also important to specify the genetic background of the transgenic mice since both C57BL/6 N and C57BL/6 J ES cells have been used [41]. If applicable, also the extend of backcrossing in the C57BL/6 N and C57BL/6 J strains should be mentioned or even quantified [42]. Indeed, genetic differences between C57BL/6 J (B6J) and C57BL/6 N (B6N) sub-strains affect the susceptibility to and pathology associated with IAV infection, with B6J mice being significantly more susceptible to A/California/04/09 H1N1 virus (H1N1), A/Vietnam/1203/2004 (H5N1), and A/Anhui/1/2013 (H7N9) virus strains compared to 6NJ [43].
To address the issue of different genetic background of various transgenic mice we wondered whether pooling the different littermate controls from our previous study [16] and comparing them with the different genetically modified mice challenged with the same IAV dose, would affect the interpretation of the experimental outcomes. To illustrate the effect of genetic backgrounds, we merged the IAV infection results from all control mice (Ripk3+/+, Ripk3 KD-KI+/+, Mlkl+/+, and Ripk3+/+Fadd +/+) and compared these for each IAV challenge dose (0.1x LD50/8 pfu/mouse; 0.2x LD50/16 pfu/mouse; 0.5x LD50/40 pfu/mouse) with the separate groups of Ripk3−/−, Ripk3 KD-KIK51A/K51A, Mlkl−/−, and Ripk3−/−Fadd −/− transgenic mice (Fig. 1 A). By doing so, we found that, compared to the pooled control mice, Ripk3−/− mice showed a non-significant difference for the medium dose, contrarily to the control mice as shown previously (Fig. 1A). This is probably observed because the Ripk3 KD-KI+/+ and Mlkl+/+ mice apparently are more resistant to the same IAV dose compared to the Ripk3+/+ mice [16]. The variance in survival between the different wild type littermates for all IAV challenge doses are shown in Fig. 1B−D [16]. The results of Fig. 1 emphasize again the need to include proper control mice to compare the experimental outcome of a genetic modification.
Conclusions
The complexity of the multiple cell autonomous (cell survival, apoptosis, necroptosis, pyroptosis) and intercellular responses by immune cells, and the amplification of a virus should be carefully evaluated in vivo when comparing the outcome of loss-of-function mutations through by comparing the outcome of IAV challenge in genetically modified with control mice. First, a defined set of different viral doses should be tested in genetically modified mice to investigate their susceptibility. Second, standardized viral preparation, titer assessment, dose-response challenge experiments and a standardized route of administration are crucial to enable cross-comparisons of reported data in order to draw solid conclusions. Finally, the use of littermate controls and matched housing background for the mice are the appropriate controls in these settings to avoid inaccurate conclusions.
References
Short KR, Kasper J, Van Der Aa S, Andeweg AC, Zaaraoui-Boutahar F, Goeijenbier M, et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur Respir J. 2016;47:954–66.
Kesavardhana S, Kanneganti TD. ZBP1: A STARG^°TE to decode the biology of Z-nucleic acids in disease. J Exp Med. 2020;217:1–4.
Rosli S, Harpur CM, Lam M, West AC, Hodges C, Mansell A, et al. Gasdermin D promotes hyperinflammation and immunopathology during severe influenza A virus infection. Cell Death Dis. 2023;14:1–11.
Thomas PG, Shubina M, Balachandran S. ZBP1/DAI-Dependent Cell Death Pathways in Influenza A Virus Immunity and Pathogenesis. Curr Top Microbiol Immunol. 2023;442:41–63.
Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;9:243–50.
Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;71:233–6.
Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–35.
Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes through intracellular M2 channel. Nat Immunol. 2010;11:404–10.
Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Ragan KB, et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell. 2020;180:1115–29.
Kesavardhana S, Subbarao Malireddi RK, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J Biol Chem. 2020;295:8325–30.
Wu XL, Hu H, Dong XQ, Zhang J, Wang J, Schwieters CD, et al. The amyloid structure of mouse RIPK3 (receptor interacting protein kinase 3) in cell necroptosis. Nat Commun. 2021;12:1–14.
Yang ZH, Wu XN, He P, Wang X, Wu J, Ai T, et al. A Non-canonical PDK1-RSK Signal Diminishes Pro-caspase-8-Mediated Necroptosis Blockade. Mol Cell. 2020;80:296–310.
Nogusa S, Thapa RJ, Dillon CP, Oberst A, Green DR, et al. RIPK3 activates parallel pathways of MLKL-Driven Necroptosis and FADD-mediated apoptosis to protect against influenza A virus article RIPK3 activates parallel pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis. Cell Host Microbe. 2016;13:13–24.
Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1 / DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:1–10.
Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses Influenza A Virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. 2016;20:674–81.
Oltean T, Van San E, Divert T, Vanden Berghe T, Saelens X, Maelfait J, et al. Viral dosing of influenza A infection reveals involvement of RIPK3 and FADD, but not MLKL. Cell Death Dis. 2021;12:471.
Downey J, Pernet E, Allard B, Meunier I, Jaworska J, Qureshi S, et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017;3:1–22.
Xu Y-L, Tang H-L, Peng H-R, Zhao P, Qi Z-T, Wang W. RIP3 deficiency ameliorates inflammatory response in mice infected with influenza H7N9 virus infection. Oncotarget. 2017;8:27715–24.
Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J Exp Med. 2020;217:e20191259.
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–95.
Lei X, Chen Y, Lien E, Fitzgerald KA. MLKL-Driven Inflammasome Activation and Caspase-8 Mediate Inflammatory Cell Death in Influenza A Virus Infection. mBio. 2023;14:e0011023.
Gautam A, Boyd DF, Nikhar S, Zhang T, Siokas I, Van de Velde LA, et al. Necroptosis blockade prevents lung injury in severe influenza. Nature. 2024;628:835–43.
Yi-Han L, Platt MP, Gilley RP, Brown D, Dube PH, Yu Y, et al. Influenza causes MLKL-driven cardiac proteome remodeling during convalescence. Circ Res. 2021;128:570–84.
Zhang H, Wu X, Li X, Li M, Li F, Wang L, et al. Crucial roles of the RIP homotypic interaction Motifs of RIPK3 in RIPK1-dependent cell death and lymphoproliferative disease. Cell Rep. 2020;31:107650.
Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66.
Rodrigue-Gervais IG, Labbé K, Dagenais M, Dupaul-Chicoine J, Champagne C, Morizot A, et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe. 2014;15:23–35.
Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to Influenza A virus via the regulation of Caspase-1. Immunity. 2009;30:566–75.
Ren R, Wu S, Cai J, Yang Y, Ren X, Feng Y, et al. The H7N9 influenza A virus infection results in lethal inflammation in the mammalian host via the NLRP3-caspase-1 inflammasome. Sci Rep. 2017;7:1–13.
Mansell A, Tate MD. In vivo infection model of severe influenza a virus. Methods Mol Biol. 2018;1725:91–9.
Xue J, Chambers BS, Hensley SE, López CB. Propagation and characterization of influenza virus stocks that lack high levels of defective viral genomes and hemagglutinin mutations. Front Microbiol. 2016;7:1–15.
Kim E, Kwon H, Park S, Kim Y, Si Y, Lee I, et al. Generation of a high-growth Influenza vaccine strain in MDCK cells for vaccine preparedness. J Microbiol Biotechnol. 2018;28:997–1006.
Momota M, Lelliott P, Kubo A, Kusakabe T, Kobiyama K, Kuroda E, et al. ZBP1 governs the inflammasome-independent IL-1α and neutrophil inflammation that play a dual role in anti-influenza virus immunity. Int Immunol. 2019;3:203–12.
Gerber P, Hambre D, Loosli CG. Antigenic variants of influenza A virus. J Exp Med. 1955;103:413–24.
Sanders CJ, Johnson B, Frevert CW, Thomas PG. Intranasal influenza infection of mice and methods to evaluate progression and outcome. Methods Mol Biol. 2013;1031:177–88.
Miller DS, Kok T, Li P. The virus inoculum volume influences outcome of influenza A infection in mice. Lab Anim. 2013;47:74–7.
Smith CA, Kulkarni U, Chen J, Goldstein DR. Influenza virus inoculum volume is critical to elucidate age-dependent mortality in mice. Aging Cell. 2019;18:1–4.
Basavaraju S, Mishra S, Jindal R, Kesavardhana S. Emerging role of ZBP1 in Z-Rna sensing, influenza virus-induced cell death, and pulmonary inflammation. MBio. 2022;13:1–14.
Vanden Berghe T, Hulpiau P, Martens L, Roosmarijn E, Van Wonterghem E, Perry SW, et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity. 2015;43:200–9.
Skarnes WC, Rosen B, West AP, Koutsourakis M, Iyer V, Mujica AO, et al. A conditional knockout resource for the genome – wide study of mouse gene function. Nature. 2013;474:337–42.
Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:1–22.
Tanimoto Y, Iijima S, Hasegawa Y, Suzuki Y, Daitoku Y, Mizuno S, et al. Embryonic stem cells derived from C57BL/6J and C57BL/6N mice. Comp Med. 2008;58:347–52.
Mekada K, Yoshiki A. Substrains matter in phenotyping of c57bl/6 mice. Exp Anim. 2021;70:145–60.
Eisfeld AJ, Gasper DJ, Suresh M, Kawaoka Y, Perez DR, Gasper DJ. C57BL / 6J and C57BL / 6NJ mice are differentially susceptible to inflammation-associated disease caused by Influenza A virus. Front Microbiol. 2019;9:1–15.
Newton K, Dugger DL, Wickliffe KE, Kapoor N, De Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60.
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J, Alvarez-diaz S, et al. The Pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–53.
Acknowledgements
Research in the Vandenabeele unit is supported by the FWO research projects (3G033120, 3G0A9322), EOS research consortium (EOS 30826052, 3G0I5722), UGent Special Research Fund Methusalem (01M00709), iBOF (01IB3920), grants from the Foundation against Cancer (365A08921), CRIG and GGIG consortia, and VIB. We thank the VIB for training, support, and availability of core facilities. We thank Prof. Koen Augustyns from Antwerp University for critical discussion.
Author information
Authors and Affiliations
Contributions
TO and PV performed study concept and design. TO made the figures. All authors read, wrote or provided comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oltean, T., Maelfait, J., Saelens, X. et al. Need for standardization of Influenza A virus-induced cell death in vivo to improve consistency of inter-laboratory research findings. Cell Death Discov. 10, 247 (2024). https://doi.org/10.1038/s41420-024-01981-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-01981-w
- Springer Nature Limited