
Abstract
As a newly identified regulated cell death, ferroptosis is a metabolically driven process that relies on iron and is associated with polyunsaturated fatty acyl peroxidation, elevated levels of reactive oxygen species (ROS), and mitochondrial damage. This distinct regulated cell death is dysregulated in various cancers; activating ferroptosis in malignant cells increases cancer immunotherapy and chemoradiotherapy responses across different malignancies. Over the last decade, accumulating research has provided evidence of cross-talk between non-coding RNAs (ncRNAs) and competing endogenous RNA (ceRNA) networks and highlighted their significance in developing and progressing malignancies. Aside from pharmaceutical agents to regulate ferroptosis, recent studies have shed light on the potential of restoring dysregulated ferroptosis-related ceRNA networks in cancer treatment. The present study provides a comprehensive and up-to-date review of the ferroptosis significance, ferroptosis pathways, the role of ferroptosis in cancer immunotherapy and chemoradiotherapy, ceRNA biogenesis, and ferroptosis-regulating ceRNA networks in different cancers. The provided insights can offer the authorship with state-of-the-art findings and future perspectives regarding the ferroptosis and ferroptosis-related ceRNA networks and their implication in the treatment and determining the prognosis of affected patients.
Similar content being viewed by others
Facts
-
Ferroptosis is a newly identified regulated cell death that has anti-tumoral properties.
-
Restoring ferroptosis enhances cancer immunotherapy and chemoradiotherapy responses.
-
The dysregulation of lncRNA and circRNA-mediated ceRNA networks is highly implicated in tumor development and progression.
-
Restoring dysregulated ferroptosis-related ceRNAs not only stimulates ferroptosis but also decreases tumor growth and oncogenesis and increases anti-neoplastic treatment efficacy.
Open questions
-
What are the regulated cell deaths, and what is ferroptosis?
-
What are the signaling pathways of ferroptosis?
-
What is the significance of ferroptosis in oncogenesis and anti-neoplastic treatments?
-
What is the concept behind the competing endogenous RNA networks?
-
Can regulating ferroptosis-related competing endogenous RNA networks be a therapeutic option for cancer treatment?
Introduction
Solid cancers are a prominent contributor to cancer-related mortality, as identified by tumor recurrence and metastasis [1]. The development and progression of solid cancers is a complex process influenced by genetic mutations within cancer cells and the surrounding microenvironment [2].
The significance of ncRNA in cellular physiology is well-established. It has been reported that organims like worm and plant have approximately comparable protein-coding genes with humans [3]; however, the extant and magnitude of ncRNA is much larger than these lower organisms [4]. This highlights the significance of ncRNA in maintaining homeostasis and developing higher eukaryotes. Consistent with this, accumulating evidence has shed light on the importance of dysregulated ncRNA expression profiles in developing various solid cancers [5]. Also, restoring and modulating the expression of ncRNAs have shown promising results in treating various malignancies in pre-clinical studies [6]. In the era of targeted therapies, the complex interactions between miRNA, lncRNA, and circRNA and the concept of ceRNA networks have opened in new chapter in studying various aspects of oncogenesis and developing novel treatments.
Cell death is a physiological process that occurs during the development of multicellular organisms. Accidental cell death (ACD) and regulated cell death (RCD) are two types of cell death that differ in their underlying causes and mechanisms [7]. ACD is a type of cell death that occurs due to external factors that cause physical or chemical damage to the cell. On the other hand, RCD is a controlled form of cell death that occurs through internal signaling pathways and is tightly regulated. Apoptosis, autophagy, necroptosis, pyroptosis, parthanatosis, ferroptosis, and cuproptosis are among the regulated cell deaths, each with distinct molecular mechanisms and characteristics [7,8,9].
The present study aimed to review the biology of miRNA, lncRNA, circRNA, and ceRNA concepts along with ferroptosis, ferroptosis pathways, the significance of ferroptosis in cancer and anti-neoplastic treatments. Furthermore, the current review provides the authorship with the current evidence on the ferroptosis-regulating ceRNA networks in solid cancers and sheds light on the novel approach to regulating tumor ferroptosis, i.e., thorough ferroptosis-regulating ceRNA networks.
Non-coding RNAs and ceRNA networks
Approximately 80% of the human genome is transcribed into RNAs that do not code for proteins; these ncRNAs have critical regulatory functions in the initiation and progression of different types of cancer [10]. NcRNAs can be categorized into various classes based on their size and function. Among them, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) have been demonstrated to play significant roles in ferroptosis, thereby impacting tumor growth [11].
MiRNAs are a class of small non-coding RNA molecules that regulate gene expression by binding to complementary sites on targeted mRNA. The biogenesis of miRNAs in humans follows a two-step process involving nuclear and cytoplasmic cleavage events [12]. MiRNA genes are first transcribed by RNA polymerase II in the nucleus, resulting in primary miRNA transcripts (pri-miRNAs) that contain stem-loop structures [13]. The microprocessor complex, consisting of the RNase III enzyme Drosha and its cofactor DGCR8, identifies and cleaves these stem-loop structures to release hairpin-shaped precursor miRNAs (pre-miRNAs) [14]. Pre-miRNA is transported into the cytoplasm by interacting with RanGTP/Exportin-5 [15]. In the cytoplasm, the pre-miRNA undergoes cleavage mediated by the RNase III enzyme Dicer-1, together with TRBP/PACT proteins [16, 17]. This cleavage process results in a short double-stranded miRNA comprising both a guide and passenger strand. Once these two miRNA strands are separated, the guide strand is loaded into the RNA-induced silencing complex (RISC) to form the miRISC, which includes Dicer1 and Argonaute proteins. The miRNA guides RISC to identify complementary sequences on target mRNAs, resulting in the repression of gene expression [17, 18]. In light of their significant regulatory roles, it is apparent that modifications in miRNA expression have been associated with human diseases, including various cancers [17].
LncRNAs are transcripts that lack the protein-coding ability and possess a minimum length of 200 nucleotides [19]. They play essential roles in a wide range of physiological and pathological processes. Aberrant lncRNA expression levels have been observed in various cancers [10]. lncRNAs can undergo a variety of processing mechanisms, such as cleavage by ribonuclease P (RNase P) to create mature 3’ ends, capping by small nucleolar RNA (snoRNA) and proteins (snoRNP) complexes at their ends, or the formation of circular structures that protect them from degradation [19]. In contrast to mRNAs, many lncRNAs transcribed by RNA polymerase II remain in the nucleus due to inefficient processing, while others are spliced and exported to the cytoplasm [20]. The cytoplasmic export of lncRNAs (and mRNAs) that contain one or a few exons is mediated by nuclear RNA export factor 1 (NXF1) [21]. In the cytoplasm, lncRNAs can undergo specific sorting processes that assign different lncRNAs to specific organelles (e.g., mitochondria, exosomes) or be distributed in the cytoplasm and interact with diverse RNA-binding proteins [20]. Based on their genomic localization, lncRNAs are divided into sense, anti-sense, bidirectional, intronic, and intergenic lncRNAs [22].
CircRNAs are single-stranded RNA transcripts derived from a noncanonical type of alternative splicing known as back-splicing. Unlike linear RNA, circRNA creates a covalently closed loop structure lacking 5′ and 3′ ends [23]. This closed-loop structure renders circRNAs less susceptible to exonuclease degradation [24]. CircRNAs can be categorized into four distinct groups: exonic circRNAs (EcRNAs), exon-intron circRNAs (EIciRNAs), circular intronic RNAs (ciRNAs), and tRNA intronic circular RNAs (TricRNAs). Back-splicing in most circRNAs involves the spliceosome machinery joining the downstream 5′ splice-donor site to the upstream 3′ splice-acceptor site, creating a closed-loop structure with a specific junction site. The process is regulated by both cis-acting elements and trans-acting splicing factors [25]. Cis-acting regulatory elements that contain reverse complementary sequences (e.g., Alu repeats) flanking an intron through complementary pairing drive exon cyclization to form EIciRNAs and EcRNAs [26]. Trans-acting factors, such as RNA-binding proteins (e.g., QKI, MBL), can identify and attach to specific sites on the pre-mRNA flanking introns, forming EIciRNAs and EcRNAs [25, 26]. Another mechanism for circRNA biogenesis is lariat-driven circularization, which occurs during exon-skipping events or intron removal from pre-mRNAs. In exon-skipping events, the skipped exonic and intronic sequences are excised from pre-mRNA and form a loop-like structure called a lariat. The lariat further undergoes internal splicing to eliminate intronic sequences, producing an EcRNA [27]. During intron removal from pre-mRNA, the processing of the intronic lariat relies on a motif that comprises a seven nucleotide GU-rich element near the 5′ splice site and an 11 nucleotide C-rich element close to the branchpoint site, and these sites are resistant to degradation by de-branching enzymes, resulting in the formation of circRNA [28]. Most circRNAs originate from pre-mRNA, whereas a minor part of intron-derived circRNAs is from pre-tRNA. In the process of pre-tRNA maturation, the cleavage of an intron-containing pre-tRNA can occur through the tRNA splicing endonuclease (TSEN) complex at the bulge-helix-bulge (BHB) motif. Then, the enzyme RtcB ligase joins the resultant intron termini, forming a stable circRNA known as TricRNAs [29]. The aberrant regulation of circRNAs has been implicated in developing various diseases like cancers [30].
In 2011, Salmena et al. described a hidden language between RNAs mediated by miRNA response elements [31]; this hypothesis was driven by their findings showing that pseudogenes could compete with ancestral protein-coding genes to bind with miRNAs [32]. As ncRNAs, circRNAs and lncRNAs possess miRNA response elements that lead to establishing a dynamic competition with mRNAs. If the expression of circRNAs and lncRNAs are upregulated, they bind to miRNAs, which leads to the liberation of mRNA expression. However, If the expression levels of circRNAs and lncRNAs are downregulated, mRNA binds to the miRNAs, which results in mRNA degradation [33]. This hypothesis provides logical justification for the lncRNA/miR/mRNA and lncRNA/miR/mRNA axes [34]. This ceRNA hypothesis also provides novel insights into another side of mRNA, i.e., its non-coding function in regulating other RNAs [31]. It is worth mentioning that although miRNAs are the central point of this competition, RNA-biding proteins can also regulate miRNAs, adding to the complexity of RNA regulation [35].
Regulated cell death and ferroptosis
As a regulated cell death, pyroptosis is initiated by activating multiprotein complexes known as inflammasomes and subsequent activation of inflammatory caspases, such as caspase 1/4/5 in humans, leading to the cleavage of the N-terminal of gasdermin D (GSDMD) [36, 37]. Necroptosis is characterized by the activation of receptor-interacting protein kinase 1 (RIPK1; also known as RIP1), receptor-interacting protein kinase 3 (RIPK3; also known as RIP3), and their substrate is mixed-lineage kinase domain-like protein (MLKL), which facilitates its oligomerization and activation [38]. Parthanatos is a caspase-independent cell death initiated by over-activated poly (ADP-ribose) polymerase1 (PARP1) and the production of poly (ADP-ribose) (PAR) polymers. PAR polymers promote the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus, which causes DNA degradation and ultimately results in cell death [7, 39]. Autophagy-dependent cell death is a type of RCD mediated by the molecular machinery of autophagy [40]. Cuproptosis occurs through the binding of copper to lipoylated components in the tricarboxylic acid (TCA) cycle, leading to the aggregation of lipoylated proteins and subsequent reduction in Fe–S cluster proteins, which induces proteotoxic stress and, eventually, cell death [8, 41].
Ferroptosis is a newly identified RCD characterized by the oxidation of polyunsaturated fatty acids (PUFAs) and the accumulation of lipid peroxides. The term ferroptosis was first coined in 2012 by Dr. Brent R. Stockwell’s lab [42]. According to their original study, comparing this iron-dependent cell death to other forms of RCD reveals distinct morphological, biochemical, and genetic differences [43]. The morphology of ferroptotic cells is characterized by smaller mitochondria, increased mitochondrial membrane density, decreased or absent cristae, outer mitochondrial membrane rupture, and the nuclei are normal in size without chromatin aggregation [44]. Accumulating studies have demonstrated that ferroptosis plays a crucial role in the development and progression of various diseases, including neurological disorders [45], cardiovascular diseases [46], ischemia-reperfusion injury [47], and cancer [48]. Regulation of ferroptosis primarily relies on the balance between glutathione (GSH) and ROS [49]. Ferroptosis inducers can be classified into two main groups. The first group, i.e., class I ferroptosis inducers, includes erastin, sorafenib, sulfasalazine, and glutamate, which function as system xc- blockers, causing a decrease in GSH levels. The second group, i.e., class II ferroptosis inducers, includes RAS synthetic lethal 3 (RSL3), ferroptosis-inducing agent 56 (FIN56), ferroptosis-inducing peroxide compound (FINO2), and ML162 (also known as DPI7), which inhibit the activity of glutathione peroxidase 4 (GPX4) [49,50,51]. Table 1 summarizes the morphological features, biochemical features, regulators, and inhibitors of well-known regulated cell deaths.
Ferroptosis pathways
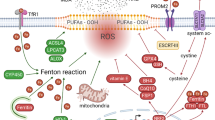
Ferroptosis regulation can be classified into two primary pathways. The first pathway involves metabolic processes associated with lipid, iron, and amino acid metabolism. The second pathway involves signaling pathways that regulate ferroptosis, including the tumor suppressor TP53, nuclear factor erythroid 2-related factor 2 (NRF2), and ferroptosis suppressor protein 1 (FSP1) pathways [52] (Fig. 1).

The main metabolisms of ferroptosis can be categorized into three groups: iron metabolism, the GSH/GPX4 pathway, and lipid peroxidation. The System xc- antiporter mediates the exchange of intracellular glutamate and extracellular cystine. Cystine is converted to cysteine, a critical building block for GSH synthesis. GSH is an important substrate for the functioning of GPX4, which converts lipid hydroperoxides into their alcohol forms. GSH and GPX4 function as scavengers of reactive oxygen species to avert oxidative stress and lipid peroxidation. Another inhibitory mechanism of ferroptosis is regulated by FSP1–ubiquinol. Ferritinophagy, iron input, and output can employ the Fenton reaction to trigger lipid peroxidation and induce ferroptosis. The production and membrane deposition of PUFA-PLs requires the involvement of the enzymes ACSL4 and LPCAT3. SLC7A11, Solute carrier family 7 member 11; SLC3A2, Solute carrier family 3 member 2 ; TFR1, Transferrin receptor 1; RSL3, RAS synthetic lethal 3; FIN56, Ferroptosis-inducing agent 56; FINO2, Ferroptosis-inducing peroxide compound; GPX4, Glutathione Peroxidase 4; GSH, Glutathione; GSSH, glutathione counterpart; FSP1, Ferroptosis inhibitor protein 1; NADPH, Nicotinamide adenine dinucleotide phosphate; STEAP3, Six-transmembrane epithelial antigen of the prostate3; DMT1, Divalent metal transporter; NCOA4, Nuclear receptor co-activator 4; PFUA, polyunsaturated fatty acid; ACSL4, Acyl-CoA synthetase long-chain family member 4; LPCAT3, Lysophosphatidylcholine acyltransferase 3; LOX, Lipoxygenases; PUFA-PL, Polyunsaturated fatty acid phospholipids; PUFA-PL-OOH, Polyunsaturated fatty acid phospholipid hydroperoxides.
Lipid metabolism
Increased lipid peroxidation is a distinctive feature, and lipid peroxide metabolism is critical in ferroptosis [53]. Lipidomic analysis revealed that PUFAs, such as arachidonic acid (AA) and adrenoyl (ADA) that are phosphatidylethanolamines (PEs), are prime targets for peroxidation [54]. Ferroptosis is likely activated by the peroxidation of membrane phospholipids to produce phospholipid hydroperoxides (PLOOH) and the breakdown of PLOOH to generate 4-hydroxynonenal or malondialdehyde. Lipid peroxidation products cause membrane instability and permeabilization, ultimately leading to cell death. Lipid peroxidation occurs in both non-enzymatic and enzymatic ways. In non-enzymatic lipid peroxidation, PUFAs are ligated to coenzyme A (CoA) by operating acyl-CoA synthase long-chain family member 4 (ACSL4), which produces acyl-CoA. Following this, acyl-CoA could be re-esterified in phospholipids through lysophosphatidylcholine acyltransferases (LPCATs) to generate phospholipids [55]. Acyl-CoA synthetase ACSL4 and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are critical drivers of ferroptosis [56, 57]. In enzymatic lipid peroxidation, lipoxygenase (LOX) catalyzes the oxidation of polyunsaturated fats into their corresponding hydroperoxides. Lipoxygenases are non-heme iron-containing dioxygenase enzymes [58]. Previous research has found that overexpression of LOX-5, LOX-12, and LOX-15 causes cells to become more vulnerable to ferroptosis. Additionally, inhibitors of LOX activity can protect cells from ferroptotic cell death [53].
Iron metabolism
Iron is an indispensable trace element in the human body involved in various biological processes, including oxygen transport, ATP production, DNA biosynthesis, and cell proliferation [59]. However, excess iron can generate ROS and lead to oxidative damage. Therefore, iron homeostasis is tightly regulated to maintain a balance between iron absorption, utilization, and storage [60]. Iron regulatory protein (IRP) is a crucial regulator of endogenous iron homeostasis. IRP binds to specific RNA sequences called iron-responsive elements (IREs) located in the 5’- or 3’-untranslated regions of mRNAs encoding proteins involved in iron metabolism [61]. Cancer cells exhibit higher metabolic requirements than normal cells, and their survival and proliferation largely depend on iron [59]. Fe3+ in the peripheral circulation binds to transferrin and forms a complex; then, the complex is imported into the endosome through endocytosis mediated by transferrin receptor 1 (TFR1). Acidic conditions in endosomes cause the release of Fe3+ from transferrin, which is then converted to Fe2+ by the ferrireductase activity of the six-transmembrane epithelial antigen of the prostate 3 (STEAP3) [62]. Following this, Fe2+ is released from endosomes into a labile iron pool (LIP) under the control of divalent metal transporter 1 (DMT1, also known as SLC11A2 or Nramp2) [62, 63]. Increased intracellular LIP can produce free radicals (hydroxyl radicals) through the Fenton reaction and participate in the peroxidation of phospholipids to form PLOOH [64]. Ferritin is a protein complex composed of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), which is capable of storing excess iron. The nuclear receptor co-activator 4 (NCOA4) directly binds to FTH1 to form the ferritin complex targeting lysosomes for “ferritinophagy”. Ferritinophagy causes increased intracellular iron levels and triggers ROS accumulation through the Fenton reaction, resulting in ferroptosis [65, 66]. Ferroportin (FPN1, also known as SLC40A1) is the only well-known iron exporter and plays a pivotal role in the export of Fe2+ from cells to the blood [67].
Amino acid metabolism
Ferroptosis regulation is closely related to amino acid metabolism. Two key molecules in amino acid metabolism are GPX4 and system xc- [68]. GPX4 belongs to the glutathione peroxidase (GPX) family and contains selenocysteine in its catalytic center, which converts toxic lipid hydroperoxides (L-OOH) to a non-toxic form of lipid alcohol (L-OH) [69]. Glutathione (GSH) is a necessary substrate for the physiological functioning of GPX4. GSH is a critical antioxidant synthesized from glutamate, cysteine, and glycine; among these amino acids, cysteine is regarded as the rate-limiting precursor. System xc- is a sodium-independent antiporter composed of two subunits, i.e., the transporter subunit, solute carrier family 7 member 11 (SLC7A11), and the regulatory subunit, solute carrier family 3 member 2 (SLC3A2). This transporter system mediates the exchange of intracellular glutamate and extracellular cystine in a ratio of 1:1 [49, 70].
p53
p53 is a human tumor suppressor gene activated in response to various forms of cellular stress, such as DNA damage, oxidative stress, and dysregulated metabolism [71]. P53 has a dual function in ferroptosis. p53 acts as a transcriptional repressor for SLC7A11, leading to reduced cellular cystine import, increased ROS accumulation, and ferroptosis induction [72]. Additionally, p53 can induce ferroptosis by increasing the expression of glutaminase 2 (GLS2) and spermidine/spermine N1 acetyltransferase 1 (SAT1) [73, 74]. On the other hand, p53 can suppress ferroptosis either by inhibiting dipeptidyl peptidase 4 (DPP4) or by inducing the activity of the cell cycle protein-dependent kinase inhibitor 1A (CDKN1A, also known as p21) [75, 76].
Nuclear factor erythroid 2-related factor 2
NRF2 is a transcription factor crucial for regulating cellular antioxidant activity [77]. Under physiological circumstances, the expression of NRF2 is low, and KEAP1 closely controls its activity. However, in response to oxidative stress, NRF2 dissociates from KEAP1 and moves to the nucleus to activate the transcriptional gene, which functions as an antioxidant against oxidative stress and inhibits ferroptosis [78, 79].
Ferroptosis suppressor protein 1
Ferroptosis suppressor protein (FSP1) is in the cytoplasmic membrane, and its anti-ferroptotic activity relies on N-myristoylation [80]. A reduced form of coenzyme Q (CoQ), known as ubiquinol, is maintained by FSP1 at the plasma membrane. Ubiquinol acts as an antioxidant by capturing lipid peroxyl radicals and preventing the diffusion of lipid peroxides, thereby inhibiting ferroptotic cell death [81].
Ferroptosis in cancer
Growing studies have investigated the significance of ferroptosis in solid cancer models and affected patients. It has been reported that the expression levels of NRF2 and SLC7A11 are upregulated in esophageal squamous cell carcinoma tissues; NRF2 is associated with metastasis, TNM stage, and lymph node metastasis in patients with esophageal squamous cell carcinoma, and NRF2 overexpression leads to radioresistance development [82]. In gastric cancer patients, NRF2 upregulation is associated with inferior prognosis of patients [83]. Increased expression of NRF2 has been positively associated with lymph node metastasis and poor differentiation; also, high expression of NRF2 is associated with poor overall survival of patients with non-small cell lung cancer (NSCLC) [84]. Besides, increased expression of NRF2 is associated with poor recurrence and disease-free survival of breast cancer patients, and NRF2 knockdown decreases the proliferation of breast cancer cells [85].
It has been reported that SLC7A11 is upregulated in renal cell carcinoma, and its increased expression is associated with the inferior survival of affected patients; SLC7A11 silencing is associated with decreased migration, invasion, and proliferation of malignant cells [86]. In patients with epithelial ovarian cancers, high co-expression of SLC7A11 and GPX4 has been substantially associated with poor overall survival and progression-free survival of affected patients [87]. Increased expression of SLC7A11 has been associated with inferior 5-year survival of patients with NSCLC, and SLC7A11 silencing decreases the tumor volume in nude animal models [88]. In patients with oral cavity squamous cell carcinoma, SLC7A11 upregulation is considerably associated with perineural and lymphovascular invasion, recurrence-free survival, disease-specific survival, and overall survival [89]. Also, Sugano et al. have reported that SLC7A11 is associated with lymphatic vessel invasion, and its expression is associated with the relapse-free survival of patients with colorectal cancer [90]. Also, high expression of SLC7A11 is associated with worse overall survival of laryngeal squamous cell carcinoma patients [91].
GPX4 upregulation is significantly associated with poor disease-specific survival and overall survival of gastric cancer patients and its silencing decreases the cell proliferation of malignant cells [92]. Increased expression of GPX4 is associated with large invasion depth, advanced tumor stage, and high grade of tumors in patients with oral squamous cell carcinoma [93]. In thyroid cancer, low expression of GPX4 is associated with improved overall survival of affected patients, and GPX4 knockdown decreases the clonogenicity of thyroid cancer cells [94]. It has been reported that increased expression of GPX4 is associated with poor overall survival of NSCLC patients as well [95]. Also, GPX4 inhibition decreases the spheroid formation, cell viability, and migration of thyroid cancer cells [96]. Collectively, tumoral ferroptosis activation can lead to the suppression of oncogenesis in solid cancers. The following discusses the significance of ferroptosis in cancer chemotherapy, radiotherapy, and immunotherapy.
Ferroptosis and chemotherapy
Chemoresistance is a considerable obstacle in cancer treatment. In this regard, ferroptosis dysregulation is involved in developing chemoresistance in various cancers. Cisplatin-resistant ovarian cancer cells stimulate autophagy, increase the autophagic degradation of FTH1, and suppress ferroptosis. Of interest, erastin-induced ferroptosis decreases the cell viability of cisplatin-resistant ovarian cancer cells [97]. In hepatocellular carcinoma, the knockout of GSTZ1, an inhibitor enzyme of NRF2, suppresses ferroptosis and increases sorafenib resistance [98]. In addition, etoposide-mediated metabolic reprogramming can increase lactate production in NSCLC cells, leading to GPX4 ubiquitination and ferroptosis resistance [99]. Ferroptosis stimulation via GPX4 knockdown or FIN56 administration can eliminate chemoresistant NSCLC cells [100]. In line with this, it has been shown that KLF11 suppresses GPX4 in lung adenocarcinoma, stimulates ferroptosis, and enhances their chemosensitivity to cisplatin [101]. In gastric cancer, the beta-catenin/TCF4 transcription complex promotes GPX4 expression and inhibits ferroptosis. Of interest, TCF4 overexpressed gastric cells are highly resistant to cisplatin [102].
In addition, chemotherapeutic agents can induce ferroptosis, and the dysregulation of this process can lead to chemoresistance [53]. Among these agents, temozolomide, a DNA alkylating agent, has shown effectiveness in treating glioblastoma multiforme [103]. Temozolomide induces ferroptosis through various pathways; Chen et al. have discovered that the androgen receptor induces resistance to temozolomide treatment in glioblastoma. They have reported that the curcumin analog induces the ubiquitination of the androgen receptor, which suppresses GPX4 and causes ferroptosis, thereby reversing temozolomide resistance in glioblastoma [104]. Furthermore, temozolomide exerts ferroptosis-inducing effects by activating NRF2/ATF4 at the mRNA and protein levels, thus increasing the expression of SLC7A11 and enhancing cystathionine gamma-lyase activity [105]. Moreover, temozolomide stimulates ferroptosis in glioblastoma cells by upregulating DMT1, a crucial protein in regulating iron homeostasis [106]. Therefore, the additive effect of ferroptosis activation on the anti-tumoral effect of chemotherapeutic agents on cell viability can be of interest and open a new era in cancer medicine. Recent findings have evaluated the efficacy of biomimetic/biocompatible formulations and pH-responsive liposomal nanoreactors for inducing ferroptosis in cancer models [107, 108].
Ferroptosis and radiotherapy
Radiotherapy employs high-energy ionizing radiation to cause DNA double-strand breakage, which is a mainstream therapeutic approach for treating various cancers. Apart from the direct DNA damage, ionizing radiation can also trigger indirect cellular effects by radiolysis of cellular water and activating oxidases, producing ROS such as hydroxyl radicals and hydrogen peroxide, which can damage nucleic acids, proteins, and lipids. Recent findings have demonstrated that ionizing radiation induces ferroptosis in cancer cells [109]. Shibata et al. have reported that treatment with erastin, as a ferroptosis inducer, decreases the levels of glutathione and GPX4 and increases radiotherapy-mediated anti-tumoral effect in vivo [110]. It has been reported that the activating effect of radiotherapy on ferroptosis is dependent on p53; radiotherapy activates p53 and suppresses radiotherapy-mediated SLC7A11 expression, leading to glutathione synthesis inhibition and ferroptosis stimulation. Of interest, ferroptosis inducers that suppress SLC7A11 enhance the radiosensitivity of p53 mutated malignant cells [111]. Besides, ferroptosis activation via inhibiting system xc- or GPX4 inhibition can synergize the radiotherapy-mediated ferroptosis stimulation in cancers. In murine xenograft and patient-derived models, ferroptosis inducers increase the anti-tumoral effect of radiotherapy [112]. As mentioned above, KEAP1 closely controls the expression of NRF1, and KEAP1 mutation leads to continuous expression of NRF2. It has been reported that KEAP1 deletion increases the expression of SLC7A11, leading to radioresistance. However, the combined deletion of KEAP1 and SLC7A11 sensitized the malignant cells to radiotherapy [109]. Koppula et al. have reported that CoQ-FSP1 is another downstream effector from the KEAP1-NRF2 axis, and the FSP1 inhibition has been associated with increased radiotherapy response of KEAP1 deficient lung cancer cells via inducing ferroptosis [113]. In addition, estrogen receptor 1 can activate SLC7A11 expression, and estrogen receptor 1 knockdown increases ferroptosis in malignant cells; also, NEDD4L ubiquitinates SLC7A11 in estrogen receptor-positive breast cancer cells during radiation [114]. Consistent with this, Liu et al. have also reported that estrogen receptor 1 knockdown enhances radiation-induced ferroptosis in breast cancer cells [115].
Ataxia telangiectasia mutated (ATM) is an indispensable component of the DNA repair system activated following cytotoxic chemotherapy or radiotherapy. ATM mediates the downregulation of SLC7A11 after radiotherapy, resulting in ferroptosis; this anti-neoplastic effect of radiotherapy is enhanced by the combination of immune checkpoint inhibitors, e.g., anti-PD-L1 or anti-CTLA4 antibodies, in tumor models [116]. In line with this, it has been shown that radiotherapy can activate ATM, leading to the inhibition of SLC7A11, reduced cysteine uptake, ferroptosis activation, and tumor growth retardation [117]. Radiotherapy induces ACSL4 expression and triggers PUFA-PL peroxidation, subsequently causing ferroptosis. Of interest, suppression of SLC7A11 and GPX4 has been associated with sensitizing radioresistant malignant cells [109]. Irradiated cancer cells also release microparticles that can enhance radiotherapy by propagating signals that trigger ferroptosis or increase the expression of proteins involved in oxidative stress [118].
Ferroptosis and immunotherapy
Cancer immunotherapy often encounters low response rates and treatment resistance in various tumors, which leads to unsatisfactory therapeutic outcomes [119]. Therefore, there is a need to increase the efficacy of cancer immunotherapy. In this regard, recent studies have shown that ferroptosis inhibits tumor growth and enhances the efficacy of immunotherapy through multiple signaling pathways, suggesting that ferroptosis induction may contribute to diminishing cancer immunotherapy resistance [120]. Yang et al. showed that the combination of GPX4 inhibition and anti-PD1 treatment enhanced tumor ferroptosis and augmented antitumor immune responses in triple-negative breast cancer [121]. Combined treatment of cyst(e)inase with PD-L1 blockade synergistically induces ferroptosis, suggesting that it could be a promising approach to enhance antitumor immunotherapy [122]. It has been reported that the tumor microenvironment of ACSL4−/− animal models displays decreased infiltration of CD8+, IFNγ+CD8+, TNFα+CD8+, and CD4+ T cells compared to the tumor microenvironment of ACSL4+/+ animal models. Of interest, the combined treatment of PD-L1 blockade with the supplementation of arachidonic acid, as a ferroptosis-promoting agent, has enhanced the efficacy of PD-L1 blockade and increased the infiltration of TNFα+, IFNγ+, and granzyme B+CD8+ T-cells in animal models. Besides, arachidonic acid has no significant effect on the function or survival of T-cells in vitro [123]. Elevated GPX4 levels in Treg cells prevent lipid peroxidation and ferroptosis, while inhibiting GPX4 increases antitumor immunotherapy [124]. In addition, it has been indicated that CYP1B1 causes the metabolization of arachidonic acid to 20-HETE, leading to the PKC pathway-mediated FBXO10 upregulation. FBXO10 has been implicated in ACSL4 degradation, paving the way for ferroptosis resistance. The combined treatment of CYP1B1 inhibition increases the sensitivity of tumor cells to anti-PD-1 blockade in C57BL/6 J mice [125]. Aside from these pathways, ferroptotic stress increases the expression of PD-L1 and sensitizes head and neck cancer animal models to anti-PD-L1 treatment [126]. On the other hand, the activation of CD8+ T cells by anti-PD-L1 antibodies regulates ferroptosis in cancer cells. The IFN-γ released by CD8+ T cells in the tumor microenvironment downregulates the expression of two subunits of system xc-, consequently promoting the accumulation of lipid peroxidation and facilitating ferroptosis [122]. Cisplatin-induced ferroptosis is partly responsible for the synergistic anti-tumoral effect of the combination of cisplatin with anti-PD-1 antibody in animal models of lung cancer. Indeed, cisplatin-induced ferroptosis increases the infiltration of T-cells, N1-neutrophil polarization, and type 1 T helper differentiation [127].
However, the effect of ferroptosis stimulation should be distinctly studied on the malignant cells and tumor-infiltrating effector cells. Liu et al. have found that ferroptosis inhibition sensitized glioblastoma to anti-PD-1/anti-PD-L1 immunotherapy [128]. Within the tumor microenvironment, CD36-mediated fatty acid uptake by tumor-infiltrating CD8+ T cells leads to lipid peroxidation and ferroptosis. This results in reduced production of cytotoxic cytokines and impaired antitumor response. Therefore, blocking CD36 expression or using ferroptosis inhibitors combined with anti-PD-1 antibodies can enhance the antitumor effects of CD8+ T cells [129]. Conche et al. have reported that GPX4 deficient hepatocellular cells promote the immunosuppressive tumor microenvironment, which is characterized by the infiltration of CXCL10-related cytotoxic CD8+ T cells, tumoral PD-L1 upregulation, and HMGB1-mediated myeloid-derived suppressor cell (MDSC) infiltration; PD-1 blockade substantially increases the survival of GPX4 deficient animal models [130]. Kim et al. have shown that ferroptosis can limit human and mouse T-cell activity, and ferroptosis inhibition synergizes with immune checkpoint inhibitors and decreases cancer progression [131]. Therefore, the anti-tumoral effect of ferroptosis should be tailored to not influence the function or survival of effector immune cells and only enhance the ferroptosis of malignant cells. Overall, tumor ferroptosis modulation can be promising to increase the anti-tumoral effect of immunotherapy.
Ferroptosis-regulating ceRNA networks
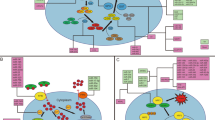
As discussed above, circRNA and lncRNA-associated ceRNA regulate a wide range of biological processes. Dysregulated ceRNA networks considerably dysregulate gene expression implicated in cancer development and progression. Given the significance of enhancing tumoral ferroptosis in improving the chemoradiotherapy and immunotherapy responses, identifying the dysregulated ferroptosis-related ceRNA networks in cancers is of substantial importance. In contrast to the pharmacological regulation of ferroptosis, gene therapy-mediated rectifying the dysregulated ferroptosis-related ceRNAs can regulate ferroptosis as well as other biological processes, like proliferation, migration, and apoptosis (Fig. 2). Because ceRNA networks regulate numerous genes both directly and indirectly. Therefore, the following discusses the available experimental evidence on the ferroptosis-related ceRNA networks in various cancers (Table 2).

Restoring ferroptosis-regulating ceRNA networks increases cancer immunotherapy and chemoradiotherapy responses, increases ferroptosis, stimulates apoptosis, and decreases proliferation, migration, invasion, and tumor growth.
CeRNA networks regulating NRF2
MT1DP downregulates NRF2 via stabilizing miR-365a-3p and sensitized NSCLC cells to erastin-induced ferroptosis. The in vivo models have indicated that the delivery of MT1DP and erastin via folate-modified liposome decreases tumor growth; the in vitro results have shown that this co-delivery increases ferroptosis and decreases the migration, invasion, and proliferation of NSCLC cells [132]. It has been reported that GMDS-AS1 and LINC01128 lncRNAs increase the chemosensitivity of lung adenocarcinoma via sponging miR-6077 and the miR-6077/KEAP1 axis. As mentioned above, KEAP1 closely regulates NRF2, and the GMDS-AS1 and LINC01128/miR-6077/KEAP1 can control NRF2 and increases ferroptosis [133]. As an upregulated circRNA in chemoresistant prolactinoma tissues, circOMA1 increases tumor growth in vivo and in vitro and inhibits ferroptosis via facilitating GXP4 and NRF2 upregulation [134].
CeRNA networks regulating ACSL4
As downregulated circRNA in cervical cancer tissues, circLMO1 overexpression decreases the proliferation and invasion and increases the ferroptosis of cervical cancer cells via the miR-4192/ACSL4 axis [135]. As downregulated circRNA in NSCLC tissues and cells, the decreased expression level of circSCN8A is associated with poor prognosis of affected patients. circSCN8A ectopic expression inhibits migration, proliferation, invasion, and tumor growth in animal models and increases ferroptosis via the circSCN8A/miR-1290/ACSL4 axis [136].
CeRNA networks regulating GPX4
As an upregulated lncRNA in cholangiocarcinoma, linc00976 increased expression is associated with lymph node metastasis and inferior overall survival of affected patients. Besides stimulating ferroptosis, linc00976 knockdown decreases the proliferation and migration of cholangiocarcinoma cells via the linc00976/miR-3202/GPX4 axis [137]. It has been reported that HCG18 knockdown decreases cell proliferation, increases apoptosis, and enhances sorafenib resistance via activating ferroptosis in hepatocellular carcinoma; these effects are mediated via the HCG18/miR-450b-5p/GPX4 axis [138]. circBLNK is an upregulated circRNA in osteosarcoma tissues and cell lines that its knockdown results in decreased proliferation, increased apoptosis, and increased intracellular free iron and lipid ROS via the circBLNK/miR-188-3p/GPX4 axis [139]. It has been shown that circDTL is upregulated in NSCLC tissues and cell lines, and circDTL knockdown increases the apoptosis, ferroptosis, chemosensitivity, and in vivo tumor growth of NSCLC via the circDTL/miR-1287-5p/GPX4 axis [140]. As an overexpressed circRNA in hepatocellular carcinoma tissues and cells, circIL4R knockdown decreases proliferation and increases ferroptosis via the circIL4R/miR-541-3p/GPX4 [141].
CeRNA networks regulating SLC7A11
Huang et al. have reported that FOXQ1 is an upregulated oncogenic transcription factor in breast cancer that binds to the promotor of circ_0000643 to increase its expression. FOXQ1 knockdown increases breast cancer ferroptosis and inhibits oncogenesis via the FOXQ1/circ_0000643/miR-153/SLC7A11 axis [142]. As an upregulated circRNA in lung adenocarcinoma, circP4HB ectopic expression increases tumor growth and suppresses ferroptosis via the circP4HB/miR-1184/SLC7A11 [143]. As an upregulated circRNA in chemoresistant ovarian cancer tissues compared to sensitive ones, circSnx12 knockdown enhances the chemosensitivity and increases apoptosis and ferroptosis of cisplatin chemoresistant ovarian cancer cells circSnx12/miR-194-5p/SLC7A11 axis [144]. CircSTIL is an increased circRNA in colorectal cancer, and its knockdown is associated with decreased proliferation and enhanced ferroptosis of colorectal cancer cells via the circSTIL/miR-431/SLC7A11 axis [145]. In thyroid cancer, circ_0067934 knockdown decreases proliferation and increases apoptosis and ferroptosis of malignant cells via the circ_0067934/miR-545-3p/SLC7A11 axis [146]. CircEPSTI1 is upregulated in cervical cancer cells, and its knockdown decreases tumor growth in vivo and in vitro and activates ferroptosis via the circEPSTI1/miR-375_miR409-3P_miR-515-5p/SLC7A11 pathway [147]. BBOX1-AS1 is an upregulated lncRNA in esophageal squamous cell cancer tissues, and its increased expression is associated with poor overall survival, lymph node metastasis, increased tumor size, and TNM stage in affected patients; BBOX1-AS1 knockdown results in decreased proliferation, invasion, migration, and increased apoptosis and ferroptosis via the BBOX1-AS1/miR-513a-3p/SLC7A11 axis [148]. In nutlin-3a-resistant SJSA1 cells, lncRNA-SNHG14 knockdown enhances the sensitivity of tumoral cells and activates the ferroptosis via the SNHG14/miR-206/SLC7A11 [149]. SLC16A1-AS1 is an upregulated lncRNA in renal cancer tissues, and its increased expression is associated with poor overall survival of patients; SLC16A1-AS1 knockdown suppresses the cell viability, migration, and invasion and enhances ferroptosis via the SLC16A1-AS1/miR-143-3p/SLC7A11 axis [150].
Future perspective and concluding remark
Ferroptosis is a newly identified regulated cell death; this metabolically-driven iron-related cell death depends on polyunsaturated fatty acyl peroxidation. Growing studies have shown that activating tumoral ferroptosis inhibits tumor development in pre-clinical models. Besides, the genes regulating ferroptosis have significant prognostic values for affected patients. Further studies have highlighted that tumoral cell ferroptosis activation displays promising results in increasing the efficacy of cancer immunotherapy and chemoradiotherapy. Therefore, activating ferroptosis in malignant cells can be promising in treating solid cancers.
The identification of the hidden language between major ncRNAs has provided valuable information about the complex interaction of RNAs in oncogenesis. The circRNA and lncRNA-mediated ceRNA networks can regulate various signaling pathways. In this regard, studies have shown that ceRNA networks are highly dysregulated in cancer development and progression, leading to oncogenes upregulation and tumor-suppressive genes downregulation. Recent studies have highlighted the significance of ceRNAs in regulating cancer ferroptosis and identified ferroptosis-regulating ceRNA networks in cancers. Since lncRNA, circRNA, and miR dramatically regulate the expression of a wide range of genes and pathways, restoring dysregulated ferroptosis-regulating ceRNA networks can provide ample opportunities to not only regulate ferroptosis and benefit from the anti-tumoral effects of ferroptosis regulation in cancer treatment, it can regulate other dysregulated pathways, like the pathways implicated in cancer migration, invasion, stemness, etc. In addition to the beneficial role of tumoral ferroptosis activation on cancer immunotherapy and chemoradiotherapy, the broad range of targets of ferroptosis-regulating ceRNA networks can be excellent targets for cancer treatment. The present study provided the available evidence on the ferroptosis-regulating ceRNA networks in different solid cancers and displays their potential for cancer treatment.
References
Pal M, Muinao T, Boruah HPD, Mahindroo N. Current advances in prognostic and diagnostic biomarkers for solid cancers: Detection techniques and future challenges. Biomed Pharmacother. 2022;146:112488.
Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78.
Baltimore D. Our genome unveiled. Nature. 2001;409:814–6.
Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009;5:e1000459.
Mohammadzadeh A, Dastmalchi N, Hussen BM, Shadbad MA, Safaralizadeh R. An updated review on the therapeutic, diagnostic, and prognostic value of long non-coding RNAs in gastric cancer. Curr Med Chem. 2022;29:3471–82.
Dastmalchi N, Safaralizadeh R, Banan Khojasteh SM, Sam MR, Latifi-Navid S, Hussen BM, et al. An updated review of the cross-talk between microRNAs and epigenetic factors in cancers. Curr Med Chem. 2021;28:8722–32.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378.
Xie LH, Fefelova N, Pamarthi SH, Gwathmey JK. Molecular mechanisms of ferroptosis and relevance to cardiovascular disease. Cells. 2022;11:2726.
Toden S, Zumwalt TJ, Goel A. Non-coding RNAs and potential therapeutic targeting in cancer. Biochim Biophys Acta Rev Cancer. 2021;1875:188491.
Balihodzic A, Prinz F, Dengler MA, Calin GA, Jost PJ, Pichler M. Non-coding RNAs and ferroptosis: potential implications for cancer therapy. Cell Death Differ. 2022;29:1094–106.
Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11:537–61.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–60.
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–5.
Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–91.
Park JE, Heo I, Tian Y, Simanshu DK, Chang H, Jee D, et al. Dicer recognizes the 5’ end of RNA for efficient and accurate processing. Nature. 2011;475:201–5.
Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323–32.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–24.
Wu H, Yang L, Chen LL. The diversity of long noncoding RNAs and their generation. Trends Genet. 2017;33:540–52.
Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22:96–118.
Zuckerman B, Ron M, Mikl M, Segal E, Ulitsky I. Gene architecture and sequence composition underpin selective dependency of nuclear export of long RNAs on NXF1 and the TREX complex. Mol Cell. 2020;79:251–67.e6.
Liu Y, Ding W, Yu W, Zhang Y, Ao X, Wang J. Long non-coding RNAs: biogenesis, functions, and clinical significance in gastric cancer. Mol Ther Oncol. 2021;23:458–76.
Bao C, Lyu D, Huang S. Circular RNA expands its territory. Mol Cell Oncol. 2016;3:e1084443.
Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34:e63.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–91.
Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12:381–8.
Barrett SP, Wang PL, Salzman J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. eLife. 2015;4:e07540.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806.
Noto JJ, Schmidt CA, Matera AG. Engineering and expressing circular RNAs via tRNA splicing. RNA Biol. 2017;14:978–84.
Sharma AR, Bhattacharya M, Bhakta S, Saha A, Lee SS, Chakraborty C. Recent research progress on circular RNAs: Biogenesis, properties, functions, and therapeutic potential. Mol Ther Nucleic Acids. 2021;25:355–71.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8.
Tang Z, Li X, Zheng Y, Liu J, Liu C, Li X. The role of competing endogenous RNA network in the development of hepatocellular carcinoma: potential therapeutic targets. Front Cell Dev Biol. 2024;12:1341999.
Yang C, Wu D, Gao L, Liu X, Jin Y, Wang D, et al. Competing endogenous RNA networks in human cancer: hypothesis, validation, and perspectives. Oncotarget. 2016;7:13479–90.
Ciafrè SA, Galardi S. microRNAs and RNA-binding proteins: a complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013;10:935–42.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–98.
Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75.
Dai W, Cheng J, Leng X, Hu X, Ao Y. The potential role of necroptosis in clinical diseases (Review). Int J Mol Med. 2021;47:89.
Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218:193–202.
Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–16.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Gong C, Ji Q, Wu M, Tu Z, Lei K, Luo M, et al. Ferroptosis in tumor immunity and therapy. J Cell Mol Med. 2022;26:5565–79.
Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit Care Med. 2019;47:410–8.
Guo Y, Zhang W, Zhou X, Zhao S, Wang J, Guo Y, et al. Roles of ferroptosis in cardiovascular diseases. Front Cardiovasc Med. 2022;9:911564.
Chen Y, Fan H, Wang S, Tang G, Zhai C, Shen L. Ferroptosis: a novel therapeutic target for ischemia-reperfusion injury. Front Cell Dev Biol. 2021;9:688605.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Zhang X, Wang L, Li H, Zhang L, Zheng X, Cheng W. Crosstalk between noncoding RNAs and ferroptosis: new dawn for overcoming cancer progression. Cell Death Dis. 2020;11:580.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–15.
Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The role of erastin in ferroptosis and its prospects in cancer therapy. OncoTargets Ther. 2020;13:5429–41.
Yu S, Jia J, Zheng J, Zhou Y, Jia D, Wang J. Recent progress of ferroptosis in lung diseases. Front Cell Dev Biol. 2021;9:789517.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Dang Q, Sun Z, Wang Y, Wang L, Liu Z, Han X. Ferroptosis: a double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022;13:925.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10:1617.
Song H, Wang P, Li C, Han S, Lopez-Baltazar J, Zhang X, et al. Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci Rep. 2016;6:35245.
Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–55.
Yang J, Zhang G, Dong D, Shang P. Effects of Iron overload and oxidative damage on the musculoskeletal system in the space environment: data from spaceflights and ground-based simulation models. Int J Mol Sci. 2018;19:2608.
Wallander ML, Leibold EA, Eisenstein RS. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta. 2006;1763:668–89.
Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866:118535.
Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA. 1998;95:1148–53.
Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82-83:969–74.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond, Engl). 2022;42:88–116.
Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142:24–38.
Shah R, Shchepinov MS, Pratt DA. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci. 2018;4:387–96.
Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303.
Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–55.
Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29:895–910.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–60.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 2016;113:E6806–e12.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22:569–75.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–6.
Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, et al. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Transl Med. 2021;19:367.
Alakuş H, Kaya M, Özer H, Eğilmez HR, Karadayı K. Nuclear factor erythroid-2-related factor 2 (Nrf2) is a potential prognostic factor in patients with gastric adenocarcinoma. Arab J Gastroenterol. 2023;24:5–10.
Tong YH, Zhang B, Yan YY, Fan Y, Yu JW, Kong SS, et al. Dual-negative expression of Nrf2 and NQO1 predicts superior outcomes in patients with non-small cell lung cancer. Oncotarget. 2017;8:45750–8.
Onodera Y, Motohashi H, Takagi K, Miki Y, Shibahara Y, Watanabe M, et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr Relat Cancer. 2014;21:241–52.
Xu F, Guan Y, Xue L, Zhang P, Li M, Gao M, et al. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 2021;10:9078–96.
Wu X, Shen S, Qin J, Fei W, Fan F, Gu J, et al. High co-expression of SLC7A11 and GPX4 as a predictor of platinum resistance and poor prognosis in patients with epithelial ovarian cancer. BJOG Int J Obstet Gynaecol. 2022;129:40–9.
Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene. 2018;37:5007–19.
Lee JR, Roh JL, Lee SM, Park Y, Cho KJ, Choi SH, et al. Overexpression of cysteine-glutamate transporter and CD44 for prediction of recurrence and survival in patients with oral cavity squamous cell carcinoma. Head Neck. 2018;40:2340–6.
Sugano K, Maeda K, Ohtani H, Nagahara H, Shibutani M, Hirakawa K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015;35:677–82.
Ma Z, Zhang H, Lian M, Yue C, Dong G, Jin Y, et al. SLC7A11, a component of cysteine/glutamate transporter, is a novel biomarker for the diagnosis and prognosis in laryngeal squamous cell carcinoma. Oncol Rep. 2017;38:3019–29.
Sugezawa K, Morimoto M, Yamamoto M, Matsumi Y, Nakayama Y, Hara K, et al. GPX4 regulates tumor cell proliferation via suppressing ferroptosis and exhibits prognostic significance in gastric cancer. Anticancer Res. 2022;42:5719–29.
Lee JR, Roh JL, Lee SM, Park Y, Cho KJ, Choi SH, et al. Overexpression of glutathione peroxidase 1 predicts poor prognosis in oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2017;143:2257–65.
Chen H, Peng F, Xu J, Wang G, Zhao Y. Increased expression of GPX4 promotes the tumorigenesis of thyroid cancer by inhibiting ferroptosis and predicts poor clinical outcomes. Aging. 2023;15:230–45.
Liu CY, Liu CC, Li AF, Hsu TW, Lin JH, Hung SC, et al. Glutathione peroxidase 4 expression predicts poor overall survival in patients with resected lung adenocarcinoma. Sci Rep. 2022;12:20462.
Sekhar KR, Hanna DN, Cyr S, Baechle JJ, Kuravi S, Balusu R, et al. Glutathione peroxidase 4 inhibition induces ferroptosis and mTOR pathway suppression in thyroid cancer. Sci Rep. 2022;12:19396.
Shi Z, Yuan H, Cao L, Lin Y. AKT1 participates in ferroptosis vulnerability by driving autophagic degradation of FTH1 in cisplatin-resistant ovarian cancer. Biochem Cell Biol. 2023;101:422–31.
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
Cheng F, Dou J, Yang Y, Sun S, Chen R, Zhang Z, et al. Drug-induced lactate confers ferroptosis resistance via p38-SGK1-NEDD4L-dependent upregulation of GPX4 in NSCLC cells. Cell Death Discov. 2023;9:165.
Golbashirzadeh M, Heidari HR, Talebi M, Yari Khosroushahi A. Ferroptosis as a potential cell death mechanism against cisplatin-resistant lung cancer cell line. Adv Pharm Bull. 2023;13:176–87.
Zhao G, Liang J, Shan G, Gu J, Xu F, Lu C, et al. KLF11 regulates lung adenocarcinoma ferroptosis and chemosensitivity by suppressing GPX4. Commun Biol. 2023;6:570.
Wang Y, Zheng L, Shang W, Yang Z, Li T, Liu F, et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 2022;29:2190–202.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-Mediated redox homeostasis. Redox Biol. 2020;30:101413.
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep. 2015;33:1465–74.
Song Q, Peng S, Sun Z, Heng X, Zhu X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med J. 2021;62:843–9.
He C, Jiang Y, Guo Y, Wu Z. Amplified ferroptosis and apoptosis facilitated by differentiation therapy efficiently suppress the progression of osteosarcoma. Small. 2023;19:e2302575.
Ji P, Wang X, Yin J, Yao Y, Du W. Amplification of ferroptosis with a liposomal nanoreactor cooperates with low-toxicity doxorubicin apoptosis for enhanced tumor chemotherapy. Biomater Sci. 2022;10:1544–53.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Shibata Y, Yasui H, Higashikawa K, Miyamoto N, Kuge Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE. 2019;14:e0225931.
Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021;40:3533–47.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020;15:469–84.
Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13:2206.
Liu R, Liu L, Bian Y, Zhang S, Wang Y, Chen H, et al. The Dual Regulation Effects of ESR1/NEDD4L on SLC7A11 in Breast Cancer Under Ionizing Radiation. Front Cell Dev Biol. 2021;9:772380.
Liu L, Zhang C, Qu S, Liu R, Chen H, Liang Z, et al. ESR1 inhibits ionizing radiation-induced ferroptosis in breast cancer cells via the NEDD4L/CD71 pathway. Arch Biochem Biophys. 2022;725:109299.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6:eaay9789.
Cai H, Ren Y, Chen S, Wang Y, Chu L. Ferroptosis and tumor immunotherapy: A promising combination therapy for tumors. Front Oncol. 2023;13:1119369.
Gong D, Chen M, Wang Y, Shi J, Hou Y. Role of ferroptosis on tumor progression and immunotherapy. Cell Death Discov. 2022;8:427.
Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35:84–100.e8.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40:365–78.e6.
Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35:109235.
Chen C, Yang Y, Guo Y, He J, Chen Z, Qiu S, et al. CYP1B1 inhibits ferroptosis and induces anti-PD-1 resistance by degrading ACSL4 in colorectal cancer. Cell Death Dis 2023;14:271.
Chung CH, Lin CY, Chen CY, Hsueh CW, Chang YW, Wang CC, et al. Ferroptosis signature shapes the immune profiles to enhance the response to immune checkpoint inhibitors in head and neck cancer. Adv Sci. 2023;10:e2204514.
Zhou Z, Zhao Y, Chen S, Cui G, Fu W, Li S, et al. Cisplatin promotes the efficacy of immune checkpoint inhibitor therapy by inducing ferroptosis and activating neutrophils. Front Pharmacol. 2022;13:870178.
Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol. 2022;24:1113–25.
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001–12.e5.
Conche C, Finkelmeier F, Pešić M, Nicolas AM, Böttger TW, Kennel KB, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774–82.
Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338–46.
Gai C, Liu C, Wu X, Yu M, Zheng J, Zhang W, et al. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis 2020;11:751.
Bi G, Liang J, Zhao M, Zhang H, Jin X, Lu T, et al. miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol Ther Nucleic acids. 2022;28:366–86.
Wu N, Zhu D, Li J, Li X, Zhu Z, Rao Q, et al. CircOMA1 modulates cabergoline resistance by downregulating ferroptosis in prolactinoma. J Endocrinol Investig. 2023;46:1573–87.
Ou R, Lu S, Wang L, Wang Y, Lv M, Li T, et al. Circular RNA circLMO1 suppresses cervical cancer growth and metastasis by triggering miR-4291/ACSL4-mediated ferroptosis. Front Oncol. 2022;12:858598.
Liu B, Ma H, Liu X, Xing W. CircSCN8A suppresses malignant progression and induces ferroptosis in non-small cell lung cancer by regulating miR-1290/ACSL4 axis. Cell Cycle. 2023;22:758–76.
Lei S, Cao W, Zeng Z, Zhang Z, Jin B, Tian Q, et al. JUND/linc00976 promotes cholangiocarcinoma progression and metastasis, inhibits ferroptosis by regulating the miR-3202/GPX4 axis. Cell Death Dis. 2022;13:967.
Li X, Li Y, Lian P, Lv Q, Liu F. Silencing lncRNA HCG18 regulates GPX4-inhibited ferroptosis by adsorbing miR-450b-5p to avert sorafenib resistance in hepatocellular carcinoma. Hum Exp Toxicol. 2023;42:9603271221142818.
Li Z, Luo Y, Wang C, Han D, Sun W. Circular RNA circBLNK promotes osteosarcoma progression and inhibits ferroptosis in osteosarcoma cells by sponging miR‑188‑3p and regulating GPX4 expression. Oncol Rep. 2023;50:192.
Shanshan W, Hongying M, Jingjing F, Yiming Y, Yu R, Rui Y. CircDTL functions as an oncogene and regulates both apoptosis and ferroptosis in non-small cell lung cancer cells. Front Genet. 2021;12:743505.
Xu Q, Zhou L, Yang G, Meng F, Wan Y, Wang L, et al. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int. 2020;44:2344–56.
Huang X, Wu J, Wang Y, Xian Z, Li J, Qiu N, et al. FOXQ1 inhibits breast cancer ferroptosis and progression via the circ_0000643/miR-153/SLC7A11 axis. Exp Cell Res. 2023;431:113737.
Pan CF, Wei K, Ma ZJ, He YZ, Huang JJ, Guo ZZ, et al. CircP4HB regulates ferroptosis via SLC7A11-mediated glutathione synthesis in lung adenocarcinoma. Transl Lung Cancer Res. 2022;11:366–80.
Qin K, Zhang F, Wang H, Wang N, Qiu H, Jia X, et al. circRNA circSnx12 confers Cisplatin chemoresistance to ovarian cancer by inhibiting ferroptosis through a miR-194-5p/SLC7A11 axis. BMB Rep. 2023;56:184–9.
Li Q, Li K, Guo Q, Yang T. CircRNA circSTIL inhibits ferroptosis in colorectal cancer via miR-431/SLC7A11 axis. Environ Toxicol. 2023;38:981–9.
Wang HH, Ma JN, Zhan XR. Circular RNA Circ_0067934 attenuates ferroptosis of thyroid cancer cells by miR-545-3p/SLC7A11 Signaling. Front Endocrinol. 2021;12:670031.
Wu P, Li C, Ye DM, Yu K, Li Y, Tang H, et al. Circular RNA circEPSTI1 accelerates cervical cancer progression via miR-375/409-3P/515-5p-SLC7A11 axis. Aging. 2021;13:4663–73.
Pan C, Chen G, Zhao X, Xu X, Liu J. lncRNA BBOX1-AS1 silencing inhibits esophageal squamous cell cancer progression by promoting ferroptosis via miR-513a-3p/SLC7A11 axis. Eur J Pharmacol. 2022;934:175317.
Li L, Zhang Y, Gao Y, Hu Y, Wang R, Wang S, et al. LncSNHG14 promotes nutlin3a resistance by inhibiting ferroptosis via the miR-206 /SLC7A11 axis in osteosarcoma cells. Cancer Gene Ther. 2023;30:704–15.
Li YZ, Zhu HC, Du Y, Zhao HC, Wang L. Silencing lncRNA SLC16A1-AS1 induced ferroptosis in renal cell carcinoma through miR-143-3p/SLC7A11 signaling. Technol Cancer Res Treat 2022;21:15330338221077803.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Wei S, Feng M, Zhang S. Molecular characteristics of cell pyroptosis and its inhibitors: a review of activation, regulation, and inhibitors. Int J Mol Sci. 2022;23:16115.
Park HH, Park SY, Mah S, Park JH, Hong SS, Hong S, et al. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp Mol Med. 2018;50:1–15.
Acknowledgements
The authors appreciate all the cancer researchers working on the ceRNA networks to decipher the molecular mechanisms of ferroptosis in oncogenesis and cancer treatment. Parts of Fig. 2 were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier licensed under CC BY 4.0 (https://smart.servier.com/).
Author information
Authors and Affiliations
Contributions
FNO wrote the manuscript, searched the literature, and helped with the visualization and conceptualization. MAS helped with editing, writing the manuscript, visualization, conceptualization, and supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by George Calin
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nejadi Orang, F., Abdoli Shadbad, M. Competing endogenous RNA networks and ferroptosis in cancer: novel therapeutic targets. Cell Death Dis 15, 357 (2024). https://doi.org/10.1038/s41419-024-06732-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06732-4
- Springer Nature Limited