
Abstract
Lung adenocarcinoma (LUAD) is most common pathological type of lung cancer. LUAD with brain metastases (BMs) usually have poor prognosis. To identify the potential genetic factors associated with BM, a genomic comparison for BM cerebrospinal fluid (CSF) and primary lung tumor samples obtained from 1082 early- and late-stage LUAD patients was performed. We found that single nucleotide variation (SNV) of EGFR was highly enriched in CSF (87% of samples). Compared with the other primary lung tissues, copy number gain of EGFR (27%), CDK4 (11%), PMS2 (11%), MET (10%), IL7R (8%), RICTOR (7%), FLT4 (5%), and FGFR4 (4%), and copy number loss of CDKN2A (28%) and CDKN2B (18%) were remarkably more frequent in CSF samples. CSF had significantly lower tumor mutation burden (TMB) level but more abundant copy number variant. It was also found that the relationships among co-occurrent and mutually exclusive genes were dynamically changing with LUAD development. Additionally, CSF (97% of samples) harbored more abundant targeted drugs related driver and fusion genes. The signature 15 associated with defective DNA mismatch repair (dMMR) was only identified in the CSF group. Cancer associated pathway analysis further revealed that ErbB (95%) and cell cycle (84%) were unique pathways in CSF samples. The tumor evolution analysis showed that CSF carried significantly fewer clusters, but subclonal proportion of EGFR was remarkably increased with tumor progression. Collectively, CSF sequencing showed unique genomic characteristics and the intense copy number instability associated with cell cycle disorder and dMMR might be the crucial genetic factors in BM of LUAD.
Similar content being viewed by others
Introduction
Lung adenocarcinoma (LUAD) is the most common histologic subtype in non-small cell lung cancers (NSCLC) and accounts for more than 38.5% of all lung cancers [1]. Brain metastasis (BM) represents an important cause of morbidity and mortality and is associated with poor prognosis [2]. BMs in lung cancer patients (20–56%) are the most commonly arising compared with other tumor types [3]. About 30% of LUAD patients are most likely to suffer from BM at the time of diagnosis, and 50% will eventually develop BMs [4]. The risk of BMs will be increased with increasing tumor grade [5], which has a negative impact on the life quality of patients with LUAD.
Cerebrospinal fluid (CSF), containing cell-free DNA (cfDNA), had been considered as a vital liquid biopsy medium for lung cancer, which provides a less-invasive and routinely accessible method to dynamically acquire genomic information of BM patients in lung cancer [6]. Previous studies had revealed CSF circulating tumor DNA (ctDNA) was more representative of brain tumor genomic alterations than plasma, and could detect brain tumor private mutations and monitor brain tumor progression [7,8,9]. Besides, CSF cfDNA could enhance the diagnostic validity for EGFR genotyping of LUAD patients with BM [10], and reveal frequent occurrence of uncommon EGFR mutations (G719A, L861Q, L703P, and G575R) in patients with leptomeningeal metastasis (LM, 54.5%) than brain parenchymal metastasis (BPM, 10%) [11]. Similarly, Ma et al. discovered that the mutation ratio of EGFR in LM (81.8%) was higher than BPM (30%) in NSCLC patients with BM, and the status of EGFR mutation was consistent between CSF ctDNA and brain lesion tissue in five patients after surgical resection [12]. Additionally, CSF DNA genotyping was associated with survival outcomes among LUAD patients with central nervous system (CNS) metastasis [13] and Osimertinib response of LM in EGFR-mutated NSCLC [14]. These studies suggested that CSF could more accurately reflect the genomic mutations of brain lesions in lung cancer patients, thereby providing targets for treatment of BM.
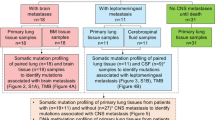
Although amounts of genes in CSF had been identified in previous researches, these studies have a limited the number of samples, and mainly focus on exploring the difference between BM lesions and primary lung tumor in advanced NSCLC, lacking of early-stage genomic comparison. Therefore, a systematic analysis of a large sample size was needed to further explore genetic alterations in the development of lung cancer. In this study, a 1021 cancer-related panel was used to detect gene mutations of 1082 unmatched samples from Chinese LUAD patients, including 135 CSF with BM (CSF group), 363 early-stage lung tumors (ESLT group), 396 late-stage lung tumors without BM (LSLT-noBM group), and 188 late-stage lung tumors with BM (LSLT-BM group) samples. To seek risk factors associated with metastasis by comparing genetic profiles between CSF and different stage primary lung tumors, thereby providing potential prognostic markers and therapeutic targets for LUAD with BMs, finally explaining the development of LUAD from a genetic perspective.
Results
Tumor mutation burden and copy number variation instability of the CSF and primary lung tumors
A 1021 panel sequencing was performed on CSF, ESLT, LSLT-noBM, and LSLT-BM groups with the average depth of 1583, 1230, 1271, and 1254×, respectively. Tumor mutation burden (TMB) and somatic copy number variation (CNV) (including amplification and deletion) count were assessed. The median TMB of CSF was 4 mutations/Mb, which was remarkably lower than the other groups (all with a median of 6 mutations/Mb) (Fig. 1A). The median CNV count of CSF was 4, while the median CNV count in ESLT, LSLT-noBM, and LSLT-BM groups were significantly reduced (2, 1, and 2 CNVs, respectively) (Fig. 1B).

A, B Show the difference of TMB and CNV between CSF and ESLT, LSLT-noBM, LSLT-BM groups, respectively. C, D Show the difference of TMB and CNV in EGFR-CSF and EGFR-ESLT, EGFR-LSLT-noBM, EGFR-LSLT-BM subgroups, respectively. Statistical analysis was performed using the Mann–Whitney test. *P < 0.05, ***P < 0.001.
We discovered EGFR was the most prevalent gene in all groups, accounting for 87% in CSF, 51% in ESLT, 64% in LSLT-noBM, and 67% in the LSLT-BM. Thus, the variation characteristics of EGFR mutant patients were further analyzed. The median TMB of EGFR mutant CSF (EGFR-CSF) and ESLT (EGFR-ESLT) samples was 5 mutations/Mb, which was lower than EGFR-LSLT-noBM (6 mutations/Mb) and EGFR-LSLT-BM samples (6 mutations/Mb) (Fig. 1C). CNV analysis showed that the median count of EGFR-CSF subgroup was significantly higher than the other three subgroups (Fig. 1D). These accumulated results suggested that CSF had a lower TMB level but higher CNV instability.
Single nucleotide variation landscapes and the differences between CSF and other groups
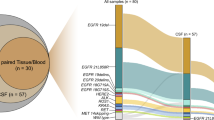
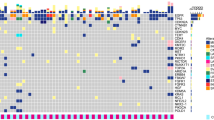
Genomic landscape of single nucleotide variation (SNV) for each group was shown in Fig. 2A. Whereafter, the comparison of mutation prevalence between CSF and the different stages lung tumors revealed that CSF harbored more prevalent EGFR, TP53, and CTNNB1, and fewer RBM10, KRAS, SMARCA4, KEAP1, FAT1, and STK11 than ESLT group. The significantly differential genes in CSF and LSLT-noBM groups were EGFR, CTNNB1, RBM10, KRAS, and FAT1. Gene EGFR, KRAS, KEAP1, FAT1, and STK11 were significantly different between CSF and LSLT-BM group. Moreover, EGFR was enriched and FAT1 was rare in CSF compared to the other three groups (Fig. 2B). Additionally, the incidence of CTNNB1 and EGFR co-mutation in the CSF group was significantly higher than that in the ESLT (11.1% vs 3.6%, P = 0.0033), LSLT-noBM (11.1% vs 4.5%, P = 0.0117), and LSLT-BM group (11.1% vs 5.3%, P = 0.0603), suggesting that the co-mutation of EGFR and CTNNB1 might be associated with BM event.

A Driver gene mutation profiles of the CSF, ESLT, LSLT-noBM, and LSLT-BM groups. Mutation frequencies in the group are shown on the left. Mutation burden (number of mutations per Mb) for each patient is shown at the top. B Comparison of mutation frequencies of driver genes between CSF and the other three groups, respectively. Significant differences of genes were calculated by two-sided Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001.
We also examined the mutation landscape in EGFR-mutant patients (Fig. S1A). The results showed that the mutation frequency of RBM10 was significantly lower in EGFR-CSF subgroup than EGFR-ESLT subgroup, while the mutation frequency of TP53 and PTCH1 in EGFR-CSF subgroup was remarkably higher than EGFR-ESLT subgroup. FAT1 was the significantly differential gene between EGFR-CSF and EGFR-LSLT-BM subgroups, while EGFR-CSF and EGFR-LSLT-noBM groups had no significantly differential genes (Fig. S1B). TP53 had an equivalent incidence in patients with advanced tumors, higher than early-stage, indicating the potential association with distant metastasis. In summary, rare gene differences were identified between EGFR CSF and other corresponding subgroups.
Co-occurrence and mutually exclusivity analyses among mutant genes showed that EGFR and STK11/KEAP1/KRAS were remarkably mutually exclusive genes shared by all lung tissue samples (Fig. 3A). Nevertheless, the gene pairs only co-occurred in CSF group included LRP1B-KDM5A (2.2%), PIK3CA-MLL (1.5%), APC-NF1 (2.2%), APC-BRCA2 (2.2%), BRCA2-NF1 (1.5%), RB1-MLL3 (1.5%), CDKN2A-KRAS (0.7%), and PTCH1-KEAP1 (0.7%), reminding their possible relation with BM event. Genes that were specifically mutually exclusive in the ESLT group included EGFR and PTCH1/BRAC2/MLL2/CDKN2A, TP53 and CTNNB1/KRAS/RBM10, and KRAS and ERBB2. The mutual exclusion between TP53 and STK11 only appeared in the LSLT-noBM group. The mutually exclusive mutations that only exist in the LSLT-BM group are EGFR and ARID1A. The above results indicated that the relationships among genes were dynamically changing with LUAD development.

A Co-occurrence and mutually exclusivity analyses of somatic mutation genes in CSF and lung tissue. Fisher’s exact test was used to identify remarkable interactions. *P < 0.05. B The somatic mutation signature analysis. From left to right: the mutation distribution profile of tumor samples and the vertical axis represents the number of mutations for each triple nucleotide type, proportion of total somatic substitutions in four groups contributed by each of the operative mutational signatures, the heatmap distribution of signatures in all samples.
A somatic mutational signature analysis was performed to describe which internal boundary or external environmental factors in the development of LUAD BM represents the most important carcinogenic factors. Signature 1 (correlates with the age of cancer diagnosis), signature 3 (associated with failure of DNA double-strand break repair by homologous recombination), and signature 13 (attributed to the activity of AID/APOBEC family cytidine deaminases) were mutual signatures in both early and late stages LUAD. Notably, signatures 5 and 15 were only identified in the CSF group (Fig. 3B). Although signature 5 has been found in various cancer types, its aetiology is still unknown. Signature 15 is associated with defective DNA mismatch repair (dMMR), an aberrant DNA repair mechanism, contributing to frequent genomic alterations and genomic instability [15], indicating dMMR signature might participate in BM event.
Somatic CNV landscapes and differences between CSF and the other groups
Next, we analyzed CNV features (Fig. 4A, B). Fifty (23.8%) genes were private in CSF group, including amplification of FANCF (6%) and FLT4 (5%) and deletion of CD274 (4%). Besides, the deletion of CDKN2A and CDKN2B, and the amplification of EGFR, CDK4, PMS2, MET, IL7R, RICTOR, FGFR4 were the most frequently observed CNV events in CSF compared with the other groups. Except for the high-frequency genes in CSF mentioned above, the significantly differential genes in CSF and ESLT group also included SDHA, FOXA1, NKX2-1, IFNG, RB1, and AXIN1. The significantly differential genes in CSF and LSLT-noBM group also included SDHA, IFNG, RB1, and AXIN1, while the significantly differential genes between CSF and LSLT-BM group were same as high-frequency genes in CSF.

A High frequently mutated genes in CSF and primary lung tumors are shown. Mutation frequencies in the group are shown on the left. CNV counts (number of CNV events) for each patient is shown at the top. B Comparison of mutation frequencies of CNV genes between CSF and the other three groups, respectively. Significantly different genes were calculated by two-sided Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001.
CNVs of EGFR-mutated patients also were assessed. The high-frequency genes in the EGFR-CSF subgroup and identified differential genes among subgroups also were consistent with the results of total samples (Fig. S2A, B). Collectively, our results demonstrated that CSF derived from BM patients suffered a remarkedly higher level of genetic disturbance events of CNV.
Brain metastasis related pathways identified using CSF
Ten pathways with statistically significant (FDR < 0.1) were enriched in CFS, including PI3K-Akt, Rap1, FoxO, Ras, ErbB, cell cycle, JAK-STAT, p53, mTOR, and AMPK pathways. Among them, only ErbB, cell cycle, JAK-STAT, mTOR, and AMPK pathways were remarkably enrolled in the CSF (Fig. 5A), suggesting these aberrant signal pathways might be associated with an increased BM risk. Meanwhile, we performed a significant analysis of alterative frequency in the pathway between CSF and the other three groups (Fig. 5B). The remarkably different pathways in CSF and ESLT groups also included Rap1, CSF vs. LSLT-noBM groups were FoxO and Ras, CSF and LSLT-BM groups were FoxO.

A Remarkable enrichment of KEGG pathways in each group according to FDR < 0.1. The vertical axis indicates gene count. Gene count means the number of mutated genes enriched in one term. B Heatmap of alteration frequency of identified pathways. The horizontal axis represents signal pathways, and the figures indicate mutation percentages. Differences with significant P values are labeled (two-sided Fisher’s exact test). **P < 0.01, ***P < 0.001. C Gene alterations (including SNV and CNV) of highly frequent and unique pathways in CSF samples. The vertical axis shows gene mutation frequency.
Next, we analyzed gene alterations of ErbB and cell cycle pathways that were highly frequent and unique in CSF samples by integrating SNVs and CNVs. It was found that the somatic mutation of TP53, the copy number loss of CDKN2A and CDKN2B, and the copy number gain of CDK4 and MDM2 played major roles in cell cycle pathway, and the somatic mutations of EGFR and PIK3CA and the amplification of EGFR, ERBB2, and MYC were the main factors affecting ErbB pathway (Fig. 5C).
Genotyping of the targeted drugs related diver genes
We examined the driver genes including EGFR, PIK3CA, BRAF, ERBB2, KRAS, and MET, as well as gene rearrangements such as ALK, RET, ROS1, and NTRK fusions. As shown in Fig. 6, these genes accounted for 97% in the CSF group, 83% in the ESLT group, 90% in the LSLT-noBM group, and 93% in the LSLT-BM group. CSF group had significantly higher druggable driver proportion than ESLT (P < 0.001) and LSLT-noBM group (P = 0.007), excluding LSLT-BM (P = 0.091) group. Furthermore, EGFR L858R, T790M, exon 19 deletion (19del), C797S, exon 20 insertion, and L861Q were identified in all groups. Besides, other rare EGFR mutations L62R, L718X, and V834L also were identified in CSF and lung tissue samples, but only L792H, G873E, and H850Y mutated in CSF. PIK3CA, ERBB2, and KRAS were similar in all groups. These results showed that CSF could be used to discover actionable drug-targets in driver genes.

The mutation landscape of several actionable driver alterations and gene rearrangements in lung cancer is shown at the top. Pie charts at the bottom indicate the proportion of these druggable genes.
Clonal architecture of mutations in CSF
PyClone analysis was performed to explore the mutation clonality in the four groups. We identified 704 mutation clusters in the CSF group with a median of 4 per sample (range, 1–16), 2952 mutation clusters in the ESLT group with a median of 6 per sample (range, 1–88), 3091 mutation clusters in the LSLT-noBM group with a median of 6 per sample (range, 1–215), and 1615 mutation clusters in LSLT-BM group with a median of 6 per sample (range, 1–122). CSF carried significantly fewer clusters than ESLT (P = 0.0002), LSLT-noBM (P < 0.0001), and LSLT-BM (P < 0.0001) group (Fig. 7A). Further analysis revealed that the difference in total clonal mutation burden was mainly due to more subclonal mutations rather than clonal mutations (Fig. 7B, C).

A–C Comparison of total clonal mutations burden (including clonal and subclonal), clonal mutation burden, and subclonal mutation burden in all somatic genes for CSF and the other three groups. Clonal mutations burden means the number of mutation clusters in each sample. Differences with significant P values are labeled (Mann–Whitney test). **P < 0.01, ***P < 0.001. D The clonal and subclonal proportion of EGFR and EGFR genotyping (L858R, T790M, and 19del) in CSF and primary lung tissue. The differences of these clonal distribution were estimated by two-sided Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001.
Clonality of the high-frequency genes among groups was further analyzed, in which the subclonal proportion of EGFR was increased in CSF. Further, the subclonal fraction of EGFR L858R and 19del in CSF was significantly higher than the other three groups, while subclonal EGFR T790M had no difference (Fig. 7D), suggesting the changes in the distribution of the main genotyping subclones might be one of the reasons for the increase in EGFR subclones. Besides, the samples with subclonal EGFR L858R and 19del were characterized by the frequently clonal TP53.
Discussion
In the current study, we analyzed genomic profile difference of Chinese LUAD patients between CSF samples and other primary lung tissues from different disease stages, and proved the unique molecular characteristics of CSF samples from multiple aspects, such as SNV, CNV, signature, clonality and pathway, and revealed CSF cfDNA was an important medium to expound the molecular features of BM LUAD patients, which provided potential prognostic markers and therapeutic targets for LUAD with BMs.
SNVs analysis showed that EGFR had a significantly higher mutation frequency in CSF, followed by the LSLT-BM, LSLT-noBM, and ESLT group, which might be contributed by a higher incidence of BMs for patients with EGFR mutation NSCLC [16]. Furthermore, mutation frequency of KRAS in the CSF group only was 0.7%, which was the lowest in all samples. This was owing to the mutual exclusivity of oncogenic KRAS and EGFR mutations in LUAD [17, 18]. Herein, CTNNB1 was highly mutated in CSF. According to reports, CTNNB1 belongs to Wnt signal pathway and is mutated in patients of lung metastasis to brain, but few or no mutations were described in original brain tumor specimens [19]. Combining with our data, CTNNB1 was indeed required for lung cancer with BM. Another study found that CTNNB1 mutations were rare in early-stage EGFR-mutant LUAD, but increased in late-stage tumors [20]. In our results, the number of EGFR and CTNNB1 co-mutation in CSF samples was significantly more than that in ESLT and LSLT-noBM lung tissue samples, but not LSLT-BM samples, indicating that the more overlap of EGFR and CTNNB1 was related to BM in LUAD.
Somatic mutations and copy number alterations possessed intratumor heterogeneity, and genome doubling and continuous dynamic chromosomal instability were related to intratumor heterogeneity, leading to parallel evolution of driver somatic copy-number alterations [21]. In our study, more abundant CNVs were observed in CSF cfDNA, suggesting that CSF had stronger genome instability, which was in accordance with the hypothesis that large-scale genomic alterations, such as copy number changes, is often evident at metastatic sites [22], and genome instability drives tumor progression and metastasis [23]. On contrast, minimum TMB was found in CSF. This was owing to highly frequent EGFR in CSF, which had been verified by a previous study that lung cancer patients with EGFR mutations had significantly lower TMB values than those with wild-type EGFR [24]. Also, the difference of somatic mutation genes among groups much less than that of copy number alteration genes, thus we considered that CNVs of CSF might play more important roles in the evolution of LUAD compared with SNVs. Besides, the frequency of NSCLC gene alterations in CSF was much higher than other groups, including CDKN2A/2B deletions, and PMS2, MET, and CDK4 amplification. Among these observations, it is known that CDKN2A/2B and CDK4, which are involved in cell cycle pathway, were high frequency mutated genes in BM tissue samples, and indicated shortened intracranial progression-free survival in CSF [4, 14, 25]. PMS2 is related to DNA mismatch repair and might accelerate tumorigenesis [26], and MET copy number gains are associated with EGFR tyrosine kinase inhibitors (TKIs) resistance in NSCLC with BMs [27]. Moreover, the amplification of FANCF and FLT4 and the deletion of CD274 only were identified in CSF samples. Studies had shown that the overexpression of FANCF and FLT4 resulted in proliferation, migration, and invasion of cancer cells [28, 29]. Deletion of CD274 was most prevalent and frequent in NSCLC, which influenced its expression levels, was associated with dismal prognosis [30]. In general, the above CNVs genes might act in promoting LUAD BMs.
Somatic mutations found that cancer genomes may be the consequence of the intrinsic slight infidelity of the DNA replication machinery, exogenous or endogenous mutagen exposures, enzymatic modification of DNA, or defective DNA repair [31]. Furthermore, the molecular mechanisms underlying genomic instability are related to processes that preserve genetic information, namely cell cycle checkpoints, DNA repair, transcription, replication, epigenetic control, chromatin remodeling, and chromosome segregation during mitosis [15]. Most of the somatic mutations in tumors are induced by exposure, and various predisposing factors have different characteristics in the mutation profile. Different mutational processes often generate different combinations of mutation types, termed “signatures” [32]. In this study, signature 5 and 15 only were detected in CSF, in which signature 5 primarily features C > T and T > C transitions, but its proposed aetiology is currently unknown [33]. A study found that signature 5 is associated with DNA repair gene ERCC2 mutations in urothelial tumors [34], which may be used for reference to lung cancer research. Signature 15 is associated with dMMR, resulting in genomic instability. Additionally, cell cycle and ErbB pathways also only were frequently enriched in CSF. cell cycle was the one of molecular mechanism contributing to genomic instability as mentioned earlier. As a key pathway of angiogenesis, the ErbB signaling pathway has been implicated in the development of NSCLC patients with BMs as evidenced by overexpression and higher activity than in corresponding primary tumors [35]. Collectively, we suggested that CSF did have much higher genomic instability. dMMR, the alterations of cell cycle and ErbB pathways might be the reasons to induce LUAD BM.
At present, several studies had confirmed that EGFR targeted therapy dramatically improved the prognosis of patients with NSCLC BMs [36, 37]. However, concomitant genetic alterations (such as CDKN2A copy number loss) [38] and the number of EGFR subclones [39] are associated with worse clinical outcomes in EGFR mutant NSCLC patients under TKIs treatment. In our study, CDKN2A loss and EGFR subclone number were frequently increased in EGFR-CSF patients, which might be one of the reasons for the poor prognosis of LUAD patients with BM. In immune checkpoint inhibitors (ICIs) monotherapy, EGFR mutation patients did not respond better than chemotherapy [40, 41], and EGFR aberrations had been considered as a risk factor for hyper-progress in immunotherapy [42]. ICI efficacy against NSCLC patients with MET amplification seemed close to that observed in unselected patients [43]. Here, EGFR and MET generally occurred in CSF samples, suggesting patients with BMs might be more not suitable for immune monotherapy.
Genomic comparison of CSF and primary lung lesions at different stages also revealed several other possible therapeutical approaches for LUAD with BM. CDK4/CDK6 inhibitor abemaciclib had been verified to enhance radiosensitivity of NSCLC in vitro and in vivo [44]. CDK4 was remarkably amplified in CSF, supporting that the CDK4 inhibitors combined with radiation might be a promising therapeutic option for LUAD patients with BM. ICIs against programmed death-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) proteins were highly effective in advanced dMMR metastatic colorectal cancer, especially combination therapy provided improved efficacy relative to anti-PD-1 monotherapy [45, 46]. We discovered dMMR mutational signature in CSF of LUAD patients. This provided a potential target for ICI combination therapy. In addition, it has recently been reported that the infiltrating immune cells in CSF could predict the response of BM patients to ICI [47], and diver gene mutations were associated with ICI response [48]. Thus, exploring the effect of special gene alterations in CSF on immune cells might provide guidance for screening patients who benefit from immunotherapy for lung cancer brain metastasis.
To conclude, a multiple perspective analysis about molecular characteristics between CSF and different stages primary tumor tissues in LUAD patients revealed that CNV might be greater than the contribution of SNV to BM. ErbB pathway alterations, and the intense copy number instability associated with cell cycle disorder and dMMR might be the crucial genetic factors in BM of LUAD.
Materials and methods
Patients and sample collection
Genomic and clinical information of 1082 LUAD patients with or without BM were analyzed, including 135 (12%) IV stage CSF with BM patients (CSF group), 363 (34%) I–III stage lung tissue patients (ESLT group), 396 (37%) IV stage lung tissue without BM patients (LSLT-noBM group), and 188 (17%) IV stage lung tissue with BM patients (LSLT-BM group). The clinical characteristics and mutated EGFR prevalence of patients were summarized in Table 1. Ten milliliter CSF was obtained by lumbar puncture. Surgical tumors fixed in formalin and embedded in paraffin (FFPE) samples were collected for DNA extraction. Peripheral blood lymphocytes (PBLs) from each patient were collected for germline DNA sequencing. All patients provided informed consent. The present study was approved by Hunan Cancer Hospital IRB Committee.
DNA extraction
CSF cfDNA was isolated using MagMAXTM Cell-Free DNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA). Lung tumor tissue DNA was extracted using Maxwell® 16 FFPE Plus LEV DNA Purification Kit (Promega, Madison, WI, USA). PBL DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). DNA concentration was measured using a Qubit fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Invitrogen, Carlsbad, CA, USA).
Library construction and panel sequencing
Methods for sequencing libraries was previously described [49]. DNA extracted from PBL and FFPE specimen sheared to 300-bp fragments with a Covaris S2 ultrasonicator (Covaris, Woburn, MA, USA). CSF DNA and fragmented PBL and tissue DNA (1.0 μg input) was added to Illumina-indexed adapters for library construction using the KAPA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA). Custom-designed probes, which covered 1.0 Mb regions for 1021 cancer-related genes were used for DNA capture [50]. Sequencing was performed on the HiSeq3000 Sequencing System (llumina, USA) with 2 × 75 bp paired-end reads.
Sequence analysis used BWA [51] (version 0.7.12-18r1039) to align clean reads to the reference human genome (hg19). SNVs and small Indels were identified by MuTect (version 1.1.4) [52]. Somatic mutations were identified by a variant allele fraction (VAF) ≥ 1.0% and at least 5 high quality reads (Phred score ≥30, mapping quality ≥30, and without paired-end reads bias). Mutations were annotated with genes using the ANNOVAR software [53]. CONTRA was used to detect CNV [54]. BreakDancer was used to detect cancer-associated gene fusion [55].
TMB and CNV count analyses
CSF-based and tissue-based TMB were defined as the total number of non-synonymous SNVs and Indels standardized by the 1.0 Mb coding region. CNV count was defined as the total number of CNV events per sample.
Subclonal analysis
Pyclone, a Bayesian clustering method, was employed to estimate the subclonal architecture of all mutations from CSF and primary tumor tissues. The SNV of each sample and its copy number information are used as the input of PyClone analysis, and the cellular prevalence was inferred and variants were clustered as previously described [56]. PyClone was run with 20,000 iterations and default parameters. Variants located in the cluster with greatest cancer cellular prevalence (CCF) mean were defined as clonal, the rest were subclonal.
Signature analysis
DeconstructSigs package (version 1.8.0) was used to identify mutational signatures within a single tumor sample based on a negative matrix factorization (NMF) algorithm [57], which relies on the Bioconductor library BSgenome.Hsapiens.UCSC.hg19 to obtain mutational context information. The unique combination of mutation types in CSF and lung tissue samples were constructed, and the mutational process was generated by COSMIC mutational signatures (version 2.0).
Pathway analysis
An online analysis tool DAVID (https://david.ncifcrf.gov/tools.jsp) was performed to identify significant gene clusters from SNVs and CNVs via annotating KEGG pathways, and 1021 cancer-related genes was considered as background gene set. The mutational frequency of pathways was obtained by computing the fraction of samples with at least one alteration in the corresponding pathway [58].
Statistical analysis
Data were analyzed using Prism 6.0 (Graph Pad Software Inc., La Jolla, CA). The Fisher’s exact test was used to compare proportions between two groups. Mann–Whitney test was used for CNV count of per sample and TMB comparison among different groups. All statistical tests were two-sided, and the result with P < 0.05 was considered as statistically significant.
Data and materials availability
The datasets supporting the conclusions of this article are available from the corresponding author on reasonable request.
References
Skřičková J, Kadlec B, Venclíček O, Merta Z. Lung cancer. Cas Lek Ceskych. 2018;157:226–36.
Patel RR, Mehta MP. Targeted therapy for brain metastases: improving the therapeutic ratio. Clin Cancer Res. 2007;13:1675–83.
Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, et al. Brain metastases. Nat Rev Dis Prim. 2019;5:5.
Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti E, et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet. 2020;52:371–7.
Consonni D, Pierobon M, Gail MH, Rubagotti M, Rotunno M, Goldstein A, et al. Lung cancer prognosis before and after recurrence in a population-based setting. J Natl Cancer Inst. 2015;107:djv059.
Offin M, Chabon JJ, Razavi P, Isbell JM, Rudin CM, Diehn M, et al. Capturing genomic evolution of lung cancers through liquid biopsy for circulating tumor DNA. J Oncol. 2017;2017:4517834.
De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015;6:8839.
Ying S, Ke H, Ding Y, Liu Y, Tang X, Yang D, et al. Unique genomic profiles obtained from cerebrospinal fluid cell-free DNA of non-small cell lung cancer patients with leptomeningeal metastases. Cancer Biol Ther. 2018;20:562–70.
Li YS, Jiang BY, Yang JJ, Zhang XC, Zhang Z, Ye JY, et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann Oncol. 2018;29:945–52.
Huang R, Xu X, Li D, Chen K, Zhan Q, Ge M, et al. Digital PCR-based detection of EGFR mutations in paired plasma and CSF samples of lung adenocarcinoma patients with central nervous system metastases. Target Oncol. 2019;14:343–50.
Ma C, Zhang J, Tang D, Ye X, Li J, Mu N, et al. Tyrosine kinase inhibitors could be effective against non-small cell Lung cancer brain metastases harboring uncommon EGFR mutations. Front Oncol. 2020;10:224.
Ma C, Yang X, Xing W, Yu H, Si T, Guo Z. Detection of circulating tumor DNA from non‐small cell lung cancer brain metastasis in cerebrospinal fluid samples. Thoracic. Cancer. 2020;11:588–93.
Wang S, Hu C, Xie F, Liu Y. Use of programmed death receptor-1 and/or programmed death ligand 1 Inhibitors for the Treatment of brain metastasis of lung cancer. Onco Targets Ther. 2020;13:667–83.
Zheng MM, Li YS, Tu HY, Jiang BY, Yang JJ, Zhou Q, et al. Genotyping of cerebrospinal fluid associated with osimertinib response and resistance for leptomeningeal metastases in EGFR-mutated NSCLC. J Thorac Oncol: Off Publ Int Assoc Study Lung Cancer. 2021;16:250–8.
Soca-Chafre G, Montiel-Dávalos A, Rosa-Velázquez IA, Caro-Sánchez CHS, Peña-Nieves A, Arrieta O. Multiple molecular targets associated with genomic instability in lung cancer. Int J Genomics. 2019;2019:9584504.
Hsu F, De Caluwe A, Anderson D, Nichol A, Toriumi T, Ho C. EGFR mutation status on brain metastases from non-small cell lung cancer. Lung Cancer. 2016;96:101–7.
Jänne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. J Clin Oncol. 2005;23:3227–34.
Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. eLife. 2015;4:e06907.
Lee CI, Hsu MY, Chou CH, Wang C, Lo YS, Loh JK, et al. CTNNB1 (beta-catenin) mutation is rare in brain tumours but involved as a sporadic event in a brain metastasis. Acta neurochirurgica. 2009;151:1107–11.
Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49:1693–704.
Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-small-cell lung cancer. N. Engl J Med. 2017;376:2109–21.
Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–75.
Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–72.
Zang YS, Dai C, Xu X, Cai X, Wang G, Wei J, et al. Comprehensive analysis of potential immunotherapy genomic biomarkers in 1000 Chinese patients with cancer. Cancer Med. 2019;8:4699–708.
Wang H, Ou Q, Li D, Qin T, Bao H, Hou X, et al. Genes associated with increased brain metastasis risk in non-small cell lung cancer: Comprehensive genomic profiling of 61 resected brain metastases versus primary non-small cell lung cancer (Guangdong Association Study of Thoracic Oncology 1036). Cancer. 2019;125:3535–44.
Meyers M, Theodosiou M, Acharya S, Odegaard E, Wilson T, Lewis JE, et al. Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2. Cancer Res. 1997;57:206–8.
Nanjo S, Arai S, Wang W, Takeuchi S, Yamada T, Hata A, et al. MET copy number gain is associated with gefitinib resistance in leptomeningeal carcinomatosis of EGFR-mutant lung cancer. Mol Cancer Ther. 2017;16:506–15.
Zhao L, Li N, Yu JK, Tang HT, Li YL, He M, et al. RNAi-mediated knockdown of FANCF suppresses cell proliferation, migration, invasion, and drug resistance potential of breast cancer cells. Braz J Med Biol Res. 2014;47:24–34.
Li J, Zhang W, Xia H, Liu Y. [High expression of VEGFR3 is positively associated with poor prognosis in lung adenocarcinoma]. Xi bao yu fen zi mian yi xue za zhi = Chin J Cell Mol Immunol. 2019;35:1023–9.
Budczies J, Bockmayr M, Denkert C, Klauschen F, Gröschel S, Darb-Esfahani S, et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274)—associations with gene expression, mutational load, and survival. Genes Chromosomes Cancer. 2016;55:626–39.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21.
Ju YS. The mutational signatures and molecular alterations of bladder cancer. Transl Cancer Res. 2017;6:S689–701.
Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016;48:600–6.
Berghoff AS, Magerle M, Ilhan-Mutlu A, Dinhof C, Widhalm G, Dieckman K, et al. Frequent overexpression of ErbB-receptor family members in brain metastases of non-small cell lung cancer patients. APMIS. 2013;121:1144–52.
Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res. 2016;22:5130–40.
Lee J, Choi Y, Han J, Park S, Jung HA, Su JM, et al. Osimertinib improves overall survival in patients with EGFR-mutated NSCLC with leptomeningeal metastases regardless of T790M mutational status. J Thorac Oncol. 2020;15:1758–66.
Chang SC, Lai YC, Chang CY, Huang LK, Chen SJ, Tan KT, et al. Concomitant genetic alterations are associated with worse clinical outcome in EGFR mutant NSCLC patients treated with tyrosine kinase inhibitors. Transl Oncol. 2019;12:1425–31.
Ai X, Cui J, Zhang J, Chen R, Lin W, Xie C, et al. Clonal architecture of EGFR mutation predicts the efficacy of EGFR-tyrosine kinase inhibitors in advanced NSCLC: a prospective multicenter study (NCT03059641). Clin Cancer Res. 2021;27:704–12.
Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated With survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. 2018;4:210–6.
Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. 2019;37:2518–27.
Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–50.
Guisier F, Dubos-Arvis C, Viñas F, Doubre H, Ricordel C, Ropert S, et al. Efficacy and safety of Anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol. 2020;15:628–36.
Naz S, Sowers A, Choudhuri R, Wissler M, Gamson J, Mathias A, et al. Abemaciclib, a selective CDK4/6 inhibitor, enhances the radiosensitivity of non-small cell lung cancer in vitro and in vivo. Clin Cancer Res. 2018;24:3994–4005.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Rubio-Perez C, Planas-Rigol E, Trincado JL, Bonfill-Teixidor E, Arias A, Marchese D, et al. Immune cell profiling of the cerebrospinal fluid enables the characterization of the brain metastasis microenvironment. Nat Commun. 2021;12:1503.
Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–8.
Nong J, Gong Y, Guan Y, Yi X, Yi Y, Chang L, et al. Circulating tumor DNA analysis depicts subclonal architecture and genomic evolution of small cell lung cancer. Nat Commun. 2018;9:3114.
Liu X, Wang F, Xu C, Chen X, Hou X, Li Q, et al. Genomic origin and intratumor heterogeneity revealed by sequencing on carcinomatous and sarcomatous components of pulmonary sarcomatoid carcinoma. Oncogene 2021;40:821–32.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma (Oxf, Engl). 2009;25:1754–60.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids Res. 2010;38:e164.
Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, et al. CONTRA: copy number analysis for targeted resequencing. Bioinforma (Oxf, Engl). 2012;28:1307–13.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat methods. 2009;6:677–81.
Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat methods. 2014;11:396–8.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31.
Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018;173:321–37. e10.
Funding
This study was supported by the Key Research And Development Funds of Hunan Cancer Hospital Climbing Plan (FY2020008 to ZH), the Key Research And Development Plans of Hunan Provincial Science and Technology Department (2017SK2131 to ZH), the Hunan Provincial subsidy health projects in 2020 of Hunan Provincial Department of Finance (30299-50502 to ZH), National Natural Science Foundation of China (81770928 to RZ), and Foundation of CSCO (Y-HR2019-0185 to RZ).
Author information
Authors and Affiliations
Contributions
ZH conceived the study and designed the experiments. ZD, NR, ZZ, ZT, and LW prepared the research materials. LC and PL did the bioinformatic analysis and performed initial exploratory analysis. JG, HC, YG, XY, and RZ provided insight in methodological approaches and analysis. ZH and XX supervised the study. ZD, LC, and PL drafted the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr. Giovanni Blandino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, Z., Cui, L., Li, P. et al. Genomic comparison between cerebrospinal fluid and primary tumor revealed the genetic events associated with brain metastasis in lung adenocarcinoma. Cell Death Dis 12, 935 (2021). https://doi.org/10.1038/s41419-021-04223-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04223-4
- Springer Nature Limited
This article is cited by
-
PMS2 amplification contributes brain metastasis from lung cancer
Biological Procedures Online (2024)
-
Combined inhibition of HER2 and VEGFR synergistically improves therapeutic efficacy via PI3K-AKT pathway in advanced ovarian cancer
Journal of Experimental & Clinical Cancer Research (2024)
-
Genetic Alterations of Melanoma Brain Metastases: A Systematic Review and Meta-Analysis
Molecular Diagnosis & Therapy (2023)
-
Leptomeningeal Metastases: New Opportunities in the Modern Era
Neurotherapeutics (2022)