
Abstract
Full-length p53 (p53α) plays a pivotal role in maintaining genomic integrity and preventing tumor development. Over the years, p53 was found to exist in various isoforms, which are generated through alternative splicing, alternative initiation of translation, and internal ribosome entry site. p53 isoforms, either C-terminally altered or N-terminally truncated, exhibit distinct biological roles compared to p53α, and have significant implications for tumor development and therapy resistance. Due to a lack of part and/or complete C- or N-terminal domains, ectopic expression of some p53 isoforms failed to induce expression of canonical transcriptional targets of p53α like CDKN1A or MDM2, even though they may bind their promoters. Yet, p53 isoforms like Δ40p53α still activate subsets of targets including MDM2 and BAX. Furthermore, certain p53 isoforms transactivate even novel targets compared to p53α. More recently, non-canonical functions of p53α in DNA repair and of different isoforms in DNA replication unrelated to transcriptional activities were discovered, amplifying the potential of p53 as a master regulator of physiological and tumor suppressor functions in human cells. Both regarding canonical and non-canonical functions, alternative p53 isoforms frequently exert dominant negative effects on p53α and its partners, which is modified by the relative isoform levels. Underlying mechanisms include hetero-oligomerization, changes in subcellular localization, and aggregation. These processes ultimately influence the net activities of p53α and give rise to diverse cellular outcomes. Biological roles of p53 isoforms have implications for tumor development and cancer therapy resistance. Dysregulated expression of isoforms has been observed in various cancer types and is associated with different clinical outcomes. In conclusion, p53 isoforms have expanded our understanding of the complex regulatory network involving p53 in tumors. Unraveling the mechanisms underlying the biological roles of p53 isoforms provides new avenues for studies aiming at a better understanding of tumor development and developing therapeutic interventions to overcome resistance.
Similar content being viewed by others

Facts
-
p53 isoforms modulate canonical functions such as in cell cycle control and apoptosis as well as non-canonical functions in DNA repair and replication.
-
p53 isoforms exert biological roles via p53α-independent transcriptional activities (p53β/γ and Δ40p53) or via hetero-oligomerization with p53α impacting on p53α activities (Δ40/133/160p53).
-
The co-existence of different p53 isoforms potentiates the complexity of their biological functions.
-
Tumor-specific expression profiles of p53 isoforms are of interest for diagnostic and prognostic implications, paving the way for tailored treatment strategies.
Open questions
Do alternative p53 isoforms impact on
-
Cancer stemness, epithelial-mesenchymal transition (EMT), metastasis and chemoresistance via non-canonical functions in DNA repair and replication?
-
The spectrum and penetrance of hereditary tumors in Li-Fraumeni patients with germline TP53 mutations?
Introduction
Human Tumor Protein 53 (TP53) gene, situated on chromosome 17p13.1 [1], comprises 13 exons, including 11 constitutive exons and 2 alternatively spliced exons [2]. Despite its identification already 40 years ago, ongoing research continues to unveil novel functions, activities, and interactions associated with this gene.
The identification of p53 isoforms
TP53 splice variants were initially identified in the 1980s [3, 4], but their presence in different species and their biological and clinical importance were only established around 15 years later [2]. Through the utilization of various mechanisms like different promoters, alternative splicing, and internal ribosome entry site (IRES), TP53 generates 12 distinct isoforms including full length p53α (also known as p53 Wild Type or p53WT) [5, 6].
As shown in Fig. 1A, canonical TP53 transcription starts from the promoter 1 while non-canonical transcription starts from promoter 2 [7]. In human cells, the transcript from promoter 1 produces p53α, alternative splicing produces variants that contain intron 2 and intron 9. Alternative splicing of intron 9 leads to the creation of mRNA variants containing exons 9β or 9γ, resulting in the β and γ subtypes. Both exon 9β and 9γ contain stop codons, causing exons 10 and 11 to remain untranslated in TP53 β and γ mRNA variants. On the other hand, the α isoform includes all exons [2, 8]. p53α binds promoter 2 (P2) upon DNA damage signals (camptothecin, doxorubicin), which induces Δ133p53/Δ113p53 in human tumor cells and zebrafish, respectively [7, 9, 10]. Others observed that not only in human cancer cells but also in human induced pluripotent stem cells (iPSCs), P2 transcription was induced by DNA damage (X-ray) as well as by cell ageing through prolonged passaging of cells, whereby site-specific CpG demethylation events in exon 5 were found to be spatially and temporally associated with transcription from the adjacent intronic P2 [11].

A The TP53 gene consists of canonical exons (represented by colored boxes) that encode different structurally and functionally defined domains of the p53 protein. Transcription from promoter 1 generates an mRNA transcript capable of translating into full-length p53 (FLp53) or ∆40p53 isoforms. ∆40p53 isoforms are only translated when intron 2 is present in the mRNA transcript. On the other hand, transcription from promoter 2 produces an mRNA transcript that codes for ∆133p53 or ∆160p53 isoforms through differential translation initiation. Alternative splicing of exon 9 modulates the production of C-terminally variant isoforms of p53 (α, β, and γ). B p53 protein consists of six distinct domains that are encoded by different exons of the TP53 gene, as depicted in (A) (with exons and coding domains shown in matching colors). Within the transactivation domain (TAD) and DNA-binding domain (DBD) arrows indicate the starting points of specific isoforms. On the bottom right, colored boxes schematically outline the two C-terminally altered isoforms: β and γ.
Different domains of p53 isoforms
p53 protein typically exists as a tetramer, composed of four monomers, with canonical functions as a transcriptionally transactivating factor [12,13,14,15,16]. The assembly of p53 tetramers, also called self-association, leads to DNA looping which links separated p53 DNA binding sites to increase p53’s concentration at one response element (RE) and/or activate additional REs [17]. Within the p53 monomer the following domains have been defined based on 3-dimensional structures and biochemical functions (Fig. 1B).
The intrinsically disordered region (IDR) within the N-terminal region of p53 has emerged as a crucial player in signaling cascades [18]. Despite having low affinity, the IDR enables highly specific interactions with other proteins [19]. TAD (transactivation domain), situated within the IDR, interacts with various proteins mediating and/or regulating p53’s functions including factors of the basal transcription machinery [20,21,22,23,24], the E3 ligase murine double minute 2 (MDM2) [25,26,27,28], or histone acetyltransferase p300-cAMP-response element binding protein (CREB) binding protein [29,30,31], as well as DNA repair and replication proteins including DNA polymerase β, α and ɩ, the single-strand binding protein Replication protein A (RPA) and the processivity factor Proliferating Cell Nuclear Antigen (PCNA) in DNA replication [32,33,34,35,36]. TAD can be divided into TAD I and TAD II (Fig. 1B) [37]. TAD I plays a critical role in p53-dependent transactivation, DNA damage-induced G1 cell cycle arrest, and apoptosis. Its functional importance outweighs that of TAD II in these processes [38] though TAD I and thereby full transcriptional transactivation is dispensable for tumor suppressive signaling pathways of p53 [2, 39, 40]. p53 and MDM2 establish an autoregulatory negative feedback loop, maintaining low p53 levels in unstressed cells. MDM2-p53 TAD interaction hinders the transcriptional ability of p53 [26,27,28, 41].
The Proline-Rich Domain (PRD) connects the TAD with the DNA-binding domain (DBD) [8]. This region contains five polyproline (PXXP) motifs, serving as binding sites for Src-Homology-3 domains that facilitate protein-protein interactions involved in signal transduction [42, 43]. Additionally, by undergoing proline isomerization, PRD alters 3-dimensional protein structure, adjusting the direction and angle of functionally interacting domains [44]. The PRD is necessary for growth inhibition and apoptosis triggered by p53 [45] and functioning as a spacer or scaffold module necessary for tumor suppression capabilities of p53 [2, 44, 46, 47].
DBD is the core domain of p53 and mediates the interaction between p53 and DNA. Within the DBD, there are several highly conserved histidines and cysteines that facilitate coordination with Zn2+ or Mg2+ ions, enabling proper p53 conformation and DNA binding ability [48,49,50]. Furthermore, the interaction between the p53 DBD and its N-terminal region contributes to the stability of p53 as a tetramer [51]. Most oncogenic mutations within the DBD result in conformational changes and/or alterations in p53’s ability to bind to target p53-specific DNA sequences. The large number of oncogenic mutations found in this domain underscores its crucial role in regulating tumor suppression [13, 15, 49, 52,53,54]. As seen in Fig. 1B, Δ40p53 isoforms possess the full-length DBD, while Δ133p53 and Δ160p53 isoforms lack significant portions of it. In particular, Δ160p53 lacks the entire first conserved cysteine motif, and is severely compromised in DNA binding, while Δ133p53 dimers still show some proficiency in binding p53-specific DNA elements [55]. Of note, the central domain with metal complexing capacity is also critical for p53´s activities in binding to RAD51 [56], recognizing DNA junctions, particularly comprising mispairings [57,58,59] and degrading such DNA structures exonucleolytically [60,61,62]. The integrity of the DBD therefore also is a prerequisite for p53´s non-canonical, i.e. transcription-independent functions in restricting low-fidelity homologous recombination and bypass of DNA replication impediments [36, 63, 64]. So far, such non-canonical functions in DNA repair have been investigated mostly for the p53α isoform.
The Hinge Domain (HD) links the DBD and Oligomerization Domain (OD) and comprises the Nuclear Localization Signal (NLS) [65]. HD enables p53 to enter the nucleus and provides structural flexibility for binding to REs [66]. Germline mutations of p53 in the HD, such as p53R306P, result in loss of transcriptional activation of p53 target genes like BCL2-Associated X (BAX) [67]. Moreover, p53 without HD is unable to recognize the consensus sequences, indicating that HD contributes to allosteric regulation of DNA binding [2, 67].
The OD is crucial for the formation of p53 tetramers and houses the nuclear export signal (NES). Tetramerization conceals the NES, keeping p53 within the nucleus to regulate target gene expression [68]. The OD assists in DNA deformation and facilitates stable DBD-DNA binding [69] such as when mediating apoptosis and cell cycle arrest [70]. Oligomerization via the OD equally promotes p53´s interactions with DNA junction structures to execute DNA recombination and replication control [36, 71].
The Carboxy-terminal domain (CTD) regulates the structure and function of the protein itself [72] and contains multiple post-translational modification sites which modulate protein degradation, tetramerization, transactivation and protein interactions [73, 74]. The extreme CTD is rich in positively charged amino acids like arginines, histidines, and lysines, enabling p53 to bind to negatively charged nucleic acids such as RNA and DNA indiscriminately [75], recognize DNA lesions and mismatches and promote DNA annealing [76,77,78]. Numerous proteins bind to the CTD, which explains why many p53 missense mutants still retain biochemical and biological activities [2]. Additionally, the nonspecific DNA binding capacity of the CTD allows p53 to diffuse along the DNA linearly or to transfer itself to another DNA molecule [79].
The biological roles of p53 isoforms and underlying mechanisms
p53 protein, with its diverse isoforms, emerges as a multifaceted player in maintaining cellular homeostasis and safeguarding the genome. In this section, we delve into the biological roles of the different p53 isoforms to unravel their intricate contributions to cellular processes. Table 1 summarizes these biological effects and the biochemical activities potentially underlying these functions.
p53 isoforms and biochemical activities
p53α-independent activities of alternative p53 isoforms
Some p53 isoforms exert their cellular roles independently of p53α (Fig. 2). Ectopically expressed p53β and p53γ were demonstrated to be incapable of activating the p53α target genes DNA Damage Inducible 1 Homolog 2 (DDI2), Arginase 2 (ARG2), CDKN1A, E2F Transcription Factor 7 (E2F7), Serpin Family E Member 1 (SERPINE1), Tumor Protein P53 Inducible Nuclear Protein 1 (TP53INP1) or TP73 through promoter reporter assays in H1299 cells [80]. In K562 and Saos 2, ectopic expression of alternative p53 isoforms in general failed to induce MDM2 or p21 [81]. Previous studies have shown that Δ40p53α shows less binding affinity to Mdm2 and Cdkn1a promoter compared to p53α in Balb/c 10.1 fibroblasts [82] and mediates no measurable p21 protein induction in H1299 and Saos 2 cells [81, 83]. Though defective in some canonical transcriptional activities of p53α, Δ40p53α binds and activates genes including BAX due to retention of its TAD II [84]. Furthermore, Δ40p53α transactivates a different set of p53-responsive genes than p53α [85], in particular, several apoptosis related genes: TIAL1 and ASPP2 which are not induced by p53α [86].

How alternative p53 isoforms exhibit biological functions.
Regarding a direct, i.e. transcription-independent function of an alternative p53 isoform in DNA repair, the only piece of information accumulated so far has been that Δ40p53α has lost the capability to form complexes with the key homologous recombination factors RAD51 and BRCA1 in the nucleus of doxorubicin-treated cells [87]. Yet it is unclear why TAD I would be required for formation of these complexes, as the physical interaction of p53 with RAD51 requires central [56] and with BRCA1 C-terminal binding sites on p53 [88]. Therefore, either p53 recognizes the same lesions on DNA as RAD51 and BRCA1 and/or even cooperates with RAD51 and BRCA1 in fork remodeling at DNA replication barriers which requires the N-terminal POLɩ binding site [36, 89]. If the latter were true, this observation would be more relevant for non-canonical activities of p53 in DNA replication than DNA repair (see Chapter 2.2).
The Δ133p53α isoform responds to γ radiation and binds to novel p53 REs in the promoters of DNA double strand break (DSB) repair genes (including RAD51, RAD52 and LIG4), which upregulates the transcription of these genes independently of p53α [90]. In H1299 cells which are p53α negative, the overexpression of Δ133p53α augments DSB repair, whereby depletion of p73 revealed dependency on this p53 family member. Chromatin immunoprecipitation experiments demonstrated that the isoform Δ133p53α and p73 collaborate to upregulate the expression of DSB repair genes including RAD51, RAD52 and LIG4 through synergistic occupancy of the corresponding REs independently of p53α [91].
Δ160p53 was reported to mediate molecular functions of oncogenic p53 gain of function mutants towards proliferation and invasion of tumor cells [92]. Accordingly, it was described that the Δ160p53α isoform is commonly involved in the gain of function phenotype of mutant p53 proteins like p53(273H), p53(175H) or p53(248W). Stable and transient expression of p53(273H) as well as some endogenously mutant p53 expressing cell lines exhibit Δ160p53α expression [92]. Moreover, the authors causally linked the gain of function activities of the p53 mutants with the expression of Δ160p53α revealing the pro-oncogenic property of Δ160p53α [92], which might also be due to fork stalling caused by Δ160p53α independently of p53α upon DNA damage [81].
Modulation of transcriptional activities of p53α by alternative p53 isoforms
Alternative p53 isoforms can alter the transcriptional activities of p53α (Fig. 2). Though lacking the typical OD partially, p53β can still form a complex with p53α at the BAX promoter in an incompletely understood fashion and augment p53α´s intrinsic transactivation activity specifically on the BAX promoter [93, 94].
Δ40p53α and Δ133p53α isoforms retain the full OD for hetero-oligomerization with p53α or other α isoforms. Forming part of such hetero-oligomeric complexes Δ40p53α, lacking the N-terminal MDM2-binding site, protects complexed p53α from degradation and shifts transcriptional transactivation activity from CDKN1A to BAX [2, 83, 84]. In ESCs from transgenic mice, the Δ40p53α/p53α complex controls the transition from pluripotency to differentiation, whereby elevated Δ40p53α levels prolong pluripotency by upregulating Nanog as well as Insulin-like growth factor 1 receptor (IGF-1R) [95]. Thereby, Δ40p53α sequesters p53α within dimers and tetramers in the cytoplasm diminishing binding of p53α to Cdkn1a, Nanog, and IGF-1R significantly reducing transcriptional activity of p53α on Cdkn1a and de-repressing transcriptional activity of p53α on Nanog. In human breast cancer (BRCa), Δ40p53α expression was also reported to associate with upregulation of stemness genes, confirming a species-independent role of this isoform in inducing stemness [96].
In transgenic mice, Δ122p53α (mouse ortholog of human Δ133p53α) decreases transactivation of Mdm2 and Cdkn1a by p53α [97]. In human neonatal foreskin and normal prostate tissue, Δ133p53α similarly reduces expression of p53α targets CDKN1A, PUMA, NOXA and BAX [98].
In conclusion, while C-terminally altered p53 isoforms cooperate with p53α during transcription, Δ133p53α exerts an inhibitory effect and Δ40p53α modulates transcription, i.e. promotes and mitigates p53α activities depending on the particular target gene. The impact of alternative isoforms, Δ40p53α in particular, on p53α transcriptional activities can vary depending on the cellular context, such as cell type, cellular differentiation program, availability of specific cofactors and the composition of the specific isoforms.
Dominant negative effects of alternative p53 isoforms
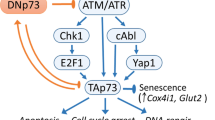
A dominant negative effect of oncogenic mutant p53 exerted via hetero-oligomerization was initially reported in 1991 [99]. More than a decade later, Δ133p53α was proven to exert a dominant negative effect on transcriptional activities of p53α towards apoptosis induction, concomitantly unleashing p53α-repressed genes in DNA repair and cell cycle progression [9, 97, 98, 100, 94, 101]. In human fibroblasts from patients with the premature aging disorder Hutchinson-Gilford Progeria Syndrome (HGPS), hetero-oligomerization as the basis of the dominant negative effect was proven by co-immunoprecipitation of p53α with FLAG-labeled Δ133p53α. Further evidence from these HGPS, but also normal fibroblasts showed that Δ133p53α blocks p53α-mediated induction of CDKN1A mRNA and miR-34a, and thereby ultimately replicative senescence [102, 103]. In the same cell model Δ133p53α also induced RAD51 via de-repression of the p53α target E2F1. p53α and Δ133p53α induction and subsequent Δ133p53α-dependent upregulation of RAD51 was also observed during induced pluripotent stem cells reprogramming, and may serve to ensure DNA repair and genome stability in the absence of apoptosis in these primitive cells [104]. Interestingly, in different cell lines genotoxic treatment with doxorubicin was shown to induce Δ133p53α by p53α-mediated transactivation of the internal TP53 promoter. In this scenario Δ133p53α, again co-precipitating with p53α, dampens p53α-dependent apoptosis and G1 but not G2 cell cycle arrest as part of a feedback loop modulating the cellular response to DNA damage [7]. Altogether, Δ133p53α dominant negatively affects transcriptional programs towards the canonical functions of p53α in cell cycle control, senescence, apoptosis and DNA repair.
The idea of a transcription-independent role of p53 in DNA replication emerged already in 1993, when p53 was found to directly bind to RPA and to stimulate Bovine Papilloma Virus 1 (BPV-1) replication [32, 105]. Multiple pieces of evidence, notably also the discovery of p53´s intrinsic 3´-5´exonuclease activity acting both in nuclei and the mitochondria [60, 106], established the concept that p53 executes non-canonical functions in DNA replication processes [107,108,109]. While the mechanistic details in mitochondria are still underexplored [110, 111], the precise role of p53α in nuclear DNA replication has been unveiled through the discovery of the p53α-POLι dependent DNA damage tolerance (DDT) pathway: When replication forks encounter replication barriers such as crosslinks, p53α collaborates with the highly error-prone POLι in a process known as idling events, i.e. iterative insertion and exonucleolytic removal of misincorporated nucleotides. This cooperative mechanism slows down DNA synthesis and enables bypass of the barrier through the involvement of the fork reversal enzymes HLTF and ZRANB3 [64]. A major clue leading to this model came from the separation-of-function mutant p53(H115N), fully functional in transcription but lacking exonuclease activity [112], and inactive in this DDT pathway. This pathway serves to safeguard DNA replication in human stem cells, including cancer stem cell-like cells, by preventing fast and error-prone bypass mechanisms [64, 113].
In crosslinker treated K562 and Saos 2 cells, human cell types devoid of p53α, expression of each of five alternative p53 isoforms (p53β, p53γ, Δ40p53α, Δ133p53α, Δ160p53α) indicated loss-of-function in this DDT pathway [81]. This phenotype can be explained by loss of critical domains necessary for oligomerization (p53β, p53γ), PCNA and POLι binding (Δ40p53α, Δ133p53α, Δ160p53α), RPA binding and exonucleolytic activity (Δ133p53α, Δ160p53α) [36, 64], respectively. Both co-expression of p53α and of one of the alternative isoforms (K562, Saos 2) as well as selective depletion of specific isoforms (HSPCs) revealed a dominant negative effect of Δ133p53α and Δ160p53α on p53α regardless of the treatment conditions [81]. Of interest with regard to the underlying mechanism, Δ133p53α and Δ160p53α expressed in unperturbed K562 and Saos 2 cells caused severe stalling of replication. From this, the dominant negative effect exerted by Δ133p53α and Δ160p53α may be due to the formation of non-productive hetero-oligomers with p53α and/or occupation of DNA replication sites through DNA and protein interactions via the residual DBD and CTD. Therefore, dominant negative effects of severely N-terminally truncated p53 isoforms are critical mechanisms affecting both canonical and non-canonical functions of p53α (Fig. 2).
The composition of p53 isoform hetero-oligomers determining the biological activities is influenced by the relative levels of the different isoforms. Such relative levels are subject to a plethora of parameters, ranging from differences in the protein stability due to loss or retention of the MDM2-binding site [84], expression changes during dedifferentiation as discussed above [104] or to DNA damage-induced splicing or alternative translation initiation, which have recently been reviewed by Avery-Kiejda and colleagues [114]. Titration experiments suggest that gradual changes of the alternative isoform/p53α ratio in mixed oligomers do not necessarily entail a linear relationship with transcription activities. Specifically, increasing the ratio of Δ40p53α/p53α will stabilize p53α through Δ40p53α-mediated protection from MDM2-mediated degradation already at one to two Δ40p53α monomers/tetramer and antagonize the transactivation capacity of p53α at higher ratios, generating a window of maximum canonical activities at intermediate Δ40p53α/p53α ratios [83]. However, the functions of hybrid oligomers consisting of three or more types of p53 isoforms are currently not understood.
Dominant negative effects can also be generated through subcellular mislocalization (Fig. 2). Δ40p53α, for example, is primarily located in the cytoplasm and can influence the localization of p53α by complexing it and shifting it from the nucleus to the cytoplasm [115]. More recently, protein aggregation has been understood to represent even another dominant negative mechanism acting on p53α (Fig. 2). The TAD of p53α was shown to significantly inhibit aggregation of the p53α DBD which explains why Δ40p53 possesses a higher aggregation tendency [116]. Δ133p53 aggregates much faster than p53α or p53C (p53 fragment engineered to cover amino acids 93-393) [117]. Thus, Δ133p53 could exert its dominant negative effects on canonical and non-canonical p53α functions by forming non-productive aggregates with p53α or by blocking access of p53α to the response elements of target genes [8] and to DNA replication fork structures [81].
As our understanding of p53 isoforms expands, more isoforms and corresponding biochemical activities may emerge on the surface. It is becoming increasingly clear that the complexity of p53-mediated cellular responses is finely tuned through the interplay between these isoforms. Collectively p53 isoforms exert non-binary rheostat-like functions in response to stress or during differentiation impacting on the maintenance of genome integrity, cell cycle progression, senescence, apoptosis, and tumor development. Exciting research continues to unravel the functional diversity and nuances of these isoforms, shedding light on their potential as therapeutic targets for cancer.
The role of p53 isoforms in cancers
Accumulating evidence indicates that dysregulation of p53 isoforms co-expressed in cells may change not only cellular p53 responses, but indeed can drive oncogenesis and modify sensitivity to certain cancer treatments [2, 114, 118, 119]. Table 2 summarizes the observations made on the dysregulations of p53 isoforms in different tumor types and their clinical impact.
p53 isoforms influence tumor development
The transcriptional activities of p53β, p53γ, and Δ40p53α play a critical role in tumor development and progression, as they regulate gene expression through collaboration with (p53β, p53γ, Δ40p53α) and modulation of (Δ40p53α) p53α´s transcriptional activities as well as through p53α-independent gene regulation (Δ40p53α) (see Table 1). Of note, regulation of p53α-dependent activities encompasses both transcriptional transactivation as well as repression of p53α target gene expression. In line with these regulatory principles, increased expression of p53β was found to correlate with better disease-free survival (DFS) and/or overall survival (OS) in breast cancer (BRCa) (mRNA), renal cell carcinoma (RCC) (mRNA), acute myeloid leukemia (AML) (protein) and melanoma (mRNA and protein) patients [120,121,122,123] and was detected in several other tumor types [102, 124] (Table 2). Overexpression of p53β in normal fibroblasts that endogenously co-express p53 isoforms feature induction of apoptosis and cell senescence via up-regulation of genes including BAX, CDKN1A and miR-34 in a p53α-dependent manner [102]. In cancer cell lines, namely MCF7 (BRCa) and H1299 (lung cancer), p53β enhanced p53α transcriptional activity of p53α on CDKN1A and/or BAX promoters [93]. p53γ expression correlated with better DFS and/or OS in BRCa (mRNA) and AML (protein) while the opposite result was found in uterine serous carcinoma (USC), yet for the mRNA level [122, 125, 126]. Moreover, its mRNA expression was elevated in tumor tissue of squamous cell carcinoma of the head and neck (HNSCC) but not in non-tumor control tissue. Most interestingly with regard to diagnostic applications, aiming at early detection of tumors, seroreactivity of p53 was elevated in individuals with premalignant colorectal cancer (CRC) lesions [124, 127]. Similar to p53β, p53γ increased transcriptional activity of p53α on the BAX promoter which possesses pro-apoptotic capacity in MCF7 cells [93]. In analogy to p53α, this canonical function most plausibly connects with the association of p53β/p53γ expression with better DFS/OS. In p53 mutant expressing BRCa, p53β and p53γ may compensate the loss of function of p53α and result in low cancer recurrence and an OS as good as that of BRCa expressing wild-type p53α [121, 125].
Δ40p53α mRNA expression levels in melanoma, glioblastoma and BRCa are higher than those in corresponding normal tissues [121, 128, 129]. It is also found to be associated with aggressive tumor growth and triple-negative BRCa with mutated TP53 [121, 130]. Along this line, a higher Δ40p53α/p53α ratio associates with worse DFS of BRCa, however, in mucinous/serous ovarian cancer with better recurrence-free survival (RFS) [131,132,133]. In BRCa, a high Δ40p53α/p53α ratio was reported to increase tumor growth by promoting the stemness phenotype [96], which indeed has been linked with basal-like and triple-negative BRCa [134]. However, the same research group also discovered that in the luminal human BRCa cell lines MCF-7 and ZR75-1, Δ40p53α at basal level decreases migration and invasion mimicking the role of p53α in suppressing cellular mobility and proliferation [135]. In the same cell lines, other researchers attributed a DNA repair stimulatory role to a high Δ40p53α versus p53α level by de-repressing RAD51 [116]. In transgenic mice, Δ40p53α collaborates with p53α to transactivate Cdkn1a, Mdm2, and Igfbp-3 to regulate the cell cycle [136]. In lung cancer, CRC, melanoma cancer cells, Δ40p53α regulates the miR-4671-5p-SGSH axis, BAX and PIDD to control cell cycle and apoptosis [84, 137, 138]. Additionally, Δ40p53α inactivates the PKR-elF2α pathway to inhibit autophagy in lung cancer and CRC cells [139]. In summary, Δ40p53α executes canonical functions regulating expression of multiple transcriptional targets with pro- and anti-tumorigenic effects, in collaboration with p53α but in some cases also in opposition to it. The final outcome of the p53α-modulatory impact of Δ40p53α might depend on the cellular context, e.g. the TP53 mutation status, the absolute and relative levels of Δ40p53α and p53α, cancer stemness, and finally genotoxic treatment of the cells. Underscoring the relevance of treatment, testing non-canonical functions of Δ40p53α in DNA replication revealed a dominant negative function towards co-expressed p53α in K562 leukemia cells upon exposure to the DNA cross-linking agent Mitomycin C but not in cells during unperturbed growth [81].
The alternative p53 isoform Δ133p53, in particular, impacts on signaling pathways related to tumor development. In mice Δ122p53/Δ133p53 promotes inflammatory responses often preceding the onset of tumor formation [97, 140,141,142,143]. Luciferase reporter assay demonstrated that Δ133p53 stimulates NF-κB activity following Helicobacter pylori infection, a significant risk factor for gastric cancer. This activation leads to the upregulation of NF-κB target genes including anti-apoptotic protein BCL-2 and pro-inflammatory factors IL-6 and IL-8 [144]. Treatment with the NF-κB inhibitor pyrrolidine dithiocarbamate resulted in the downregulation of Δ133p53 mRNA levels in MKN45 gastric cancer cells [145]. This suggests a regulatory feedback loop between Δ133p53 and NF-κB, a key regulatory factor during tumorigenesis and tumor progression via cell-intrinsic and extrinsic mechanisms ranging from cell death prevention [146], epithelial-mesenchymal transition (EMT) triggering metastasis [147], and anticancer drug resistance [148] to the regulation of tumor-associated macrophages [149]. Increased mRNA expression of the Δ133p53α isoform relative to p53α correlated with poor DFS in CRC patients. Here, the underlying mechanism was found to be tumor invasion promoted by the Δ133p53α activated JAK-STAT3 and RhoA-ROCK signaling [143]. Besides, Δ122p53/Δ133p53 facilitates tumor migration and invasion by downregulating E-cadherin and β1-integrin, upregulating Itgb7 and Vcam1, or activating RhoA-ROCK signaling [97, 143, 150]. In support of a role in tumor-associated inflammatory responses, Δ133p53α activates the expression of IFN-γ signaling genes in ER+ BRCa with mutant p53 [151]. Moreover, overexpression of Δ133p53β enhances the expression of genes associated with the IFN-γ signaling pathway in prostate cancer [152].
The dominant negative effect of Δ133p53 is central to its role in tumor development and progression underscored by the fact that Δ133p53 isoforms were never expressed alone as the only p53 isoform in cancer or normal cells [102]. Δ133p53α isoforms were overexpressed in tumor tissues of cholangiocarcinoma, lung cancer, colon cancer, ovarian cancer, melanoma and esophageal squamous cell carcinoma (ESCC) [102, 123, 131, 153,154,155]. Elevated Δ133p53α/β versus p53α mRNA ratio was correlated with poorer OS or DFS in various cancer patients, namely in cholangiocarcinoma, ESCC, melanoma, prostate cancer [123, 152, 153, 155] (Table 2). Δ133p53α (Δ122p53α/Δ113p53α, mouse/zebrafish orthologs of human Δ133p53) dominant negatively regulates the transactivation of MDM2, CDKN1A, PUMA, NOXA, BAX and miR-34a by p53α to control cell cycle and apoptosis [97, 98, 103].
Δ160p53α was found to be upregulated in melanoma compared to non-tumor tissue [123]. As shown for Δ133p53α in cell lines [7], Δ160p53α/β can be induced by doxorubicin, cisplatin as well as etoposide in melanoma [156]. Different from the p53α-mediated transactivation of the internal TP53 promoter in case of Δ133p53α in cell lines [7], translation of Δ160p53α is induced by oncogenic mutant p53 proteins and in this way involved in their gain of function phenotype [92] promoting tumor proliferation and migration. Importantly, Δ160p53α can be imported into the nucleus and bind chromatin [156].
Both Δ133p53 and Δ160p53, i.e. the p53 isoforms with prominent dominant negative biological effects, were shown to affect the usage of the error-free p53- and POLɩ-dependent DDT pathway triggered by co-expressed p53α [81]. Strikingly also, Δ133p53 and Δ160p53 caused fork stalling during DNA replication [81]. Such fork stalling was already noticeable under unperturbed growth conditions and already without co-expression of p53α suggesting an inhibitory effect on p53α co-factors of the DDT pathway [64, 81]. Indeed, the formation of POLɩ foci was downregulated in the presence of Δ133p53 or Δ160p53, which could be explained by the non-productive occupation of forks blocking access to replication factors such as POLɩ. Independently of the precise mechanism affecting such non-canonical function of p53α, both fork stalling and dysregulated DDT pathway usage are known to cause genomic instability and thereby tumorigenesis [89]. Moreover, loss of fork protection and resulting DNA damage are known to be sufficient to induce EMT genes and thereby invasiveness and metastasis [157].
Taken together, both canonical and non-canonical functions of alternative p53 isoforms can promote initiation and progression of cancer. Among the many factors determining the fate of a cell on the route towards malignancy, the expression levels of the p53 isoforms are essential. Evidence above demonstrates that most cancer types listed in Table 2 feature high expression levels of N-terminally truncated isoforms correlating with poorer prognosis while the C-terminally altered isoforms cause heterogenous effects.
p53 isoforms and responsiveness to cancer therapy
Cancer therapy resistance remains a significant challenge in the management of various malignancies. Radiation therapy and chemotherapy are important components of cancer therapy, which often exert their anti-tumor effects by interfering with DNA replication processes and/or inducing DNA damage [158]. While p53α is known to be a key modulator of these processes [159], whether isoforms that are C-terminally altered or N-terminally truncated are involved in these processes and impact on cancer therapy resistance has remained underexplored.
So far, it has been reported that C-terminally altered isoforms of p53 affect the cancer therapy resistance via altered transcriptional activity. Despite lacking the OD, p53β was shown to form a complex with p53α and exhibit a preference for specific p53 response elements (e.g. BAX promoter) after treatment with actinomycin D, which leads to apoptosis [94]. Furthermore, the ectopic expression of p53β in melanoma cell lines augmented transcription of CDKN1A and PUMA mediated by p53α upon cisplatin treatment [129]. H1299 cells with stable transduction of p53β displayed heightened susceptibility to DNA-damaging agents including doxorubicin and camptothecin in a p53α-independent manner [80]. Upon exposure to ionizing radiation (IR), alternative splicing of p53 pre-mRNA results in the generation of p53β mRNA and protein, which is essential for the activation of genes involved in cellular senescence [160]. Similar to p53β, H1299 cells transduced with p53γ exhibited heightened sensitivity to doxorubicin and camptothecin, which correlated with increased p21 and BAX expression. However, the protein level of p53γ was found to decrease after doxorubicin treatment. Additionally, treatment with a combination of doxorubicin and dicoumarol (an NAD(P)H quinone oxidoreductase 1 inhibitor) further escalated the degradation of p53γ [80]. Regardless of these p53γ isoform-specific features regarding protein stability, both p53β and p53γ have the potential to sensitize cancer cells to therapy.
The p53α-POLι dependent DDT pathway bypasses DNA damage and confers resistance to DNA stressors such as Mitomycin C or cisplatin [36, 64, 81, 161]. Singly expressed p53β and p53γ failed to mediate this pathway after treatment, and also did not exert a dominant negative effect on co-expressed p53α in DNA fiber assays [81]. From this, a major role of p53β/p53γ in chemotherapy resistance via usage of such a DDT pathway is unlikely. Taken together, C-terminally altered isoforms seem to sensitize cancer cells via transcriptional activities without additional influence of canonical functions in DNA replication. Given that multiple DNA repair factors bind to the C-terminus of p53α, it will be interesting to see whether canonical functions in DNA repair are affected, such as observed for Δ40p53 [87].
N-terminally truncated p53 isoforms were reported to influence resistance to cancer therapy via a dominant negative effect. In melanoma cell lines, Δ40p53α impedes cisplatin-induced p53α-dependent transcriptional activation of the PUMA and CDKN1A promoters [129]. Along this line, Δ40p53α antagonized p53α-mediated transcription of BAX, NOXA, PUMA, and CDKN1A post-treatment with doxorubicin but not with cisplatin reducing the fraction of cells arrested in G1-phase and undergoing apoptosis [87]. When exposed to 5-fluorouracil, the presence of Δ40p53 similarly hindered the canonical transcriptional activity of p53α [162]. Consistently, a high Δ40p53α/p53α ratio was shown not only to promote MCF7 cell growth and stemness features but also resistance to doxorubicin treatment after transplantation into immunodeficient mice [130].
DNA repair-related functions of p53α are also modulated by overexpression of Δ40p53α facilitating the transcription of RAD51 otherwise repressed by p53α [87, 163]. However, such result might also be explained at least in part by the Δ40p53α-induced cell cycle shift, since RAD51 is expressed during S- and G2-phase in the cell cycle [164]. Moreover, in BRCa cell lines, doxorubicin treatment led to the formation of nuclear p53α-RAD51-BRCA1 but not to Δ40p53α-RAD51-BRCA1 complexes [87], suggesting that unlike p53α, Δ40p53α is not involved in the non-canonical function of the fidelity control of homologous recombination [56, 74, 165, 166]. Accordingly, aberrant DNA repair processes are predicted to become increasingly de-restricted with increasing Δ40p53α/p53α ratio, which however, needs to be substantiated experimentally.
During replication stress caused by DNA damage-inducing agents, the dominant negative effect of the N-terminally truncated isoforms Δ133/160p53α affects the p53α-POLι dependent DDT pathway which facilitates the successful bypass of DNA damage and prevents the replication fork from collapsing [81]. As this pathway has been linked with resistance to Mitomycin C and to cisplatin [64, 161], Δ133p53α and Δ160p53α are predicted to sensitize cancer cells to DNA crosslinking agents via this transcription-independent mechanism. On the other hand, Δ133p53α affects canonical p53 functions in the DNA damage response post-treatment with doxorubicin by forming a complex with p53α, antagonizing induction of the G1 arrest (via repressing CDKN1A transactivation) and of apoptosis (de-repressing transcription of BCL-2). Of note, Δ133p53α is upregulated by p53α-dependent transcriptional transactivation at the internal promoter in response to a low dose of this DNA intercalating agent triggering such a negative feedback loop [7]. Furthermore, Δ113p53 (zebrafish ortholog of human Δ133p53) collaborates with p73 to promote DNA double-strand repair via rad51, Lig4 and rad52 upregulation. In 10.1 cells (mouse p53-null fibroblast cell line), Δ122p53β (murine Δ133p53β isoform) reduced sensitivities to temozolomide and tert-butyl hydroperoxide, two treatments inducing base damage [167]. In CRC cells, Δ133p53β hindered camptothecin, i.e. topoisomerase I inhibitor-induced apoptosis by binding to RhoB [168]. Altogether, N-terminally truncated p53 isoforms have the potential to cause chemoresistance of cancer cells by both canonical and non-canonical functions. The net outcome of these different responses to genotoxic drugs might depend not only on the cellular context and on p53 isoform levels but also on the type and severity of DNA damage.
p53 isoforms play crucial roles in tumor development and cancer therapy resistance. Understanding the roles and mechanisms of these p53 isoforms is essential for developing effective strategies to combat cancer. Dominant negative effects on transcriptional activities and on transcription-independent DNA replication regulatory activities are key mechanisms through which p53 isoforms exhibit their biological functions and influence tumor development and cancer therapy resistance. Hence, integrating such knowledge from different cancer entities and treatment regimens will help to develop refined strategies to overcome therapy resistance to cancer treatment.
Conclusion
The study of p53 isoforms has provided significant insights into the complex biological roles of the p53 protein in both normal cellular functions and cancer development. The diverse isoforms of p53 exhibit canonical functions in transcriptionally transactivating or repressing target genes, impacting on cell cycle control, apoptosis induction and DNA repair, in particular. Aside from these canonical functions, non-canonical, i.e. transcription-independent functions of these isoforms in DNA repair and replication have been discovered more recently. In a concerted action, integrating cooperative, antagonistic and independent activities of the different isoforms, these biological functions modulate tumor suppressor activities and influence the sensitivity of cancer cells to therapies.
Aside from the structural diversity of p53 isoforms abnormal expression or dysregulation of p53 isoforms have been implicated in various types of cancers, contributing to tumor progression, metastasis, and drug resistance. It is known that the level of p53α plays a critical role in differentially activating different gene sets [169], and can rapidly be induced by genotoxic treatment impacting on the cellular program [170]. Intriguingly, it has also been observed that the level of p53 determines the DDT pathway chosen to bypass replication impediments (L.W., personal communication). Given that in Guo et al. Δ133p53α and Δ160p53α were found to abrogate such p53α functions, a change in cellular levels of p53α, Δ133p53α and/or Δ160p53α will affect the pathway choice by mixed oligomer formation or competition for DDT pathway components [81]. Thus, both for canonical as well as noncanonical functions the levels of p53 isoforms are relevant, fine-tuning the outcome of activating specific gene sets and specific DDT pathways, respectively.
However, despite all these advances further research is warranted to elucidate the precise molecular mechanisms by which p53 isoforms exert their effects and to reveal their potential as diagnostic and prognostic biomarkers. First, non-canonical functions of alternative p53 isoforms in DNA repair are underexplored, though structural differences between p53 isoforms affecting the interaction sites with crucial DNA repair factors like POLβ [33], RAD51 [56, 74, 165, 166] or BRCA1 [87] must impact on base excision repair and homologous recombination, respectively. Second, it remains to be determined in how far non-canonical functions of alternative isoforms in DNA replication and repair modify stemness features/EMT of cancer cells and thereby invasiveness, metastasis and chemoresistance. Third, results from functional assays have become valuable tools for the classification of germline variants of TP53 with regard to their pathogenicity [171,172,173]. Yet, the impact of alternative splicing as well as of alternative transcription or translation start sites have been largely ignored. Understanding the functional diversity and interplay of p53 isoforms may pave the way for personalized approaches towards cancer prevention and the development of innovative therapies.
References
Joruiz SM, Bourdon JC. p53 isoforms: key regulators of the cell fate decision. Cold Spring Harb Perspect Med. 2016;6:a026039.
Anbarasan T, Bourdon JC. The emerging landscape of p53 isoforms in physiology, cancer and degenerative diseases. Int J Mol Sci. 2019;20:6257.
Matlashewski G, Lamb P, Pim D, Peacock J, Crawford L, Benchimol S. Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. EMBO J. 1984;3:3257–62.
Wolf D, Harris N, Goldfinger N, Rotter V. Isolation of a full-length mouse cDNA clone coding for an immunologically distinct p53 molecule. Mol Cell Biol. 1985;5:127–32.
Grover R, Ray PS, Das S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle. 2008;7:2189–98.
Khoury MP, Bourdon J-C. The isoforms of the p53 protein. Cold Spring Harb Perspect Biol. 2010;2:a000927.
Aoubala M, Murray-Zmijewski F, Khoury MP, Fernandes K, Perrier S, Bernard H, et al. P53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011;18:248–58.
Vieler M, Sanyal S. p53 isoforms and their implications in cancer. Cancers (Basel). 2018;10:288.
Chen J, Ng SM, Chang Q, Zhang Z, Bourdon JC, Lane DP, et al. P53 isoform Δ113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009;23:278–90.
Marcel V, Vijayakumar V, Fernández-Cuesta L, Hafsi H, Sagne C, Hautefeuille A, et al. p53 regulates the transcription of its Δ133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene 2010;29:2691–700.
Blackburn J, Roden DL, Ng R, Wu J, Bosman A, Epstein RJ. Damage-inducible intragenic demethylation of the human TP53 tumor suppressor gene is associated with transcription from an alternative intronic promoter. Mol Carcinog. 2016;55:1940–51.
Raycroft L, Wu H, Lozano G. Transcriptional activation by wild-type but not transforming mutants of the p53 anti-oncogene. Science (80-). 1990;249:1049–51.
Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 1991;65:1083–91.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817–25.
Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–11.
Fischer M. Census and evaluation of p53 target genes. Oncogene 2017;36:3943–56.
Stenger JE, Tegtmeyer P, Mayr GA, Reed M, Wang Y, Wang P, et al. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J. 1994;13:6011–20.
Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inf Ser Workshop Genome Inf. 2000;11:161–71.
Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29.
Seto E, Usheva A, Zambetti GP, Momand J, Horikoshi N, Weinmann R, et al. Wild-type p53 binds to the TATA-binding protein and represses transcription. Proc Natl Acad Sci USA. 1992;89:12028–32.
Chen X, Farmer G, Zhu H, Prywes R, Prives C. Cooperative DNA binding of p53 with TFIID (TBP): a possible mechanism for transcriptional activation. Genes Dev. 1993;7:1837–49.
Thut CJ, Chen JL, Klemm R, Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science. 1995;267:100–4.
Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci USA. 1995;92:5154–8.
Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier JL, et al. Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol Cell Biol. 1994;14:7013–24.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992;69:1237–45.
Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–14.
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996;274:948–53.
Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–08.
Raj N, Attardi LD. The transactivation domains of the p53 protein. Cold Spring Harb Perspect Med. 2017;7:a026047.
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997;89:1175–84.
Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997;387:823–7.
Dutta A, Ruppert JM, Aster JC, Winchester E. Inhibition of DNA replication factor RPA by p53. Nature 1993;365:79–82.
Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO J. 2001;20:914–23.
Melle C, Nasheuer H-P. Physical and functional interactions of the tumor suppressor protein p53 and DNA polymerase alpha-primase. Nucleic Acids Res. 2002;30:1493–9.
Banks D, Wu M, Higa LA, Gavrilova N, Quan J, Ye T, et al. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle. 2006;5:1719–29.
Biber S, Pospiech H, Gottifredi V, Wiesm L. Multiple biochemical properties of the p53 molecule contribute to activation of polymerase iota-dependent DNA damage tolerance. Nucleic Acids Res. 2020;48:12188–203.
Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85:375–404.
Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011;145:571–83.
Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, et al. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci USA. 2011;108:17123–8.
Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25:133–43.
Haupt Y, Mayat R, Kazazt A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296–9.
Walker KK, Levine AJ. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA. 1996;93:15335–40.
Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 1994;76:933–45.
Opitz R, Müller M, Reuter C, Barone M, Soicke A, Roske Y, et al. A modular toolkit to inhibit proline-rich motif-mediated protein-protein interactions. Proc Natl Acad Sci USA. 2015;112:5011–6.
Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998;17:4668–79.
Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 2002;21:9–21.
Toledo F, Krummel KA, Lee CJ, Liu C-W, Rodewald L-W, Tang M, et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9:273–85.
Pavletich NP, Chambers KA, Pabo CO. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993;7:2556–64.
Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (80-). 1994;265:346–55.
Xue Y, Wang S, Feng X. Influence of magnesium ion on the binding of p53 DNA-binding domain to DNA-response elements. J Biochem. 2009;146:77–85.
Natan E, Baloglu C, Pagel K, Freund SMV, Morgner N, Robinson CV, et al. Interaction of the p53 DNA-binding domain with its n-terminal extension modulates the stability of the p53 tetramer. J Mol Biol. 2011;409:358–68.
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–76.
Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M, Fersht AR. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci USA. 1999;96:8438–42.
Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene 2007;26:2145–56.
Lei J, Qi R, Tang Y, Wang W, Wei G, Nussinov R, et al. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. 2019;33:4225–4235.
Buchhop S, Gibson MK, Wang XW, Wagner P, Stürzbecher HW, Harris CC. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res. 1997;25:3868–74.
Yoon D, Wang Y, Stapleford K, Wiesmüller L, Chen J. P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol. 2004;336:639–54.
Dudenhöffer C, Rohaly G, Will K, Deppert W, Wiesmüller L. Specific mismatch recognition in heteroduplex intermediates by p53 suggests a role in fidelity control of homologous recombination. Mol Cell Biol. 1998;18:5332–42.
Yang Q, Zhang R, Wang XW, Spillare EA, Linke SP, Subramanian D, et al. The processing of Holliday junctions by BLM and WRN helicases is regulated by p53. J Biol Chem. 2002;277:31980–7.
Mummenbrauer T, Janus F, Müller B, Wiesmüller L, Deppert W, Grosse F. p53 protein exhibits 3’-to-5’ exonuclease activity. Cell 1996;85:1089–99.
Süsse S, Janz C, Janus F, Deppert W, Wiesmüller L. Role of heteroduplex joints in the functional interactions between human Rad51 and wild-type p53. Oncogene 2000;19:4500–12.
Janus F, Albrechtsen N, Knippschild U, Wiesmüller L, Grosse F, Deppert W. Different regulation of the p53 core domain activities 3’-to-5’ exonuclease and sequence-specific DNA binding. Mol Cell Biol. 1999;19:2155–68.
Akyüz N, Boehden GS, Süsse S, Rimek A, Preuss U, Scheidtmann K-H, et al. DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol Cell Biol. 2002;22:6306–17.
Hampp S, Kiessling T, Buechle K, Mansilla SF, Thomale J, Rall M, et al. DNA damage tolerance pathway involving DNA polymerase ι and the tumor suppressor p53 regulates DNA replication fork progression. Proc Natl Acad Sci USA. 2016;113:E4311–9.
Liang SH, Clarke MF. The nuclear import of p53 is determined by the presence of a basic domain and its relative position to the nuclear localization signal. Oncogene 1999;18:2163–66.
Aurelio ON, Cajot JF, Hua MLH, Khwaja Z, Stanbridge EJ. Germ-line-derived hinge domain p53 mutants have lost apoptotic but not cell cycle arrest functions. Cancer Res. 1998;58:2190–5.
Kong XT, Gao H, Stanbridge EJ. Mechanisms of differential activation of target gene promoters by p53 hinge domain mutants with impaired apoptotic function. J Biol Chem. 2001;276:32990–33000.
Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999;18:1660–72.
D’Abramo M, Bešker N, Desideri A, Levine AJ, Melino G, Chillemi G. The p53 tetramer shows an induced-fit interaction of the C-terminal domain with the DNA-binding domain. Oncogene 2016;35:3272–81.
Beckerman R, Yoh K, Mattia-Sansobrino M, Zupnick A, Laptenko O, Karni-Schmidt O, et al. Lysines in the tetramerization domain of p53 selectively modulate G1 arrest. Cell Cycle. 2016;15:1425–38.
Dudenhöffer C, Kurth M, Janus F, Deppert W, Wiesmüller L. Dissociation of the recombination control and the sequence-specific transactivation function of P53. Oncogene 1999;18:5773–84.
Graupner V, Schulze-Osthoff K, Essmann F, Jänicke RU. Functional characterization of p53β and p53γ, two isoforms of the tumor suppressor p53. Cell Cycle. 2009;8:1238–48.
Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950.
Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16.
Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, et al. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res. 2008;36:1900–12.
Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 1995;81:1013–20.
Bakalkin G, Yakovleva T, Selivanova G, Magnusson KP, Szekely L, Kiseleva E, et al. p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc Natl Acad Sci USA. 1994;91:413–7.
Brain R, Jenkins JR. Human p53 directs DNA strand reassociation and is photolabelled by 8-azido ATP. Oncogene 1994;9:1775–80.
McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol Cell. 2004;16:413–24.
Silden E, Hjelle SM, Wergeland L, Sulen A, Andresen V, Bourdon JC, et al. Expression of TP53 isoforms p53β or p53γ enhances chemosensitivity in TP53null cell lines. PLoS One. 2013;8:e56276.
Guo Y, Rall-Scharpf M, Bourdon J-C, Wiesmüller L, Biber S. p53 isoforms differentially impact on the POLι dependent DNA damage tolerance pathway. Cell Death Dis. 2021;12:941.
Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, et al. ΔN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002;21:6722–8.
Hafsi H, Santos-Silva D, Courtois-Cox S, Hainaut P. Effects of Δ40p53, an isoform of p53 lacking the N-terminus, on transactivation capacity of the tumor suppressor protein p53. BMC Cancer. 2013;13:134.
Yin Y, Stephen CW, Luciani MG, Fåhraeus R. p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–7.
Sharathchandra A, Katoch A, Das S. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscip Rev RNA. 2014;5:131–9.
Ohki R, Kawase T, Ohta T, Ichikawa H, Taya Y. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Sci. 2007;98:189–200.
Steffens Reinhardt L, Zhang X, Groen K, Morten BC, De Iuliis GN, Braithwaite AW, et al. Alterations in the p53 isoform ratio govern breast cancer cell fate in response to DNA damage. Cell Death Dis. 2022;13:907.
Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, et al. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998;16:1713–21.
Berti M, Cortez D, Lopes M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol. 2020;21:633–51.
Gong L, Gong H, Pan X, Chang C, Ou Z, Ye S, et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015;25:351–69.
Gong H, Zhang Y, Jiang K, Ye S, Chen S, Zhang Q, et al. p73 coordinates with Δ133p53 to promote DNA double-strand break repair. Cell Death Differ. 2018;25:1063–79.
Candeias MM, Hagiwara M, Matsuda M. Cancer-specific mutations in p53 induce the translation of Δ160p53 promoting tumorigenesis. EMBO Rep. 2016;17:1542–51.
Marcel V, Fernandes K, Terrier O, Lane DP, Bourdon JC. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014;21:1377–87.
Bourdon J, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37.
Ungewitter E, Scrable H. Δ40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010;24:2408–19.
Steffens Reinhardt L, Groen K, Zhang X, Morten BC, Wawruszak A, Avery-Kiejda KA. P53 isoform expression promotes a stemness phenotype and inhibits doxorubicin sensitivity in breast cancer. Cell Death Dis. 2023;14:1–16.
Slatter TL, Hung N, Campbell H, Rubio C, Mehta R, Renshaw P, et al. Hyperproliferation, cancer, and inflammation in mice expressing a Δ133p53-like isoform. Blood 2011;117:5166–77.
Mondal AM, Zhou H, Horikawa I, Suprynowicz FA, Li G, Dakic A, et al. Δ133P53α, a natural P53 isoform, contributes to conditional reprogramming and long-term proliferation of primary epithelial cells. Cell Death Dis. 2018;9:750.
Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991;65:765–74.
Xie N, Chen M, Dai R, Zhang Y, Zhao H, Song Z, et al. SRSF1 promotes vascular smooth muscle cell proliferation through a Δ133p53/EGR1/KLF5 pathway. Nat Commun. 2017;8:16016.
Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72.
Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009;11:1135–42.
von Muhlinen N, Horikawa I, Alam F, Isogaya K, Lissa D, Vojtesek B, et al. p53 isoforms regulate premature aging in human cells. Oncogene 2018;37:2379–93.
Gong L, Pan X, Chen H, Rao L, Zeng Y, Hang H, et al. p53 isoform Δ133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci Rep. 2016;6:37281.
Li R, Botchan MR. The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell 1993;73:1207–21.
Bakhanashvili M, Grinberg S, Bonda E, Rahav G. Excision of nucleoside analogs in mitochondria by p53 protein. AIDS 2009;23:779–88.
Park J-H, Zhuang J, Li J, Hwang PM. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016;590:924–34.
Gottifredi V, Wiesmüller L. The tip of an iceberg: replication-associated functions of the tumor suppressor p53. Cancers (Basel). 2018;10:1–18.
Ho T, Tan BX, Lane D. How the other half lives: what p53 does when it is not being a transcription factor. Int J Mol Sci. 2020;21. https://doi.org/10.3390/ijms21010013.
Gupta S, De S, Srivastava V, Hussain M, Kumari J, Muniyappa K, et al. RECQL4 and p53 potentiate the activity of polymerase γ and maintain the integrity of the human mitochondrial genome. Carcinogenesis 2014;35:34–45.
Schreier HK, Wiehe RS, Ricchetti M, Wiesmüller L. Polymerase ζ is involved in mitochondrial DNA maintenance processes in concert with APE1 activity. Genes (Basel). 2022;13:879.
Ahn J, Poyurovsky MV, Baptiste N, Beckerman R, Cain C, Mattia M, et al. Dissection of the sequence-specific DNA binding and exonuclease activities reveals a superactive yet apoptotically impaired mutant p53 protein. Cell Cycle. 2009;8:1603–15.
Ihle M, Biber S, Schroeder IS, Blattner C, Deniz M, Damia G, et al. Impact of the interplay between stemness features, p53 and pol iota on replication pathway choices. Nucleic Acids Res. 2021;49:7457–75.
Steffens Reinhardt L, Groen K, Newton C, Avery-Kiejda KA. The role of truncated p53 isoforms in the DNA damage response. Biochim Biophys Acta - Rev Cancer. 2023;1878:188882.
Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987–97.
Dos Santos NM, De Oliveira GAP, Rocha MR, Pedrote MM, Da Silva Ferretti GD, Rangel LP, et al. Loss of the p53 transactivation domain results in high amyloid aggregation of the δ40p53 isoform in endometrial carcinoma cells. J Biol Chem. 2019;294:9430–39.
Kovachev PS, Banerjee D, Rangel LP, Eriksson J, Pedrote MM, Martins-Dinis MMDC, et al. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J Biol Chem. 2017;292:9345–57.
Vlašić I, Horvat A, Tadijan A, Slade N. p53 family in resistance to targeted therapy of melanoma. Int J Mol Sci. 2022;24:65.
Steffens Reinhardt L, Groen K, Xavier A, Avery-Kiejda KA. p53 dysregulation in breast cancer: insights on mutations in the TP53 network and p53 isoform expression. Int J Mol Sci. 2023;24:10078.
Zhang H, Zhao Y, Sun P, Zhao M, Su Z, Jin X, et al. p53β: A new prognostic marker for patients with clear-cell renal cell carcinoma from 5.3 years of median follow-up. Carcinogenesis 2018;39:368–74.
Avery-Kiejda KA, Morten B, Wong-Brown MW, Mathe A, Scott RJ. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014;35:586–96.
Ånensen N, Hjelle SM, Van Belle W, Haaland I, Silden E, Bourdon JC, et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012;31:1533–45.
Ozretić P, Hanžić N, Proust B, Sabol M, Trnski D, Radić M, et al. Expression profiles of p53/p73, NME and GLI families in metastatic melanoma tissue and cell lines. Sci Rep. 2019;9:12470.
Boldrup L, Bourdon JC, Coates PJ, Sjöström B, Nylander K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur J Cancer. 2007;43:617–23.
Bourdon JC, Khoury MP, Diot A, Baker L, Fernandes K, Aoubala M, et al. P53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011;13:R7.
Bischof K, Knappskog S, Stefansson I, McCormack EM, Trovik J, Werner HMJ, et al. High expression of the p53 isoform γ is associated with reduced progression-free survival in uterine serous carcinoma. BMC Cancer. 2018;18:1–10.
Montero-Calle A, Garranzo-Asensio M, Torrente-Rodríguez RM, Ruiz-Valdepeñas Montiel V, Poves C, Dziaková J et al. p53 and p63 proteoforms derived from alternative splicing possess differential seroreactivity in colorectal cancer with distinct diagnostic ability from the canonical proteins. Cancers (Basel). 2023;15. https://doi.org/10.3390/cancers15072102.
Takahashi R, Giannini C, Sarkaria JN, Schroeder M, Rogers J, Mastroeni D, et al. p53 isoform profiling in glioblastoma and injured brain. Oncogene 2013;32:3165–74.
Avery-Kiejda KA, Xu DZ, Adams LJ, Scott RJ, Vojtesek B, Lane DP, et al. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin Cancer Res. 2008;14:1659–68.
Steffens Reinhardt L, Groen K, Zhang X, Morten BC, Wawruszak A, Avery-Kiejda KA. p53 isoform expression promotes a stemness phenotype and inhibits doxorubicin sensitivity in breast cancer. Cell Death Dis. 2023;14:509.
Hofstetter G, Berger A, Schuster E, Wolf A, Hager G, Vergote I, et al. Δ133P53 is an independent prognostic marker in P53 mutant advanced serous ovarian cancer. Br J Cancer. 2011;105:1593–9.
Hofstetter G, Berger A, Berger R, Zorić A, Braicu EI, Reimer D, et al. The N-terminally truncated p53 isoform Δ40p53 influences prognosis in mucinous ovarian cancer. Int J Gynecol Cancer. 2012;22:372–9.
Morten BC, Scott RJ, Avery-Kiejda KA. Comparison of the QuantiGene 2.0 assay and real-time RT-PCR in the detection of p53 isoform mRNA expression in formalin-fixed paraffin-embedded tissues—a preliminary study. PLoS One. 2016;11:e0165930.
Le Du F, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F, et al. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget 2015;6:12890–908.
Zhang X, Groen K, Morten BC, Steffens Reinhardt L, Campbell HG, Braithwaite AW, et al. Effect of p53 and its N-terminally truncated isoform, Δ40p53, on breast cancer migration and invasion. Mol Oncol. 2022;16:447–65.
Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–19.
Pal A, Khan D, Tripathi SK, Ghosh PK, Ghosh S, Patra S, et al. p53 translational isoform Δ40p53 orchestrates cellular SGSH levels via microRNA-4671-5p to modulate cell cycle. bioRxiv. 2023.04.04;535506.
Takahashi R, Markovic SN, Scrable HJ. Dominant effects of Δ40p53 on p53 function and melanoma cell fate. J Invest Dermatol. 2014;134:791–800.
Zang Y, Shi Y, Liu K, Qiao L, Guo X, Chen D. Δ40p53 is involved in the inactivation of autophagy and contributes to inhibition of cell death in HCT116-Δ40p53 cells. Oncotarget 2017;8:12754–63.
Sawhney S, Hood K, Shaw A, Braithwaite AW, Stubbs R, Hung NA, et al. Alpha-enolase is upregulated on the cell surface and responds to plasminogen activation in mice expressing a Δ133p53α mimic. PLoS One. 2015;10:e0116270.
Slatter TL, Hung N, Bowie S, Campbell H, Rubio C, Speidel D, et al. Δ122P53, a mouse model of Δ133P53Α, enhances the tumor-suppressor activities of an attenuated P53 mutant. Cell Death Dis. 2015;6:e1783.
Campbell HG, Slatter TL, Jeffs A, Mehta R, Rubio C, Baird M, et al. Does Δ133p53 isoform trigger inflammation and autoimmunity? Cell Cycle. 2012;11:446–50.
Campbell H, Fleming N, Roth I, Mehta S, Wiles A, Williams G, et al. Δ133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signaling. Nat Commun. 2018;9:254.
Wei J, Noto J, Zaika E, Romero-Gallo J, Correa P, El-Rifai W, et al. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc Natl Acad Sci USA. 2012;109:E2543–50.
Zhang H-M, Sang X-G, Wang Y-Z, Cui C, Zhang L, Ji W-S. Role of Δ133p53 isoform in NF-κB inhibitor PDTC-mediated growth inhibition of MKN45 gastric cancer cells. World J Gastroenterol. 2017;23:2716–22.
Wu X, Sun L, Xu F. NF-κB in cell deaths, therapeutic resistance and nanotherapy of tumors: recent advances. Pharmaceuticals (Basel). 2023;16. https://doi.org/10.3390/ph16060783.
Oh A, Pardo M, Rodriguez A, Yu C, Nguyen L, Liang O, et al. NF-κB signaling in neoplastic transition from epithelial to mesenchymal phenotype. Cell Commun Signal. 2023;21:291.
Bou Antoun N, Chioni A-M. Dysregulated signalling pathways driving anticancer drug resistance. Int J Mol Sci. 2023;24. https://doi.org/10.3390/ijms241512222.
He R, He Y, Du R, Liu C, Chen Z, Zeng A, et al. Revisiting of TAMs in tumor immune microenvironment: insight from NF-κB signaling pathway. Biomed Pharmacother. 2023;165:115090.
Gadea G, Arsic N, Fernandes K, Diot A, Joruiz SM, Abdallah S, et al. TP53 drives invasion through expression of its Δ133p53β variant. Elife 2016;5:e14734.
Mehta SY, Morten BC, Antony J, Henderson L, Lasham A, Campbell H, et al. Regulation of the interferon-gamma (IFN-γ) pathway by p63 and Δ133p53 isoform in different breast cancer subtypes. Oncotarget 2018;9:29146–61.
Kazantseva M, Mehta S, Eiholzer RA, Gimenez G, Bowie S, Campbell H, et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019;10:631.
Nutthasirikul N, Limpaiboon T, Leelayuwat C, Patrakitkomjorn S, Jearanaikoon P. Ratio disruption of the Δ133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int J Oncol. 2013;42:1181–8.
Fragou A, Tzimagiorgis G, Karageorgopoulos C, Barbetakis N, Lazopoulos A, Papaioannou M, et al. Increased δ133p53 mRNA in lung carcinoma corresponds with reduction of p21 expression. Mol Med Rep. 2017;15:1455–60.
Tu Q, Gong H, Yuan C, Liu G, Huang J, Li Z, et al. Δ133p53/FLp53 predicts poor clinical outcome in esophageal squamous cell carcinoma. Cancer Manag Res. 2020;12:7405–17.
Tadijan A, Precazzini F, Hanžić N, Radić M, Gavioli N, Vlašić I et al. Altered expression of shorter p53 family isoforms can impact melanoma aggressiveness. Cancers (Basel). 2021;13. https://doi.org/10.3390/cancers13205231.
Heitmeir B, Deniz M, Janni W, Rack B, Schochter F, Wiesmüller L. Circulating tumor cells in breast cancer patients: a balancing act between stemness, EMT features and DNA damage responses. Cancers (Basel). 2022;14:997.
Reuvers TGA, Kanaar R, Nonnekens J. DNA damage-inducing anticancer therapies: from global to precision damage. Cancers (Basel). 2020;12:2098.
Abuetabh Y, Wu HH, Chai C, Al Yousef H, Persad S, Sergi CM, et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp Mol Med. 2022;54:1658–69.
Chen J, Crutchley J, Zhang D, Owzar K, Kastan MB. Identification of a DNA damage-induced alternative splicing pathway that regulates p53 and cellular senescence markers. Cancer Discov. 2017;7:766–81.
Mansilla SF, Bertolin AP, Venerus Arbilla S, Castaño BA, Jahjah T, Singh JK, et al. Polymerase iota (Pol ι) prevents PrimPol-mediated nascent DNA synthesis and chromosome instability. Sci Adv. 2023;9:eade7997.
Levandowski CB, Jones T, Gruca M, Ramamoorthy S, Dowell RD, Taatjes DJ. The Δ40p53 isoform inhibits p53-dependent eRNA transcription and enables regulation by signal-specific transcription factors during p53 activation. PLOS Biol. 2021;19:e3001364.
Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–24.
Yamamoto A, Taki T, Yagi H, Habu T, Yoshida K, Yoshimura Y, et al. Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol Gen Genet. 1996;251:1–12.
Janz C, Süsse S, Wiesmüller L. p53 and recombination intermediates: role of tetramerization at DNA junctions in complex formation and exonucleolytic degradation. Oncogene 2002;21:2130–40.
Bertrand P, Saintigny Y, Lopez BS. p53’s double life: transactivation-independent repression of homologous recombination. Trends Genet. 2004;20:235–43.
Kazantseva M, Eiholzer RA, Mehta S, Taha A, Bowie S, Roth I, et al. Elevation of the TP53 isoform Δ133p53β in glioblastomas: an alternative to mutant p53 in promoting tumor development. J Pathol. 2018;246:77–88.
Arsic N, Ho-Pun-Cheung A, Evelyne C, Assenat E, Jarlier M, Anguille C, et al. The p53 isoform delta133p53β regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS One. 2017;12:e0172125.
Lokshin M, Tanaka T, Prives C. Transcriptional regulation by p53 and p73. Cold Spring Harb Symp Quant Biol. 2005;70:121–8.
Eldridge CB, Allen FJ, Crisp A, Grandy RA, Vallier L, Sale JE. A p53-dependent checkpoint induced upon DNA damage alters cell fate during hiPSC differentiation. Stem Cell Rep. 2020;15:827–35.
Kato S, Han S-Y, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA. 2003;100:8424–9.
Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7.
Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178–90.e8.
Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J Clin Invest. 2013;123:5247–57.
Pehar M, O’Riordan KJ, Burns‐Cusato M, Andrzejewski ME, Del Alcazar CG, Burger C, et al. Altered longevity‐assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9:174–90.
Gambino V, De Michele G, Venezia O, Migliaccio P, Dall’Olio V, Bernard L, et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell. 2013;12:435–45.
Parkar S, López-Iniesta MJ, Ramalho AC, Kimura K, Zhao J, da Silva Rita F, et al. Δ246p53 is a new p53 isoform that responds to DNA damage and regulates tumour growth. bioRxiv. 2023.04.07;536059.
Ou Z, Yin L, Chang C, Peng J, Chen J. Protein interaction between p53 and δ113p53 is required for the anti-apoptotic function of δ113p53. J Genet Genom. 2014;41:53–62.
Yamanaka S, Campbell NR, An F, Kuo SC, Potter JJ, Mezey E, et al. Coordinated effects of microRNA-494 induce G2/M arrest in human cholangiocarcinoma. Cell Cycle. 2012;11:2729–38.
Arsic N, Gadea G, Lagerqvist EL, Bußon M, Cahuzac N, Brock C, et al. The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Rep. 2015;4:531–40.
Mondal S, Bhattacharya K, Mandal C. Nutritional stress reprograms dedifferention in glioblastoma multiforme driven by PTEN/Wnt/Hedgehog axis: a stochastic model of cancer stem cells. Cell Death Discov. 2018;4:110.
Steffens Reinhardt L, Groen K, Morten BC, Bourdon J-C, Avery-Kiejda KA. Cytoplasmic p53β isoforms are associated with worse disease-free survival in breast cancer. Int J Mol Sci. 2022;23. https://doi.org/10.3390/ijms23126670.
Song W, Huo SW, Lü JJ, Liu Z, Fang XL, Jin XB, et al. Expression of p53 isoforms in renal cell carcinoma. Chin Med J (Engl). 2009;122:921–6.
Haaland I, Hjelle SM, Reikvam H, Sulen A, Ryningen A, McCormack E et al. p53 protein isoform profiles in AML: correlation with distinct differentiation stages and response to epigenetic differentiation therapy. Cells. 2021;10. https://doi.org/10.3390/cells10040833.
Groen K, Steffens Reinhardt L, Bourdon J-C, Avery-Kiejda KA. It is not all about the alpha: elevated expression of p53β variants is associated with lower probability of survival in a retrospective melanoma cohort. Cancer Cell Int. 2023;23:228.
Bischof K, Knappskog S, Hjelle SM, Stefansson I, Woie K, Salvesen HB, et al. Influence of p53 isoform expression on survival in high-grade serous ovarian cancers. Sci Rep. 2019;9:5244.
Knezović Florijan M, Ozretić P, Bujak M, Pezzè L, Ciribilli Y, Kaštelan Ž, et al. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. Urol Oncol Semin Orig Investig. 2019;37:578.e1–10.
Jesus ANB, Taha A, Wang D, Mehta PM, Mehta S, Reily-Bell A et al. Increased expression of the Δ133p53β isoform enhances brain metastasis. Int J Mol Sci. 2023;24. https://doi.org/10.3390/ijms24021267.
Saha T, Kar RK, Sa G. Structural and sequential context of p53: a review of experimental and theoretical evidence. Prog Biophys Mol Biol. 2015;117:250–63.
Acknowledgements
The authors acknowledge BioRender.com for offering image developing tool, National Natural Science Foundation of China (#82203151 to YG), Scientific Research Talent Cultivation Project of Zhongda Hospital Southeast University (to YG and HW), and the German Cancer Aid (Priority Program ‘Translational Oncology’, project HerediVar´, #70114178 to LW) for offering funding for this review.
Funding
This review was funded by National Natural Science Foundation of China (#82203151 to YG) and Scientific Research Talent Cultivation Project of Zhongda Hospital Southeast University (Jiangsu Province High-Level Hospital Construction Funds, #CZXM-GSP-RC56 to YG and #CZXM-GSP-RC112 to HW). LW was financially supported by the German Cancer Aid, Priority Program ‘Translational Oncology’, project HerediVar´, #70114178.
Author information
Authors and Affiliations
Contributions
YG, LW and MC made the outline of the review. YG, HW, LW and MC wrote the review. YG and HW produced the figures and tables. LW optimized the figures and tables. All authors revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Giovanni Blandino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, Y., Wu, H., Wiesmüller, L. et al. Canonical and non-canonical functions of p53 isoforms: potentiating the complexity of tumor development and therapy resistance. Cell Death Dis 15, 412 (2024). https://doi.org/10.1038/s41419-024-06783-7
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06783-7
- Springer Nature Limited