
Abstract
An important epigenetic switch marks the onset and maintenance of senescence. This allows transcription of the genetic programs that arrest the cell cycle and alter the microenvironment. Transcription of endogenous retroviruses (ERVs) is also a consequence of this epigenetic switch. In this manuscript, we have identified a group of ERVs that are epigenetically silenced in proliferating cells but are upregulated during replicative senescence or during various forms of oncogene-induced senescence, by RAS and Akt, or after HDAC4 depletion. In a HDAC4 model of senescence, removal of the repressive histone mark H3K27me3 is the plausible mechanism that allows the transcription of intergenic ERVs during senescence. We have shown that ERVs contribute to the accumulation of dsRNAs in senescence, which can initiate the antiviral response via the IFIH1-MAVS signaling pathway and thus contribute to the maintenance of senescence. This pathway, and MAVS in particular, plays an active role in shaping the microenvironment and maintaining growth arrest, two essential features of the senescence program.
Similar content being viewed by others

Introduction
Cellular senescence is controlled by a complex genetic program, aimed at achieving permanent growth arrest and altering the microenvironment [1]. Once established, cellular senescence contributes to aging and progressive dysfunctions of tissues and organs [2, 3]. Senescent cells secrete various cytokines and modulators of the ECM (extracellular matrix) defined as SASP (senescence associated secretory phenotype). SASP exerts several effects, including reinforcing senescence and activating the immune clearance of senescent cells. SASP may also promote proliferation of adjacent cells and stem cell maintenance to support tissue regeneration [4, 5].
To fulfill the genetic program of senescence, cells undergo timely accurate epigenetic resetting [6]. This reorganization involves the restructuring of proximal and distal regulatory elements (promoters and enhancers) with the stabilization of new super-enhancers (SEs) [7,8,9].
Transposable elements make up nearly 50% of the human genome, and about 8% of them are endogenous retroviruses (ERVs) [10, 11]. ERVs are retrotransposons that contain long-terminal-repeats (LTRs). ERVs are normally inactive but under conditions such as DNA damage, viral infection or aging, they become activated [12, 13]. ERVs expression is normally silenced by epigenetic mechanisms that maintain a repressed chromatin state. DNA hypomethylating agents and HDACs inhibitors can upregulate ERVs expression [14,15,16].
HDACs may be part of repressive complexes responsible for silencing proviruses, also involved in regulating senescence [17,18,19,20,21,22]. Notably, the class IIa HDAC family member HDAC4, acts as a brake on senescence by supervising H3K27 acetylation at specific SEs. During senescence, HDAC4 is degraded by the ubiquitin-proteasome system in a GSK3β-dependent manner [8]. This epigenetic switch could contribute to the resurgence of certain ERVs. Indeed, previous studies have shown that epigenetic perturbations elicit the transcription of ERVs during cellular senescence in aging and cancer [15, 23,24,25]. In this manuscript, we investigated whether ERVs are regulated during different forms of senescence and what role these retroviral elements might play in the senescence program.
Results
Epigenetic resetting and upregulation of ERVs transcription during senescence
We hypothesized that the epigenetic perturbations associated with senescence might also be involved in the upregulation of some ERVs. To investigate this hypothesis, we selected a set of ERVs (ERV9-1, ERVW-1, ERVFC1-1, ERVH48-1, Table S1), that are upregulated after pharmacologically induced epigenetic switch [15]. These ERVs are good candidates for upregulation during the epigenetic switch that characterizes senescence [6].
First, we examined the expression levels of these ERVs during replicative senescence. IMR90 diploid fibroblasts harvested at increasing population doubling were chosen as replicative senescent model [8]. All four ERVs were upregulated during replicative senescence (Fig. 1A). The SA β-gal staining confirmed the induction of senescence (Fig. 1B). Transcriptional activation and chromatin remodeling at specific ERV sequences during senescence is coupled to upregulation of the interferon (IFN) response [26]. More generally, reactivation of ERVs can trigger the innate immune response [27, 28]. Induction of the IFN response during replicative senescence has been studied using a signature of IFN-stimulated genes (ISGs): OAS1, IFI6, RIG1, IFIH1. The upregulation of OAS1, IFI6, IFIH1, and RIG1 was coupled to the induction of ERVs expression (Fig. 1C).

A Expression levels of the indicated ERVs in IMR90 cells undergoing replicative senescence. RNAs were extracted at the indicated passages of cultured cells and processed for qRT-PCR. Data are relative to passage 12. Mean ± SD; n = 2. *p < 0.05, **p < 0.01, ***p < 0.001, t-test relative to PD12. B SA β-gal positivity in IMR90 cells undergoing replicative senescence. Cells were fixed processed for SA β-gal assay and scored at the microscope. C Expression levels of the indicated ISGs in IMR90 cells undergoing replicative senescence. RNAs were extracted at the indicated passages of cultured cells and processed for qRT-PCR. Data are relative to passage 12. Mean ± SD; n = 2. *p < 0.05, **p < 0.01, ***p < 0.001, t-test relative to PD12. D SA β-gal positivity in SK-LMS-1 cells knocked-out for HDAC4 and re-expressing a 4OHT inducible, PAM mutated version of HDAC4 (SK-LMS-1HDAC4-/-/HDAC4PAM-ER). Bar = 50 µm. E Quantitative analysis of SA-β-gal positivity in SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT. Mean ± SD; n = 3. **p < 0.01, ***p < 0.001, t-test relative to -4OHT condition. F Immunofluorescence analysis of LMNA (red) and H1.2-GFP (green) distribution in SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown of not in the presence of 4OHT for 48 h. Confocal images are shown in pseudocolors. Bar = 10 µm. G Expression levels of the indicated ERVs in SK-LMS-1 cells and SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT. Mean ± SD; n = 3. **p < 0.01, ***p < 0.001, t-test relative to -4OHT condition. H Expression levels of the indicated ISGs in SK-LMS-1 cells and SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, t-test relative to -4OHT condition. I Representative confocal pictures of SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown for 48 h in the presence or not of 4OHT. Immunofluorescence was performed to visualize dsRNAs (red) and nuclei (blue). Confocal images are shown in pseudocolors. Bar 10 µM. J Expression levels of the indicated ERVs and ISGs in SK-LMS-1HDAC4-/-/HDAC4PAM-ER cells grown in the absence of 4OHT for the indicate hours. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, t-test relative to time zero.
As a second model of cellular senescence, we chose low-grade leiomyosarcoma cells SK-LMS-1 that undergo senescence after HDAC4 knock-out. In SK-LMS-1 cells, HDAC4 is required to suppress the activity of typical enhancers (TEs) and SEs that control the senescence program [8]. Therefore, we could refer to this model as epigenetically induced senescence. SK-LMS-1HDAC4-/-/HDAC4PAM-ER, in which HDAC4 is knocked-out and which re-expresses a PAM mutant inducible version of HDAC4 fused to ER (estrogen receptor), was selected as an inducible model for senescence [8]. In this cell line, HDAC4 can be turned on and off by removing tamoxifen (4OHT) and senescence induced, as proved by SA β-gal staining (Fig. 1D, E) and Lamin B1 downregulation (Fig. S1A).
Histone H1-mediated heterochromatinization is important to silence noncoding RNA derived from heterochromatic repeats or endogenous retroviruses to avoid a nonspecific IFN response [29]. Abrogation of HDAC4 expression after removal of 4OHT leads to downregulation of H1 (Fig. 1F), upregulation of ERVs, except ERVW-1, which contains a coding gene (syncytin1) (Fig. 1G), and IFN genes (Fig. 1H). Immunofluorescence analyses using the J2 antibody [30] demonstrate the accumulation of dsRNAs species in SK-LMS-1HDAC4-/-/HDAC4PAM-ER cells after depletion of HDAC4 (Fig. 1I).
To better capture the timing of activation of ERVs expression and IFN response, we performed a time-course analysis. Samples were collected at different time points after removal of 4OHT. Figure 1J shows that induction of ERVs and upregulation of IFN genes are time-coupled responses.
In summary, these results show that epigenetically regulated ERVs are induced in two different models of senescence.
Senescence induced after HDAC4 knock-out elicits the expression of ERVs and of the IFN response
To further confirm that HDAC4 is associated with upregulation of ERVs during senescence, we chose a classical model of OIS: BJ human diploid fibroblasts co-expressing RAS (G12V). In these cells, RAS triggers senescence and co-expression of the viral oncoprotein E1A stop the program. BJ-RAS/E1A cells, after elimination of HDAC4 by CRISPR/Cas9, underwent senescence. This model was previously fully characterized [8]. Here we quantified the expression levels of a group of ERVs. Entry into senescence is associated with upregulation of all ERVs tested, including ERVW-1, which was not upregulated in LMS cells, and IFN response genes (Fig. 2A). We confirmed the upregulation of ERVs in another model of HDAC4-dependent senescence to exclude lineage-specific effects. In A375 melanoma cells silenced for HDAC4, we expressed a Dox-inducible version of HDAC4. In this way, the cell line can be propagated in culture. After removal of Dox, cells enter senescence (Fig. S1B). Also in this model, senescence is coupled to upregulation of the selected ERVs and genes of the IFN response (Fig. 2B).

A (left) SA β-gal positivity in BJ-hTERT/E1A/RAS WT or knocked out for HDAC4; (right) Expression levels of the indicated ERVs, ISGs and SASP genes in BJ-hTERT/E1A/RAS knocked out for HDAC4. RNAs were extracted and processed for qRT-PCR. Data are relative BJ-hTERT/E1A/RAS expressing HDAC4 (set to 1). Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001 t-test. B Expression levels of the indicated ERVs, ISGs and SASP genes in A375 melanoma cells knocked out for HDAC4. RNAs were extracted and processed for qRT-PCR. Data are relative to wt A375 cells. Mean ± SD; n = 2 for each clone. *p < 0.05, **p < 0.01, ***p < 0.001 paired t-test between plus and minus DOX conditions. C SA β-gal positivity in BJ-hTERT cells expressing inducible versions of AKT or RAS. Cells grown for the indicated days in the presence of 4OHT were fixed, processed for SA-β-gal assay and scored at the microscope. n = 3, **p < 0.01, ***p < 0.001, Dunn’s Multiple Comparison Test. D Expression levels of the indicated ERVs and of IL1B in BJ-hTERT/GFP, BJ-hTERT/AKT and BJ-hTERT/RAS grown for the indicated days in the presence of 4OHT. RNAs were extracted and processed for qRT-PCR. Data are relative to BJ-hTERT/GFP without 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, Dunn’s Multiple Comparison Test.
Finally, we investigated whether these ERVs are also upregulated in additional models of OIS triggered by strong oncogenes. We used BJ-hTERT cells expressing inducible versions of HRAS-G12V or myr-AKT1 (Fig S1C). Following oncogene induction, after an initial proliferative burst, these cells enter senescence 8 days later. SA-β-gal assay (Fig. 2C), IL1B upregulation (SASP gene) (Fig. 2D) and the upregulation of CDKN1A (Fig. S1C) confirmed the induction of OIS. Also in this model, all ERVs tested were upregulated (Fig. 2D). Altogether, these results demonstrate that ERV9-1, ERVW-1, ERVFC1-1 and ERVH48-1 are part of a common genetic program that is engaged in different forms of senescence.
Interestingly, in MCF10A cells, knockout of HDAC7, the most abundantly expressed class IIa HDAC, induces expression of ERV9-1 and ERVH48-1 but not ERVW-1 and ERVFC1-1. These cells do not enter senescence in response to the deletion of HDAC7 [31], possibly because of the deletion within the CDKN2A locus [32]. Interestingly, RAS also triggered the upregulation of ERVFC1-1, and the concomitant deletion of HDAC7 further enhanced the upregulation of these three ERVs and the IFN response (Fig. S2).
Therefore, the link to ERVs is not limited to HDAC4 but also involves other class IIa HDACs as general regulators to achieve directly or indirectly the silencing of defined ERVs.
Deletion of HDAC4 leads to a decrease in H3K27me3 at several intergenic ERVs
In cells, ERVs are often silenced to prevent their abnormal transcription. Epigenetic markers for silenced chromatin such as H3K9me3, H3K27me3, and DNA methylation are deposited at ERVs loci [33,34,35]. To better understand the mechanisms that trigger the expression of ERVs during OIS, we examined genome-wide variations in H3K27me3 distribution in SK-LMS-1HDAC4-/-/HDAC4PAM-ER cells undergoing senescence. ChIP-seq analysis revealed that deletion of HDAC4 does not cause overt changes in H3K27me3 (Fig. 3A) [8]. Instead, a selected number of genomic regions show a consistent reduction in H3K27me3, as shown by the heat-map (H3K27me3 signal >2 folds). To understand whether these regions contained ERVs, they were crossed with distal intergenic LTRs (n = 316.185), which revealed 146 regions containing ERVs whose H3K27me3 levels were reduced after HDAC4 KO (Fig. 3B and Table S2). Importantly, significant H3K27me3 demethylation was observed after HDAC4 depletion in the genomic loci of ERV9-1 and ERVFC1-1, but not in the ORF containing ERVW-1 and ERVH48-1 (Table S2).

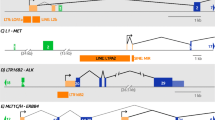
A Heat-maps of the H3K27me3 signal distribution in SK-LMS-1HDAC4-/-/HDAC4PAM-ER WT or KO for HDAC4. (left) a region of ±5 kb around intergenic LTRs and (right) a region of ±5 kb around the intergenic LTRs which show a reduction of at least 2 folds in H3K27me3 signal after HDAC4 knock-out. B Venn diagram showing the number of intergenic LTRs where H3K27me3 is reduced by at least 2 folds after the knockout of HDAC4. C Expression levels of the indicated ERVs in SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or absence of 4OHT. Data are relative to the presence of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test between plus and minus 4OHT conditions. D Expression levels of the indicated ERVs in BJ-hTERT/RAS-ER grown in the presence or absence of 4OHT. Data are relative to the absence of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test between plus and minus 4OHT conditions. E–G Detailed view of H3K27me3 and H3K27ac tracks at three representatives intergenic ERVs (MER21c, MLT1c, and MER57b1), upregulated after HDAC4 knock-out and showing a reduction of the H3K27me3 signal. Gene structure and chromosomal location are shown.
The up-regulation of the transcripts of three of these ERVs (MER57B1, MER21C and MLT1C) was validated by qRT-PCR (Fig. 3C). Moreover, their upregulation also occurred in BJ-hTERT fibroblasts during RAS-induced OIS (Fig. 3D).
Accordingly, genomic loci containing MER57B1, MER21C and MLT1C show a dramatic reduction of H3K27me3 signals after HDAC4 depletion, whereas H3K27ac levels remain largely unchanged (Fig. 3E–G). Importantly, upregulation of ERVs during senescence may also involve additional epigenetic mechanisms. For example, the METTL9 locus is characterized by an increase in H3K27ac during senescence triggered by HDAC4 depletion (Fig. S3).
In conclusion, these results confirm that the upregulation of ERVs during senescence is a general and conserved phenomenon involving epigenetic resetting.
MER57B1, MER21C, ERV9-1 and ERVFC-1 are not upregulated in response to DNA damage
It has been reported that induction of ERVs is associated with the presence of DNA damage and that senescence is associated with the accumulation of DNA damage [36, 37]. Similarly, the activation of IFN response was previously correlated to the activation of cGAS/STING pathway as a consequence of the accumulation of cytoplasmic DNA scars [1, 2, 6]. To clarify the correlation between ERVs upregulated in senescent cells and the DNA damage response (DDR), SK-LMS-1 cells were treated with various DNA-damaging agents: aphidicolin (APH), camptothecin (CPT) and doxorubicin (DOX). All compounds induced DNA damage (γH2AX positivity) and activation of the DDR, which was confirmed by TP53 phosphorylation and stabilization (Fig. 4A). The expression of the investigated ERVs was analyzed within 24 h after treatment to exclude their modulation as a secondary event after the activation of senescence by chemotherapeutic agents [37]. Their expression remained unchanged during DDR (Fig. 4B). Therefore, we can affirm that upregulation of these ERVs is specifically correlated with senescence.

A Immunoblot analysis of TP53 activation and DNA damage (γH2AX). Lysates obtained from SK-LMS-1 cells, treated for the indicated times with 6.25 μM Camptothecin (CPT), 1 μM Aphidicolin (APH) or 100 nM Doxorubicin (DOX), were immunoblotted using the indicated antibodies. Actin and SMC3 were used as loading control. B Expression levels of the indicated ERVs in SK-LMS-1 treated for 24 h with 6.25 μM CPT, 1 μM APH or 100 nM DOX. Data are relative to untreated cells. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, Dunn’s Multiple Comparison Test.
dsRNAs and the regulation of senescence
Transcription by LTRs can generate dsRNAs that are recognized by selected receptors in the cytoplasm, endosomal compartment, or cell surface. These receptors activate signaling pathways that induce type I and type III interferon (IFN) expression and increased immunogenicity [38, 39]. TLR3 is the major receptor for dsRNAs at the cell surface, whereas RIG1 (retinoic acid-inducible gene I) and IFIH1/MDA5 (interferon induced with helicase C domain 1/melanoma differentiation-associated gene 5) are the major cytosolic receptors. RIG1 mainly recognizes 5’-triphosphate sRNAs and short dsRNA (<1 kb). IFIH1, on the other hand, preferentially binds long dsRNAs (>2 kb) [40]. Engagement of these receptors leads to activation of MAVS (mitochondrial antiviral signaling) [41].
Several studies have addressed the contribution of IFN response in senescence and aging [42]. The upregulation of ERVs during senescence may be responsible for the onset of the IFN response, thus contributing to senescence.
First, we transfected the synthetic poly(I:C) (polyriboinosinic:polyribocytidylic acid), as a mimic of the dsRNA generated by ERVs transcription. In SK-LMS-1 cells, poly(I:C)-HMW or poly(I:C)-LMW trigger dramatic upregulation of IFN genes IFIH1, IFNB1 and OAS1. Pretreatment with RNAse III impaired the upregulation of IFN response, proving the specificity. No differences were detectable between LMW and HMW poly(I:C), suggesting the integrity of both IFIH1 and RIG1 signaling (Fig. 5A). We next examined proliferative potential (Fig. 5B), induction of cell death (Fig. 5C) and accumulation of senescent cells (Fig. 5D). Poly(I:C) transfections lead to cell cycle arrest, cell death, but also accumulation of SA β-gal positive cells. The CDK4 inhibitor palbociclib was used as a positive control for senescence. Induction of cell death (Fig. 5E) and of senescence (Fig. 5F) in response to transfection of LMW and HMW poly(I:C), was confirmed in immortalized human fibroblasts (BJ-hTERT).

A Expression levels of the indicated ISGs. SK-LMS-1 cells were transfected for 48 h with 10 ng/ml of low molecular (LMW) and high molecular (HMW) weight poly (I:C), pre-digested or not for 30 min with 50U of RNAse III. Data are relative to untreated cells. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. B SK-LMS-1 cells were treated as in (A) and the proliferative potential was evaluated as doubling. Mean ± SD; n = 5. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. C SK-LMS-1 cells were treated as in (A) and cell death was evaluated by Trypan blue staining. Mean ± SD; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. D SK-LMS-1 cells were treated as in (A) and senescence was evaluated by SA β-gal staining. Palbociclib was used 3 µM for 48 h. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. E BJ-hTERT cells were treated as in (A) and cell death was evaluated by Trypan blue staining. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. F BJ-hTERT cells were treated as in (A) and senescence was evaluated after 11 days from transfection by SA β-gal staining. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to control condition. G Quantification of the enrichment over input of the indicated species of RNAs in the preparation of double-stranded enriched RNAs (dsRNAs) from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the absence of 4OHT for 4 days. The J2 antibody was used for the purification of dsRNAs. H Analysis of SA β-gal positivity in SK-LMS-1 cells transfected for 72 h with 30 pmoles of dsRNAs obtained as described in G), and pre-digested or not for 30’ with 50U of RNAse III. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. I Cell death as determined by Trypan blue assay positivity in SK-LMS-1 cells transfected for 72 h with 30 pmoles of dsRNAs obtained as described in (G), and pre-digested or not for 30’ with 50U of RNAse III. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. J Enrichment (expressed as multiplication factor of total read counts) of the indicated non-coding elements in dsRNA-seq samples obtained from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or absence of 4OHT for 4 days, as indicated. *p < 0.05, Mann–Whitney. K Volcano plot that shows statistical significance (-log10(P value)) versus magnitude of change (log2(fold change)) of the abundance of ERVs in dsRNA-seq samples obtained from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the absence of 4OHT for 4 days, in respect to cells grown in the presence of 4OHT. L Volcano plot that shows statistical significance (-log10(P value)) versus magnitude of change (log2(fold change)) of the abundance of ERVs in dsRNA-seq samples obtained from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the absence of 4OHT for 4 days, in respect to the total RNA extracted from cells grown in the presence of 4OHT.
Finally, dsRNAs were immunopurified from senescent SK-LMS -1HDAC4-/-/HDAC4PAM- ER cells grown in the absence of HDAC4 (-4OHT) using the J2 antibody. The enrichment of ERVs is shown in Fig. 5G. Transfection of cellular dsRNAs generated during senescence was sufficient to induce senescence (Fig. 5H). Of note, a proportion of cells that received dsRNAs isolated from senescent cells activated an apoptotic program (Fig. 5I). Treatment with RNAse III confirmed the specificity of dsRNA activity.
To characterize the dsRNAs present in senescent cells, we performed RIP-seq using the J2 antibody in proliferating SK-LMS -1HDAC4-/-/HDAC4PAM- ER cells and in cells entering senescence after the depletion of HDAC4 (96 h -4OHT). PCA (principal component analysis) confirmed the high enrichment of specific dsRNA species compared to the input during immunoprecipitation and the good reproducibility of the results (Fig. S4A). By restricting the analysis to ERVs alone, the distinction between cells undergoing senescence and proliferating cells becomes even clearer, suggesting that the differential expression of ERVs and their folding to dsRNA are meaningful parameters to distinguish the two states (Fig. S4B). Consistently with previous data [43], retrotransposons are the most enriched elements in dsRNA preparations purified from cells, whether proliferating or senescent (Fig. 5J). ERVs are the second most enriched class of elements in dsRNA purifications but, importantly, the only class that is significantly enriched in dsRNA species isolated from senescent cells (Fig. 5J). Specifically, 32 ERV entities were identified that are selectively enriched in dsRNA preparations from senescent cells compared to proliferating cells (Fig. 5K and Table S3). Of the ERVs examined throughout the manuscript (ERV9-1, ERVW-1, ERVFC1-1, ERVH48-1), ERVFC1-1 and ERVH48-1 were among the most upregulated ERVs folded as dsRNA in SK-LMS-1 senescent cells, whereas ERVW-1 was not upregulated in this model of senescence, as expected (Fig. 1J).
ERV9-1 was not found to be significantly enriched as dsRNA during senescence. However, when the upregulation of ERV9-1 family entities was analyzed, upregulation of most of its entities was observed, although not significantly in virtue of the scattering of data (Fig. S5). ERVFC1-1 and ERVH48-1 are also among the ERVs that most efficiently produce dsRNAs (Fig. 5L and Table S4). By contrast HERVK [44] was the most significant downregulated dsRNAs ERV under senescence induced after HDAC4 KO.
Finally, we identified a core signature of 42 ERVs (Fig. S4C and Table 1) that are significantly enriched upon dsRNAs purification and are highly induced in cells that become senescent after HDAC4 depletion compared to proliferating cells. 87% of dysregulated ERVs belong to class I ERVs and 57.5% to the HUERSP, HEPSI and HERVHF supergroups (Fig. S4D) [45].
Overall, our analysis highlights how certain supergroups of ERVs are overrepresented in senescent cells and folded as dsRNAs and therefore represent possible ligands for intracellular receptors of dsRNA species.
dsRNAs receptors and the regulation of senescence
To understand the contribution of the dsRNA signaling pathway to the induction of senescence, the expression of the components RIG1, IFIH1 and MAVS was silenced in BJ-hTERT cells during RAS-induced senescence. We optimized a specific protocol to maintain silencing of the genes throughout the experiments (Fig. 6A). RT-PCR experiments confirmed the efficiency of silencing (Fig. 6B). The activities of IFIH1 and MAVS, but not RIG1, contribute to cell cycle arrest (Fig. 6C) and the appearance of senescence (Fig. 6D).

A Schematic representation of the experimental protocol used to evaluate the effect of gene silencing during OIS in BJ-hTERT/RAS-ER cells. B Expression levels of the indicated genes in cells silenced with the relative siRNA as schematized in (A). Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, t-test. C Analysis of BrdU incorporation in BJ-hTERT/RAS-ER cells, treated as schematized in (A). Significance is relative to cells grown without 4OHT and transfected with control siRNA. Mean ± SD; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, t-test relative to control. D SA β-gal positivity in BJ-hTERT/RAS-ER cells, treated as schematized in (A). Significance is relative to cells grown without 4OHT and transfected with control siRNA. Mean ± SD; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, t-test relative to control. E Immunoblot analysis in BJ-hTERT/RAS-ER cells silenced for RIG1, IFIH1 and MAVS. Lysates obtained from cells grown or not in the presence of 4OHT were immunoblotted and probed with the indicated antibodies. F SA β-gal positivity in BJ-hTERT/RAS-ER cells silenced for RIG1, IFIH1 and MAVS, after treatment with 4OHT. Bar = 50 µm. G Quantitative analysis of SA-β-gal positivity BJ-hTERT/RAS-ER cells knocked-down for RIG1, IFIH1 and MAVS grown in the presence or not of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, t-test relative to shRNA control. H Expression levels of the indicated genes in BJ-hTERT/RAS-ER cells silenced for RIG1, IFIH1 and MAVS, grown in the presence or not of 4OHT. RNAs were extracted and processed for qRT-PCR. Data are relative to BJ-hTERT/RAS-ER control cells grown in the absence of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, t-test.
To confirm these results, we stably knocked-down the expression of RIG1, IFIH1 and MAVS with shRNAs by lentiviral infection (Fig. 6E). RAS-induced OIS in BJ-hTERT cells is characterized by the appearance of a rapidly migrating form of IFIH1. Importantly, upregulation of p16/CDKN2A in response to RAS was dramatically downregulated in MAVS-silenced cells. When IFIH1 was downregulated p16/CDKN2A was modestly repressed. RIG1 silencing has no effect on the upregulation of p16/CDKN2A during RAS-induced senescence. SA β−gal staining confirmed the role of MAVS and IFIH1 in the regulation of OIS and excluded a contribution of RIG1 (Fig. 6F, G). Expression of CSF2, CDKN1A and IF16 was examined to monitor SASP, cell cycle arrest and activation of IFN response. Expression of MAVS and IFIH1 was required for maintenance of CSF2 expression. CDKN1A upregulation was specifically dependent on MAVS, while IF16 upregulation in response to OIS was affected by all sensors (Fig. 6H).
A MAVS dependent pathway sustains senescence
MAVS emerged as an important player of the RAS-induced senescence. To further confirm the role of MAVS, we again used the HDAC4-dependent model of senescence. SK-LMS-1HDAC4-/-/HDAC4PAM-ER cells were engineered to express the dominant negative (DN) version of MAVS (MAVSΔCARD) [46]. As positive control, we used a mutant version of TP53 (TP53R175H) which is known to escape OIS [47]. Since the role of TP53 in OIS is well established, we used only one clone.
Expression of TP53-DN and of MAVS-DN in HDAC4−/− cells reduced SA-β-gal positivity and rescued proliferation, similar to the reintroduction of HDAC4 (Fig. 7A, B). The anti-senescence effect was confirmed by the rescue of LMNB1 expression (Fig. 7C, D). To confirm the involvement of MAVS in senescence, we also silenced its expression in SK-LMS-1HDAC4−/− cells (Fig. 7E). Interfering with MAVS suppressed activation of the senescence program in terms of SASP, cell cycle genes and IFN response, similar to re-expression of HDAC4. We observed that TP53 had a much stronger effect on regulating growth arrest genes compared with HDAC4 and MAVS. In contrast, upregulation of CXCL8 is highly dependent on the activities of HDAC4 and MAVS (Fig. 7F).

A Representative microscopic images of SA β-gal stained SK-LMS-1HDAC4-/-/HDAC4PAM grown in the presence or not of 4OHT and expressing the indicated transgenes (scale bar 40 μm). B Quantitative analysis of SA β-gal positivity and BrdU incorporation in SK-LMS-1HDAC4-/-/HDAC4 grown in the presence or not of 4OHT and expressing the indicated transgenes. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to NEO condition. C Immunoblot analysis using the indicated antibodies. Whole cell extracts were obtained from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT and expressing or not TP53. D Immunoblot analysis using the indicated antibodies. Whole cell extracts were obtained from SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT and expressing or not MAVS-DN. E Quantitative analysis of SA β-gal positivity in SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the presence or not of 4OHT and transfected with the indicated siRNAs. Mean ± SD; n = 3. **p < 0.01, paired t-test relative to HDAC4−/− control condition. F Expression levels of the indicated genes in SK-LMS-1HDAC4-/-/HDAC4PAM-ER knocked out for HDAC4 and re-expressing HDAC4 or the indicated transgenes. RNAs were extracted and processed for qRT-PCR. Data are relative to SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the absence of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to NEO condition. G Immunoblot analysis using the indicated antibodies. Whole cell extracts were obtained from A375 cells grown WT or KO for HDAC4 and expressing the indicated transgenes. HDAC4 expression was maintained by adding Dox to the culture medium. H Quantitative analysis of SA β-gal positivity in A375 cells WT or KO for HDAC4 and expressing the indicated transgenes. HDAC4 expression was maintained by adding Dox to the culture medium. Mean ± SD; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to NEO condition. I Expression levels of the indicated genes in A375 cells WT or KO for HDAC4 and expressing the indicated transgenes. HDAC4 expression was maintained by adding Dox to the culture medium. RNAs were extracted and processed for qRT-PCR. Data are relative to SK-LMS-1HDAC4-/-/HDAC4PAM-ER grown in the absence of 4OHT. Mean ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, paired t-test relative to NEO condition.
To further confirm the contribution of MAVS to senescence, A375 melanoma cells in which HDAC4 was knocked-out, were engineered to express MAVS-DN. The Dox-inducible system was used to re-express HDAC4, and two different clones of knocked-out cells were selected. We also overexpressed TP53-DN as a positive control (Fig. 7G). All different transgenes counteracted LMNB1 downregulation and senescence induced by HDAC4 knock-out (Fig. 7G, H).
Expression of SASP genes IL1B and CXCL8 was efficiently repressed by HDAC4, whereas TP53-DN and MAVS-DN repressed CXCL8 less efficiently (Fig. 7I). TP53-DN strongly suppressed the upregulation of cell cycle genes CDKN1A and GADD45A compared with MAVS-DN. Instead, MAVS-DN repressed IFN response genes more efficiently, apart from RIG1 (Fig. 7I).
In summary, ERVs that are upregulated during senescence contribute to the accumulation of dsRNAs that can activate IFIH1-MAVS signaling and the antiviral response, thus contributing to the maintenance of the senescence program.
Discussion
In this manuscript, we provide evidence for upregulation of ERV transcripts in different models of cellular senescence. The ERVs here investigated were found upregulated during replicative senescence, OIS, and during HDAC4 deletion-induced senescence. However, these ERVs are not upregulated in response to genotoxic stress. This finding suggests that these ERVs are part of a central genetic program that is universally deployed in various forms of senescence in human cells.
Induction of ERVs during senescence is the result of profound epigenomic rearrangements. Global genomic analysis revealed that a selected number of intergenic LTRs exhibit a marked reduction in H3K27me3 during senescence. However, it is plausible that other epigenetic mechanisms, involved in the abrogation of ERV expression [48, 49], are discharged during senescence. Accordingly, senescence and aging are known to be characterized by methylation changes that can subvert the transcriptional landscape [50].
The upregulation of retroviral elements during senescence and aging has been reported previously [23, 24]. HERV1 and L1 and, to a lesser extent, HERVK and HERVL are deregulated during replicative senescence and in fibroblasts with Li-Fraumeni syndrome in which senescence was induced by genotoxic and oxidative drugs [23]. Similar to our observation, this upregulation is the result of a change in the heterochromatin/euchromatin compartments [23]. Recently, it has been reported that integrated human ERVK (HML-2) is activated by epigenetic resetting during senescence and can assemble into functional virions that can maintain the senescence program. HERVK, once secreted, can trigger senescence in neighboring cell [44]. In our dsRNA purification from senescent cells, HERVK is the most highly depopulated ERV. Therefore, our data suggest that HERVK is not only strongly induced but also strongly processed in senescent cells, resulting in minimal accumulation of dsRNAs.
The expression of ERVs can support a pro-inflammatory microenvironment through the expression of ISGs [26,27,28]. Accordingly, we found that ERVs upregulation in senescence is closely associated with activation of the IFN response in all models of cellular senescence examined.
Indeed, bidirectional transcription of LTRs or self-pairing between tandem ERV genome copies promotes folding of ERV transcripts into dsRNA structures [51]. Consistent with this, we observed that dsRNAs accumulate in the cytoplasm of senescent cells. These dsRNA species are mainly composed by LINE and ERVs elements and they could be recognized by RLRs family members RIG1 and IFIH1. These receptors can trigger a strong inflammatory-immune response through the engagement of MAVS [39].
Exogenous administration of dsRNA species derived from a cellular model of HDAC4-dependent senescence, induces senescence and cell death in transformed cells. Similarly, senescence and apoptosis occurred when tumor cells and normal cells were transfected with synthetic poly(I:C) molecules. Activation of the RIG-I/IFIH1/MAVS pathway and de-repression of ERVs were observed in cancer, aging, autoimmune, and neurodegenerative diseases [39]. However, the direct role of ERVs transcripts in maintaining senescence through the RLRs pathway is poorly understood [39, 52, 53].
Although RIG1 and IFIH1 have a very similar structure, they show different binding preferences for dsRNAs, recognizing short (<1 kb) and long (>2 kb) dsRNAs, respectively [54]. In this context, the use of poly(I:C) of different lengths proved that both cytoplasmic dsRNA immunosensors could be efficiently activated during the senescence program, as confirmed by the proper activation of the IFN response. However, we found that compared with RIG1, depletion of IFIH1 and MAVS strongly counteracted the senescence phenotype. The differential biological response of RIG1 and IFIH1 is not clear and requires further investigations but may depend on the altered sensitivity of these cytoplasmic immunosensors to various dsRNA elements [39].
How might the signaling axis ERVs/dsRNAs > IFIH1 > MAVS contribute to senescence? We have found that the strongest effect manifests at the level of SASP; therefore, interfering with the signaling pathway may reduce SASP intensity. Indeed, dsRNAs not only upregulated the expression of type I and type III IFNs but also affected NF-κB-dependent upregulation of proinflammatory genes by turning on MAVS [55, 56]. We confirmed that impairment of MAVS reduced the expression of IL1B and CXCL8, two known NF-kB targets. Because SASP can mediate enhancement (in an autocrine manner) or induction of senescence (in a paracrine manner) [57], the contribution of ERVs to SASP is a plausible mechanism. However, as SASP is not only involved in enhancing senescence but has other effects [57,58,59], ERVs activation might also influence other aspects of SASP.
Our results suggest that ERVs upregulated during senescence are not simply passengers of epigenetic reprogramming but contribute to the accumulation of dsRNA species, which play an active role in maintaining the program with important consequences for human aging.
Materials and methods
Cell cultures and treatments
BJ-hTERT, BJ-hTERT/HRASG12V, BJ-hTERT/myrAKT1, IMR90, SK-LMS-1, SK-LMS-1HDAC4-/-HDAC4PAM-ER, were previously described [8]. Parental BJ, IMR90 and SK-LMS-1 have been authenticated by STR DNA Profiling (MWG Eurofins, Germany). BJ-TERT fibroblasts were grown in Earle’s salts minimal essential medium (Euroclone, Milan, Italy) completed with nonessential amino acids (Sigma-Aldrich St. Louis, MO, USA). IMR90 and SK-LMS-1 cells were cultured in Dulbecco modified Eagle medium (Euroclone, Milan, Italy). All mediums were supplemented with 10% of fetal bovine (FBS), L-glutamine 2 mM; penicillin 100 U/mL, and streptomycin, 100 μg/mL; (Euroclone). All cell lines were routinely tested from Mycoplasma contamination.
Chemical compounds and antibodies
The following chemicals were used: 4-hydroxytamoxifen (4OHT), Doxycycline, Trypan Blue, Aphidicolin, Doxorubicin, BrdU (Sigma-Aldrich, Germany), Camptothecin (Enzo Life Sciences Farmingdale, NY USA), poly (I:C)-HMW/-LMW (InvivoGen, Toulouse, France). Antibodies used are provided in Table S5.
Immunofluorescence and immunoblotting
For intracellular staining of dsRNA, cells were fixed with 4% paraformaldehyde and permeabilized for 5′ with 0.5% Triton X-100. ICC blocking solution, 3% w/v BSA (Sigma-Aldrich), 3% v/v goat serum (Abcam, Cambridge UK), 0.02% v/v Tween-20 in PBS, was applied for 1 h. Next, incubation with 250 ng J2 antibody in blocking solution was performed. The secondary antibody was Alexa Fluor 546-conjugated (Thermo Fisher Scientific, Waltham, MA USA). Nuclei were stained with Hoechst 33258 (Sigma-Aldrich). Cells were imaged with a confocal microscope Leica AOBS SP8.
For S phase analysis, cells were grown for 3 h with 50 μM BrdU. Cells were fixed with 3% paraformaldehyde and permeabilized with 0.3% Triton X-100. After fixation, coverslips were treated with HCl (2%), quenched with Borate, and processed for immunofluorescence.
Cell lysates after SDS-PAGE and immunoblotting on nitrocellulose were incubated with primary antibodies. HPR-conjugated secondary antibodies were obtained from Cell Signaling Technology (Danvers, Massachusetts, USA) and blots were developed with Super Signal West Dura (Thermo Fisher Scientific).
dsRNA immunoprecipitation
Total RNA was extracted with Tri Reagent (Molecular Research Center, Cincinnati, OH, USA) from a pellet of 35*106 SKLMS-1 HDAC4 KO cells. IP was performed O/N in Polysomal Lysis Buffer in the presence of 10 μg of J2 antibody. 50 μl slurry protein A (GE Healthcare Chicago, Il, USA) was added and incubated in continuous rotation at 4 °C for 4 h. After 5 washes, RNA was recovered by phenolchloroform extraction/ethanol precipitation and resuspended in 20 μl RNase-free water. The precipitation was repeated until a final amount of 1.6 μg of purified RNA was reached. 800 ng were treated for 20’ at 37 °C with 50U ShortCut RNAse III (NEB, Ipswich, MA, USA), in the digestion buffer supplied by the manufacturer. The digestion was stopped with EDTA. The remaining 800 ng were treated in the same manner but without the addition of RNAse III. 30 pmoles of purified dsRNA treated or not with RNAse III were transfected in recipient cells, by using 15 μl of Lipofectamine 3000 and 250 μl Optimem (Thermo Fisher Scientific, USA).
dsRNA-seq and data analysis
Total RNA was extracted with Tri Reagent (Molecular Research Center) from a pellet of 35*106 SK-LMS-1HDAC4-/-/HDAC4PAM-ER cells grown for 96 h with or without 4OHT. Three biological replicates were harvested and subjected to two independent IP. IP was performed O/N in Polysomal Lysis Buffer in the presence of 3 μg of J2 antibody (Scicons, Hungary). The immune complexes were purified by incubating (O/N) each sample with 16 μl of magnetic Dynabeads coated with protein A/G (ThermoFisher). After washing the collected immune complexes three times, RNA was recovered by phenol chloroform extraction/ethanol precipitation and resuspended in 10 μl RNase-free water (Ambion, USA). Concentration, integrity and purity of RNA were checked using Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit). 10 ng of RNA was digested with DNAseI (NEB), freed from rRNA (using the ribo-zero method) and reverse transcribed to obtain cDNA. Adapters were ligated to the fragmented cDNA after end repair and A-tailing according to the DNBSEQ protocol (MGI-Tech). 18 rounds of PCR amplification were performed to enrich the cDNA fragments. The PCR products were then purified using Ampure XP beads (Agencourt). ssCirDNA was obtained after denaturation and circularization. DNA spheres were generated from each library according to BGI specifications (SOP-SS-025) and subjected to paired-end 100 sequencing at the BGI Genomics facility (BGI Genomics, China). The quality of the reads was assessed using fastqc. The expression levels of different consensus sequences of transposable elements (TEs) were calculated using TEspeX [60]. Reads were mapped to a reference transcriptome using STAR, which consists of TE consensus sequences downloaded from Dfam and the Ensembl transcript sequences containing all coding and non-coding annotated transcripts [61]. To limit the effects of TE fragments embedded in canonical coding and noncoding non-TEs transcripts and to allow quantification of autonomously expressed TEs, reads mapped to TE fragments embedded in coding and/or long noncoding transcripts were discarded. Finally, the number of reads in each TE consensus in each sample was counted to create a consensus sample count matrix. PCA was performed on variance-stabilized transformed read counts by using the varianceStabilizingTransformation function as implemented in the DESeq2 R package [62].
Differentially enriched TE consensus sequences were identified with the edgeR package using the workflow developed by Gao and colleagues [63]. Briefly, the number of reads mapped into each consensus was normalized using the TMM method implemented in edgeR. Then, the dsRIP samples were compared with the corresponding INPUT samples to identify TE in the dsRNA conformation using the generalized linear models (glmQLFTest function) implemented in edgeR with the following formula “(dsRIP+4OHT + dsRIP-4OHT) - (INPUT + 4OHT + INPUT-4OHT)”. Significant results were then identified as TE consensus with a p < 0.30 and a |log2FoldChange dsRIP / INPUT | > 0.585.
To identify dsRNA conformational changes between the two tested conditions by normalizing for INPUT samples, we then fitted a generalized linear model, as implemented in the edgeR glmQLFTest function, with the following formula “(dsRIP+4OHT + dsRIP-4OHT) - (INPUT + 4OHT + INPUT-4OHT)”. dsRNA-enriched TEs families were selected as families with a p < 0.05, while enriched entities in the ds RIP-4OHT sample were identified with respect to the INPUT + 4OHT sample with a cut-off of |log2FoldChange dsRIP+4OHT / INPUT-4OHT | > 0.585 and |log2FoldChange dsRIP+4OHT /dsRIP-4OHT | > 0.585.
SA-β-gal assay
Cells were fixed for 5′(PBS 2% formaldehyde/0.2% glutaraldehyde), washed twice and stained for 16 h at 37 °C with staining solution: 40 mM citric acid/Na phosphate buffer, 5 mM K4[Fe(CN)6]3H2O, 5 mM K3[Fe(CN)6], 150 mM sodium chloride, 2 mM magnesium chloride and 1 mg/mL X-gal (PanReac Applichem, Darmstadt, Germany). Images were acquired with Leica LD bright field optical microscope.
RNA extraction and quantitative qRT-PCR
A total of 1 μg of total RNA was retrotranscribed using 100 U of M-MLV reverse transcriptase (Invitrogen). qRT-PCR analyses were performed using Bio-Rad CFX96 and SYBR green technology (Sigma-Aldrich). The data were analysed by a comparative threshold cycle using the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase 1 (HPRT) as normalizer genes. Oligonucleotides are described in Fig. S6 and Table S6.
H3K27me3 ChIP-seq experiments and data analysis
ChIP-seq were performed previously [8]. We used the FastQC (v0.11.8) and MultiQC (v1.7) programs to evaluate the quality of the sequencing reads. Bowtie 2 (v2.3.4.3) was used to align reads to the NCBI GRCh38 human genome reference. We performed peak calling of the H3K27me3 and H3K27ac histone marks against inputs using MACS2 (v2.1.2 - broad option, other options as default). Peak heatmaps and profiles were generated using the deepTools (v3.1.3) suite and the gplots (v3.0.1.1), biomaRt (v2.38.0) and Gviz (v 1.24.0) packages, working with R/Bioconductor as previously described [64]. We identified regions showing a consistent variation of the H3K27me3 mark, using Fc > 2 as a cutoff; overlaps of at least one nucleotide between these regions and ERVs were found using the bedtools (v2.28.0) toolset [8].
Infections, transfections and silencing
Retroviral infections were performed as previously described [65]. For poly (I:C) and RNAi oligos transfections (Table S6), 24 h after plating cells were incubated with OptiMem medium containing Lipofectamine (Thermo Fisher Scientific).
Statistics
For experimental data, Student t-test was employed. Mann–Whitney test was applied when normality could not be assumed. For comparisons between more than two samples, Anova test was applied coupled to Kruskal–Wallis and Dunn’s Multiple Comparison Test. Excel and GraphPad Prism were used for statistical analysis. We marked with *p < 0.05, **p < 0.01, ***p < 0.001.
Data availability
Raw data corresponding to ChIP-seq experiments are uploaded with GEO accession GSE149644. Raw data corresponding to dsRNA-seq are uploaded with GEO accession GSE250348, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE250348. Processed data are available as supplementary tables.
References
Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–53.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46.
Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594:100–5.
Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446.
Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–83.
Paluvai H, Di Giorgio E, Brancolini C. The histone code of senescence. Cells. 2020;9:466–84.
Martínez-Zamudio RI, Roux PF, de Freitas JANLF, Robinson L, Doré G, Sun B, et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nat Cell Biol. 2020;22:842–55.
Di Giorgio E, Paluvai H, Dalla E, Ranzino L, Renzini A, Moresi V, et al. HDAC4 degradation during senescence unleashes an epigenetic program driven by AP-1/p300 at selected enhancers and super-enhancers. Genome Biol. 2021;22:129–54.
Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6:612–29.
Cardelli M. The epigenetic alterations of endogenous retroelements in aging. Mech Ageing Dev. 2018;174:30–46.
Geis FK, Goff SP. Silencing and transcriptional regulation of endogenous retroviruses: an overview. Viruses. 2020;12:884.
Grandi N, Tramontano E. Human endogenous retroviruses are ancient acquired elements still shaping innate immune responses. Front Immunol. 2018;9:2039.
Nevalainen T, Autio A, Mishra BH, Marttila S, Jylhä M, Hurme M. Aging-associated patterns in the expression of human endogenous retroviruses. PLoS One. 2018;13:e0207407.
Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genom. 2010;41:194–200.
Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284–300.
White CH, Beliakova-Bethell N, Lada SM, Breen MS, Hurst TP, Spina CA, et al. Transcriptional modulation of human endogenous retroviruses in primary CD4+ T cells following vorinostat treatment. Front Immunol. 2018;9:603.
Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell. 2015;163:230–45.
Place RF, Noonan EJ, Giardina C. HDACs and the senescent phenotype of WI-38 cells. BMC Cell Biol. 2005;6:37.
Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–97.
Pegoraro G, Kubben N, Wickert U, Göhler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;10:1261–7.
Xu CL, Sang B, Liu GZ, Li JM, Zhang XD, Liu LX, et al. SENEBLOC, a long non-coding RNA suppresses senescence via p53-dependent and independent mechanisms. Nucleic Acids Res. 2020;48:3089–102.
Warnon C, Bouhjar K, Ninane N, Verhoyen M, Fattaccioli A, Fransolet M, et al. HDAC2 and 7 down-regulation induces senescence in dermal fibroblasts. Aging (Albany NY). 2021;13:17978–8005.
Colombo AR, Elias HK, Ramsingh G. Senescence induction universally activates transposable element expression. Cell Cycle. 2018;14:1846–57.
Yu YC, Hui TZ, Kao TH, Liao HF, Yang CY, Hou CC, et al. Transient DNMT3L expression reinforces chromatin surveillance to halt senescence progression in mouse embryonic fibroblast. Front Cell Dev Biol. 2020;8:103.
Min B, Jeon K, Park JS, Kang YK. Demethylation and derepression of genomic retroelements in the skeletal muscles of aged mice. Aging Cell. 2019;6:e13042.
Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019;4:697–709.
Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;6277:1083–7.
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–73.
Izquierdo-Bouldstridge A, Bustillos A, Bonet-Costa C, Aribau-Miralbés P, García-Gomis D, Dabad M, et al. Histone H1 depletion triggers an interferon response in cancer cells via activation of heterochromatic repeats. Nucleic Acids Res. 2017;45:11622–42.
Schonborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991;19:2993–3000.
Cutano V, Di Giorgio E, Minisini M, Picco R, Dalla E, Brancolini C. HDAC7-mediated control of tumour microenvironment maintains proliferative and stemness competence of human mammary epithelial cells. Mol Oncol. 2019;13:1651–68.
Cowell JK, LaDuca J, Rossi MR, Burkhardt T, Nowak NJ, Matsui S. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet Cytogenet. 2005;163:23–9.
Liu S, Brind’Amour J, Karimi MM, Shirane K, Bogutz A, Lefebvre L, et al. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014;28:2041–55.
Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. 2016;64:1074–87.
Li P, Wang L, Bennett BD, Wang J, Li J, Qin Y, et al. Rif1 promotes a repressive chromatin state to safeguard against endogenous retrovirus activation. Nucleic Acids Res. 2017;45:12723–38.
Lee AK, Pan D, Bao X, Hu M, Li F, Li CY. Endogenous retrovirus activation as a key mechanism of anti-tumor immune response in radiotherapy. Radiat Res. 2020;193:305–17.
d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22.
Jansz N, Faulkner GJ. Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 2021;22:147.
Di Giorgio E, Xodo LE. Endogenous retroviruses (ERVs): does RLR (RIG-I-Like Receptors)-MAVS pathway directly control senescence and aging as a consequence of ERV de-repression? Front Immunol. 2022;13:917998.
Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–40.
Chen Y, Shi Y, Wu J, Qi N. MAVS: a two-sided CARD mediating antiviral innate immune signaling and regulating immune homeostasis. Front Microbiol. 2021;12:744348.
Frisch SM, MacFawn IP. Type I interferons and related pathways in cell senescence. Aging Cell. 2020;19:e13234.
Porath HT, Knisbacher BA, Eisenberg E, Levanon EY. Massive A-to-I RNA editing is common across the metazoa and correlates with dsRNA abundance. Genome Biol. 2017;18:185.
Liu X, Liu Z, Wu Z, Ren J, Fan Y, Sun L, et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell. 2023;186:287–304.
Vargiu L, Rodriguez-Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, et al. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology. 2016;13:7.
Qi N, Shi Y, Zhang R, Zhu W, Yuan B, Li X, et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun. 2017;8:15676.
Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–51.
Wang Z, Fan R, Russo A, Cernilogar FM, Nuber A, Schirge S, et al. Dominant role of DNA methylation over H3K9me3 for IAP silencing in endoderm. Nat Commun. 2022;13:5447.
Stolz P, Mantero AS, Tvardovskiy A, Ugur E, Wange LE, Mulholland CB, et al. TET1 regulates gene expression and repression of endogenous retroviruses independent of DNA demethylation. Nucleic Acids Res. 2022;50:8491–511.
Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev. 2018;174:3–17.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–86.
Ireton RC, Gale M Jr. RIG-I like receptors in antiviral immunity and therapeutic applications. Viruses. 2011;3:906–19.
Mao J, Zhang Q, Wang Y, Zhuang Y, Xu L, Ma X, et al. TERT activates endogenous retroviruses to promote an immunosuppressive tumour microenvironment. EMBO Rep. 2022;23:e52984.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5.
Chowdhury A, Witte S, Aich A. Role of mitochondrial nucleic acid sensing pathways in health and patho-physiology. Front Cell Dev Biol. 2022;10:796066.
Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, et al. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017;13:e1006720.
Pérez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14:547–58.
Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34:1565–76.
Hao X, Wang C, Zhang R. Chromatin basis of the senescence-associated secretory phenotype. Trends Cell Biol. 2022;32:513–26.
Ansaloni F, Gualandi N, Esposito M, Gustincich S, Sanges R. TEspeX: consensus-specific quantification of transposable element expression preventing biases from exonized fragments. Bioinformatics. 2022;38:4430–3.
Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016;44:D81–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Gao Y, Chen S, Halene S, Tebaldi T. Transcriptome-wide quantification of double-stranded RNAs in live mouse tissues by dsRIP-Seq. STAR Protoc 2021;2:100366.
Di Giorgio E, Dalla E, Franforte E, Paluvai H, Minisini M, Trevisanut M, et al. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020;48:646–64.
Di Giorgio E, Clocchiatti A, Piccinin S, Sgorbissa A, Viviani G, Peruzzo P, et al. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol Cell Biol. 2013;33:4473–91.
Acknowledgements
We thank Dr. Francesca D’Este for the support with the confocal microscopy.
Funding
The research leading to these results has received funding from AIRC under IG 2021 - ID. 26200 project – P.I. Brancolini Claudio and MFAG 2020- ID. 25000 to EDG.
Author information
Authors and Affiliations
Contributions
CB conceptualized and designed the research and analyzed the data; EDG, ED, LR, and NG analyzed data and provided statistical analysis; EDG, VT, LR, and MB provided acquisition, analysis and interpretation of data, and statistical analysis; CB performed microscopic studies. EDG and CB wrote the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Angelo Peschiaroli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Giorgio, E., Ranzino, L., Tolotto, V. et al. Transcription of endogenous retroviruses in senescent cells contributes to the accumulation of double-stranded RNAs that trigger an anti-viral response that reinforces senescence. Cell Death Dis 15, 157 (2024). https://doi.org/10.1038/s41419-024-06548-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06548-2
- Springer Nature Limited