
Abstract
Multiciliated cells (MCCs) in the brain reside in the ependyma and the choroid plexus (CP) epithelia. The CP secretes cerebrospinal fluid that circulates within the ventricular system, driven by ependymal cilia movement. Tumors of the CP are rare primary brain neoplasms mostly found in children. CP tumors exist in three forms: CP papilloma (CPP), atypical CPP, and CP carcinoma (CPC). Though CPP and atypical CPP are generally benign and can be resolved by surgery, CPC is a particularly aggressive and little understood cancer with a poor survival rate and a tendency for recurrence and metastasis. In contrast to MCCs in the CP epithelia, CPCs in humans are characterized by solitary cilia, frequent TP53 mutations, and disturbances to multiciliogenesis program directed by the GMNC-MCIDAS transcriptional network. GMNC and MCIDAS are early transcriptional regulators of MCC fate differentiation in diverse tissues. Consistently, components of the GMNC-MCIDAS transcriptional program are expressed during CP development and required for multiciliation in the CP, while CPC driven by deletion of Trp53 and Rb1 in mice exhibits multiciliation defects consequent to deficiencies in the GMNC-MCIDAS program. Previous studies revealed that abnormal NOTCH pathway activation leads to CPP. Here we show that combined defects in NOTCH and Sonic Hedgehog signaling in mice generates tumors that are similar to CPC in humans. NOTCH-driven CP tumors are monociliated, and disruption of the NOTCH complex restores multiciliation and decreases tumor growth. NOTCH suppresses multiciliation in tumor cells by inhibiting the expression of GMNC and MCIDAS, while Gmnc-Mcidas overexpression rescues multiciliation defects and suppresses tumor cell proliferation. Taken together, these findings indicate that reactivation of the GMNC-MCIDAS multiciliogenesis program is critical for inhibiting tumorigenesis in the CP, and it may have therapeutic implications for the treatment of CPC.
Similar content being viewed by others


Introduction
The choroid plexus (CP) in each brain ventricle consists of stromal vasculatures ensheathed by epithelia [1,2,3]. The CP is responsible for the synthesis and secretion of cerebrospinal fluid in the central nervous system. Recent studies revealed multiciliated cells (MCCs) in the CP of the mouse [1,2,3]. Unlike ependymal cells that form multiple motile cilia to drive cerebrospinal fluid flow within the central nervous system after birth, MCCs in the CP epithelia arise during embryogenesis, display increased motility of their multiple cilia until birth, and experience a gradual regression in the motility during postnatal life [3,4,5]. Tumors of the CP comprise ~20% of brain tumors diagnosed in children under 1 year of age [6, 7]. Research aimed at understanding the origin and molecular characteristics of CP carcinoma (CPC) is essential for developing new therapies to improve clinical outcomes [8,9,10,11,12].
MCCs on the epithelial lining of the brain ventricles, the airway, and reproductive tracts control fluid movement through the beating of multiple motile cilia on their apical surface. Multiciliogenesis is directed by a network of transcription factors that include two members of the Geminin family of coiled-coil containing nuclear proteins: Geminin Coiled-Coil Domain Containing (GMNC) and multi-ciliate differentiation and DNA synthesis associated cell cycle protein (MCIDAS) [13, 14]. GMNC and MCIDAS play sequential roles in the early steps of the MCC differentiation program that is triggered by NOTCH inhibition [15,16,17,18,19,20]. GMNC and MCIDAS activate the expression of downstream MCC factors, including forkhead box J1 (FOXJ1), v-myb avian myeloblastosis viral oncogene homolog (MYB), and cyclin O (CCNO), as well as TP53 family member TAp73 [21,22,23,24,25,26,27,28].
GMNC and MCIDAS both transcriptionally regulate multiciliogenesis in ependymal cells through the E2F4/5-DP1 transcription factors [15, 16, 29]. In contrast, Geminin antagonizes the transcriptional functions of GMNC and MCIDAS. And Geminin and GMNC play antagonistic roles in the maintenance of the stem and ependymal cell populations in the adult neurogenic niche, respectively [30, 31]. CP epithelial cells are derived from neuroepithelial progenitors that express orthodenticle homeobox 2 (OTX2) and Growth differentiation factor 7 (GDF7). As these progenitors exit the cell cycle to undergo multiciliogenesis and differentiation, TAp73 is activated in MCCs, while aquaporin 1 (AQP1), transthyretin (TTR), and cytokeratins are upregulated in epithelial cells [32]. Our previous work showed that, in contrast to all other MCCs, TAp73 was dispensable for multiciliogenesis in the CP, suggesting that its differentiation program may be distinct from other MCC types [26]. Therefore, further analysis of the molecular mechanisms governing multiciliogenesis in the CP, as well as the functional significance of these cilia, will be important to understand their role in the pathology of both ciliopathies and CP tumor development.
Examination of human CP tumors revealed abnormal NOTCH activity in a subset of tumors [33], and we demonstrated that sustained NOTCH1 expression in mice led to CP papilloma (CPP) that arose from monociliated progenitors in hindbrain roof plate [34, 35]. These progenitors proliferated in response to Sonic Hedgehog (SHH), but subsequently became quiescent after birth [34,35,36]. Here, we show that human CPC, and to a lesser extent CPP, display consistent defects in the GMNC-MCIDAS transcriptional program and amplifications of NOTCH pathway components. Using two distinct murine models, we found that CPCs in mice exhibit multiciliation defects and a deficient GMNC program. In addition, persistent NOTCH and SHH signals are sufficient to drive aggressive tumors in mice that resemble human CPC. These tumors display singular primary cilia resulting from the repression of the GMNC-MCIDAS multiciliation program by NOTCH. Biochemical or pharmacological disruption of the NOTCH complex restored multiciliation and suppressed tumor cell proliferation. Our findings indicate that the GMNC-MCIDAS transcriptional network is essential for MCC differentiation in the CP, and its activation can induce multiciliation and decrease CP tumor cell proliferation. These findings underscore the critical role of a compromised GMNC-MCIDAS multiciliogenesis program in CPC development and suggest that this could be exploited therapeutically to impair proliferation and promote tumor differentiation.
Results
CPCs in humans exhibit reduced multiciliation and a deficient GMNC-MCIDAS program
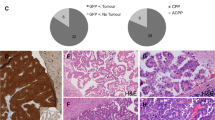
Most CP tumors in humans, especially CPCs, consist of monociliated tumor cells and frequently display large-scale genomic alterations [34, 37,38,39]. Analysis of published data revealed recurrent chromosomal changes that affect loci encompassing multiciliogenesis regulators, including GMNC on chromosome 3, that is lost in all hypodiploid CPCs, MCIDAS, CCNO, microRNA 449 (MIR449), and CDC20B, that are all located within the same locus of chromosome 5, and MYB on chromosome 6, that is lost in many CPCs (Fig. 1A). Conversely, N-acetyl galactosamine-type O-glycosylation enzyme GALNT11, a positive regulator of NOTCH signaling on chromosome 7, is gained in >80% CP tumors (Fig. 1A) [37,38,39,40]. In agreement, among 11 cases of human CPCs examined, most displayed significantly reduced or complete loss of GMNC expression, and GMNC expression was heterogeneous and only detected in a subpopulation of tumor cells (Fig. 1B, C). Decreased FOXJ1 expression was also observed in the majority of samples (Fig. 1B, C). A similar trend of reduced GMNC and FOXJ1 expression was observed in 31 human CPPs (Fig. 1B, C). Consistent with the requirement for GMNC and FOXJ1 in MCC differentiation, analysis of cilia marker ADP-ribosylation factor-like 13b (ARL13B) in six CPCs found that all were monociliated, while 11 of 17 CPPs analyzed were monociliated (Fig. 1B, D). Accordingly, RT-qPCR analysis revealed low levels of GMNC and MCIDAS expression in most human CP tumors compared to normal tissues, while FOXJ1 expression in CPCs was significantly lower than CPPs (Fig. 1E). In contrast, TAp73 expression in human CPC and CPP varied from significantly reduced to normal levels in similar proportions (Supplementary Fig. S1A). Moreover, analysis of a published dataset revealed differential expression of genes involved in ciliogenesis in human CP tumors, contributing to significant enrichment of the pathway (Supplementary Fig. S1B, C) [37]. Thus, CPCs in humans are characterized by multiciliation defects and deficiencies in the GMNC-MCIDAS program, as well as recurrent amplification of NOTCH regulators.

A Copy number analysis of human CPC (n = 23 tumors from 23 individuals) and CPP (n = 32 tumors from 32 individuals). B Representative images of human CP tumors that show GMNC and FOXJ1 expression by RNAscope (left panels), and ARL13B expression by immunostaining (brown signals in upper right panels, red signals in lower right panel). DAPI staining (blue, lower right panel) labels nuclei. Scale bars, 25 µm. Results were obtained from at least three independent experiments. C Summary of GMNC and FOXJ1 expression in human CP tumors shown in B (CPC: n = 11; CPP: n = 31). D Summary of cilia status in human CP tumors (CPC: n = 6; CPP: n = 11). E RT-qPCR analysis of the expression of GMNC, MCIDAS and FOXJ1 in CP tumors and normal control tissues in humans (CPP: n = 10; CPC: n = 8; normal tissue: n = 1 for brain, trachea, lung, testis, and epididymis, respectively; mean ± s.e.m., student t-test, **P < 0.01). Data represent two independent experiments.
Gmnc-Mcidas signaling is essential for generating multiciliated epithelia in the CP
These results suggested that suppressing the MCC fate program controlled by GMNC and MCIDAS was a key step in the genesis of CP tumors. While GMNC was implicated in the formation of MCCs in the CP [15], a detailed examination of its role in the CP has not been carried out. The Gmnc conditional allele (Gmncflox/flox) has two loxP sites located in introns 3 and 5 that allow Cre-mediated deletion of exons 4/5 to generate the null allele (Gmnc−/−) [15]. Using electron microscopy and immunostaining, we compared wild type CP to animals with a conditional deletion of Gmnc in the roof plate/CP by the Lmx1a-Cre transgene [41]. We found that both ependymal cells and the CP epithelium of Gmnc−/− animals were comprised solely of monociliated cells, compared to wild type controls that exhibited multiple basal bodies and multiciliation (Fig. 2A, B; Supplementary Figs. S2A, B, S3A). RT-qPCR using primers from exons 4/5 revealed significantly reduced Gmnc levels in the CP from Lmx1a-Cre;Gmncflox/− (Lcre;Gmncflox/−) mice at postnatal (P) day 7 (P7), consistent with efficient Gmnc disruption (Fig. 2C). Analysis of Ki-67 expression showed that both Gmnc−/− and wild type CP epithelial cells became postmitotic and the expression of epithelial markers cytokeratins, TTR, and OTX2 was comparable between Gmnc−/− and wild type CP, though Aqp1 expression was significantly increased in Gmnc−/− animals (Fig. 2C; Supplementary Fig. S3B–F).

A Transmission electron micrographs are shown of CP epithelial cells in newborn Gmnc−/− (asterisks) and wild type (arrow) mice. Boxed regions are magnified on the right. Notice that Gmnc−/− CP epithelial cell exhibits single basal body and solitary cilia compared to wild type epithelial cell with multiple basal bodies. CP choroid plexus, C cilia, BB basal body. Images are representative of at least three independent experiments. B The expression of acetylated α-tubulin (ac-α-tub, magenta) and γ-tubulin (green) is shown in the CP epithelial cells in newborn Gmnc−/− and wild type animals. Boxed regions are shown in higher magnification on the right. DAPI staining (cyan) labels nuclei. Scale bar, 20 µm. BB basal body. Images represent three independent experiments. C RT-qPCR analysis of the expression of Gmnc (primers/probe from exons 4/5 or 2/3), Aqp1 TAp73, and Foxj1 in the CP from Lcre;Gmncflox/− and wild type mice at day P7 (n = 11 animals per genotype, mean ± s.e.m., paired t-test, ****P < 0.0001). Data represent three independent experiments. D Representative images of the expression of Gmnc and Foxj1 (upper panel), Mcidas and Myb (lower panel) are shown at day E13.5 in roof plate (upper roof plate marked by dotted lines) and CP in the hindbrain and the lateral ventricle in Gmnc−/− (arrowheads) and wild type (arrows) animals. Scale bars, 50 µm. Images represent at least three independent experiments. E The expression of ARL13B (yellow) and AQP1 (green) is shown in the CP epithelial cells in the hindbrain and lateral ventricles at day P7 in Mcidas−/− (arrowheads) and wild type (arrows) animals. DAPI staining (cyan) labels nuclei. Scale bars, 10 µm. Results were obtained from three independent experiments.
Gmnc mRNA was detected in wild type CP epithelial cells adjoining the roof plate at embryonic (E) day 13.5 (E13.5), and Gmnc transcripts persisted in the epithelial cells of Gmnc−/− CP (Fig. 2D). RT-qPCR with primers from exons 2/3 showed increased Gmnc levels, and further sequencing revealed a mutant transcript with exon 3 spliced to exon 6 (designated as Gemc1Δ4-5) (Fig. 2C; Supplementary Fig. S4A). The altered splicing causes a frame shift and stop codon after a few amino acids, generating a truncated Gmnc transcript lacking crucial functional domains. The expression of GMNC targets Foxj1 and TAp73 in the CP and ependyma was markedly reduced in Gmnc−/− mice, whereas Gmnc overexpression stimulated TAp73 expression (Fig. 2C, D; Supplementary Figs. S4B, D, S5A, C, S14, Supplementary Table 1). Together, these data establish that Gmnc is essential for MCC differentiation and the expression of TAp73 and Foxj1 in the CP epithelium.
We next examined critical components of the MCC transcriptional cascade, including Mcidas, Myb, and Ccno. All 3 genes were transiently upregulated in a subpopulation of epithelial cells next to the roof plate during development and their expression was lost in Gmnc−/− mice (Fig. 2D; Supplementary Fig. S5B), indicating that GMNC activates the MCIDAS-dependent program in the CP, as has been shown in other MCC-containing tissues. Examination of CP epithelial cells in Mcidas−/− mice revealed only solitary primary cilia, and OTX2 and AQP1 expression were similar to that of the wild type CP (Fig. 2E; Supplementary Fig. S6A, B). The expression of Gmnc, Foxj1, and TAp73 remained unaltered by Mcidas loss, although Mcidas overexpression stimulated TAp73 and Ccno expression (Supplementary Figs. S4C, D, S6C, D, S14, Supplementary Table 1). Taken together, these results indicate that MCIDAS plays a critical role in multiciliogenesis downstream of GMNC in the CP.
Gmnc loss mediates cilia defects in CPCs with deficient Rb1/Trp53 signaling
CPC frequently occurs in Li-Fraumeni syndrome patients and somatic TP53 mutations in sporadic CPC predict poor outcome [37, 42]. In mice, Trp53 deletion combined with Rb1 loss or Myc overexpression leads to CPC with characteristics of their human counterparts [8,9,10, 42,43,44]. We crossed Lmx1a-Cre mice that express Cre in the roof plate/CP [41], with a mouse strain carrying conditional alleles of Trp53 and Rb1 (p53flox/flox;Rbflox/flox) to cause their deletion in CPC progenitors [45]. All Lmx1a-Cre;p53flox/flox;Rbflox/flox (Lcre;p53cko;Rbcko) mice developed CPC, characterized by higher numbers of Ki-67+ proliferative cells (Fig. 3A). As early as 9 weeks after birth, a small population of Gmnc-negative cells was detected in the CP of Lcre;p53cko;Rbcko animals, whereas OTX2+ monociliated cells were present among MCCs in the CP at 11 weeks of age (Fig. 3B, C). Tumor cells in these mice were monociliated and exhibited significantly reduced Gmnc and Foxj1 levels (Fig. 3B–D; Supplementary Fig. S7A; Supplementary Table 1). Thus, Rb1/Trp53-deficient murine CPC recapitulates the multiciliation defects and GMNC program deficiencies in CPC in humans.

A Hematoxylin and eosin (H&E) staining and Ki-67 expression are shown in CPC (arrowheads, boundary between tumor and unaffected brain region is marked by dotted lines) and the CP (arrows) from Lcre;p53cko;Rbcko and Lcre;p53cko;Rbcko;Gmnccko animals. Scale bars, 50 µm. Images are representative of at least three independent experiments. B RNAscope analysis of Gmnc and Mcidas expression in tumor cells (arrowheads, boundary between tumor and unaffected brain region is marked by dotted line) and the CP (arrows) in Lcre;p53cko;Rbcko and Lcre;p53cko;Rbcko;Gmnccko animals. Scale bar, 50 µm. Results were obtained from three independent experiments. C Representative images of immunofluorescence of ARL13B (yellow) and OTX2 (green) are shown in multiciliated epithelial cells (arrows) or monociliated tumor cells (arrowheads) in wild type and Lcre;p53cko;Rbcko animals, respectively. Scale bar, 10 µm. Data represent three independent experiments. D RT-qPCR analysis of gene expression in wild type CP and CPC from Lcre;p53cko;Rbcko and Lcre;p53cko;Rbcko;Gmnccko animals (wild type CP: n = 10; CPC: n = 11 for Lcre;p53cko;Rbcko animals, n = 10 for Lcre;p53cko;Rbcko;Gmnccko animals; mean ± s.e.m., one-way ANOVA, *P < 0.05, ****P < 0.0001, NS not significant). Data represent three independent experiments. E Representative images of immunofluorescence of ARL13B (red) are shown in tumor cells from Lcre;p53cko;Rbcko;Gmnccko animals infected with viruses expressing GMNC-myc (green) or MCIDAS-myc (green). OTX2 (green) labels tumor cells. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Three independent experiments were conducted. F Kaplan–Meier curve depicting the survival of Lcre;p53cko;Rbcko, Lcre;p53cko;Rbcko;Gmnc−/+, and Lcre;p53cko;Rbcko;Gmnccko animals compared to wild type mice.
The loss of Gmnc expression at early stages of tumorigenesis suggested that this may promote the reduced multiciliation observed in Rb1/Trp53-deficient CPC. To address this, we interbred Lcre;p53cko;Rbcko mice with Gmncflox/− animals. The resulting Lmx1a-Cre;p53cko;Rbcko;Gmncflox/− (Lcre;p53cko;Rbcko;Gmnccko) mice succumbed to CPCs that expressed OTX2 and showed proliferation levels similar to that of Lcre;p53cko;Rbcko mice (Fig. 3A; Supplementary Fig. S7B). The expression of Ttr, Aqp1, Gdf7, and Foxj1 was significantly reduced, whereas TAp73 expression was more variable in CPC in these mice (Fig. 3D; Supplementary Fig. S7B; Supplementary Table 1). A non-functional Gemc1Δ4-5 mutant transcript was detectable in the CP of the Lcre;p53cko;Rbcko;Gmnccko mice, but its presence was dramatically reduced in tumor cells in these animals, indicating similar impairment in the upstream regulation of Gmnc (Fig. 3B, D).
To determine if reactivation of the GMNC-MCIDAS program would affect tumorigenesis, we expressed Myc-tagged GMNC or MCIDAS in tumor cells from Lcre;p53cko;Rbcko;Gmnccko mice. Reintroduction of either Gmnc or Mcidas activated the expression of TAp73 and Foxj1 and caused multiciliation of tumor cells (Fig. 3E; Supplementary Fig. S7C, D). In contrast, Gmnc loss failed to accelerate tumor development in Lcre;p53cko;Rbcko;Gmnccko animals, indicating that GMNC does not play a clear tumor suppressive role (Fig. 3F). Together, these data indicate that a defective GMNC-MCIDAS program mediates cilia defect in Rb1/Trp53-deficient CPC and facilitates tumor growth.
SHH and NOTCH pathways drive CPC
Previous analyses revealed abnormal SHH and NOTCH pathway activities in human CP tumors, including CPC [34]. We previously used a Rosa26-NICD1 mouse strain that exhibits Cre-mediated expression of the intracellular domain of NOTCH1 (NICD1) and green fluorescent protein (GFP) [46]. After crossing with Lmx1a-Cre animals, Lmx1a-Cre;Rosa26-NICD1 (Lcre;NICD1) mice developed CPP that underwent transient SHH-driven proliferation [34].
To determine whether combined defects in SHH and NOTCH signaling were sufficient to drive more aggressive CPC-like tumors, we bred a mouse strain carrying a Patched1 conditional allele (Ptchflox/flox) to Lcre;NICD1 mice. Loss of Patched1 constitutively activates SHH signaling in the roof plate/CP in Lmx1a-Cre;Ptchflox/flox (Lcre;Ptchcko) animals [34, 47]. As expected, a thickened upper roof plate was observed in Lcre;Ptchcko mice at day E14.5 (Fig. 4A). Though most Lcre;NICD1 mice survived normally, all Lmx1a-Cre;Ptchflox/flox;NICD1 (Lcre;Ptchcko;NICD1) animals died at birth from abnormal cellular growth in the hindbrain region (Fig. 4A). These animals displayed an enlarged upper roof plate with an increased presence of Ki-67+ proliferative cells, an abnormal CP growth with increased Ki-67 index, and a loss of monolayer epithelial architecture (Fig. 4A; Supplementary Fig. S8A).

A Wild type, Lcre;NICD1, Lcre;Ptchcko, and Lcre;Ptchcko;NICD1 animals are shown at day E14.5. Notice the cranium defects resulting from enlarged and folded roof plate in the midbrain-hindbrain region of Lcre;Ptchcko and Lcre;Ptchcko;NICD1 animals (white arrowheads). H&E staining and Ki-67 expression are shown of roof plate (upper roof plate marked by red lines) and the CP (black arrows) in the hindbrain in wild type and Lcre;Ptchcko animals, and CPP and abnormal CP growth (black arrowheads) in Lcre;NICD1 and Lcre;Ptchcko;NICD1 animals, respectively. Enlarged roof plate disrupts the cranium in Lcre;Ptchcko and Lcre;Ptchcko;NICD1 animals (red arrows). The upper roof plate is shown in higher magnification in the right (Lcre;Ptchcko;NICD1 animal) and lower (wild type, Lcre;NICD1, Lcre;Ptchcko, and Lcre;Ptchcko;NICD1 animals) panels. Scale bars, 100 µm. Quantification of Ki-67 expression in the upper roof plate and CP in the hindbrain is shown (wild type mice: n = 11; Lcre;NICD1 mice: n = 4; Lcre;Ptchcko mice: n = 3; Lcre;Ptchcko;NICD1 mice: n = 7 for upper roof plate, n = 8 for the CP; mean ± s.e.m., one-way ANOVA, ***P < 0.001; ****P < 0.0001). Data are representative of at least three independent experiments. B Representative results of immunohistochemical staining for OTX2, and AQP1 are shown in the upper roof plate (marked by dotted lines) and the CP (arrows) in the hindbrain at day E14.5 in wild type and Lcre;Ptchcko animals, and CPP and abnormal CP growth (black arrowheads) in Lcre;NICD1 and Lcre;Ptchcko;NICD1 animals, respectively. Residual AQP1-expressing epithelial cells (red arrowhead) are mixed with tumor cells in Lcre;NICD1 animals. Scale bar, 50 µm. Images represent at least three independent experiments.
Like CPP in Lcre;NICD1 mice, the abnormal CP growth in Lcre;Ptchcko;NICD1 animals exhibited elevated expression of the SHH pathway targets Gli1 and Mycn, reduced Shh expression, as well as increased levels of NOTCH targets Hes1 and Hes5 (Supplementary Figs. S8B, S9A–C). Similar to Rb1/Trp53-deficient CPC, the abnormal CP growth in Lcre;Ptchcko;NICD1 animals expressed OTX2, and showed reduced expression of AQP1, TTR, and cytokeratins, although some CP epithelial cells were mixed due to incomplete Cre-mediated activation of NICD1 (Fig. 4B; Supplementary Fig. S8C; Supplementary Table 1). Thus, combined activation of the NOTCH and SHH pathways leads to increased proliferation and pathological cell overgrowth in the upper roof plate region, accompanied by a loss of differentiated epithelial cells in the CP. Moreover, these animals develop malignant CP tumors that closely match the characteristics of CPC in humans.
NOTCH activation leads to reduced multiciliation in CP tumors
In contrast to MCCs in the CP, NOTCH-driven CP tumors consisted of monociliated cells with decreased Foxj1 expression (Fig. 5A) [34], suggesting that NOTCH might mediate reduced multiciliation in CP tumors. To address this, tumor cells from Lcre;NICD1 mice were treated with a recombinant amino-terminal fragment of SHH (ShhN) and a small molecule Inhibitor of Mastermind Recruitment 1 (IMR-1, or IMR-1A) to block the recruitment of Mastermind-like protein 1 (MAML1) to the NOTCH transcriptional complex, or infected with viruses expressing GFP fused to dominant negative MAML1 (dnMAML1) that disrupts the complex [48, 49]. Remarkably, staining with the cilia markers ARL13B and γ-tubulin revealed GFP+ multiciliated tumor cells within 72 h of treatment (Fig. 5B, C; Supplementary Fig. S10A). Consistently, tumor cell proliferation was markedly reduced, and the expression of Foxj1 was significantly increased by IMR-1 (Fig. 5D, E; Supplementary Fig. S10B).

A Immunofluorescent staining for ARL13B (yellow) is shown at day E14.5 in the upper roof plate progenitors (marked by dotted lines and orange arrowheads) and the CP epithelial cells (arrows) in the hindbrain in wild type and Lcre;Ptchcko animals, and CPP and abnormal CP growth (white arrowheads and dotted lines) in Lcre;NICD1 and Lcre;Ptchcko;NICD1 animals, respectively. GFP (green) labels tumor cells. DAPI staining (cyan) labels nuclei. Scale bar, 10 µm. Results were obtained from at least three independent experiments. The expression of ARL13B (red) is shown in tumor cells infected with viruses expressing GFP-tagged dnMAML1 or GFP (B), or treated with vehicle, or IMR-1/IMR-1A (C). GFP (green) labels infected or treated cells. DAPI staining (blue) labels nuclei. Scale bars, 20 µm. The percentage of multiciliated tumor cells after treatment is shown (n = 3, mean ± s.e.m., two-tailed unpaired t-test, **P < 0.01, ***P < 0.001). Results were obtained from at least three independent experiments, respectively. D The expression of Ki-67 (red) is shown in GFP+ tumor cells treated with vehicle or IMR-1. Quantitation of Ki-67 expression is shown (n = 6 per treatment; mean ± s.e.m., paired t-test, ***P < 0.001). Results were obtained from at least three independent experiments. E RNAscope analysis of Foxj1 expression (red) is shown in tumor cells treated with vehicle or IMR-1. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Quantification of Foxj1 transcript is shown on the right (n = 7 per treatment; mean ± s.e.m., two-tailed unpaired t-test, **P < 0.01). Data are representative of three independent experiments.
After a 7-day in vivo IMR-1 treatment from day E10.5, multiciliated tumor cells were detected in Lcre;NICD1 and Lcre;Ptchcko;NICD1 animals at day E17.5 and day P7 (Fig. 6A, B). This was accompanied by a significant decrease in tumor cell proliferation, and a reduction of total tumor cell numbers by several folds at day P7 (Fig. 6C, D). Moreover, the expression of SOX2 in the ventricular zone, PAX6, and Atoh1 in progenitors derived from rhombic lips was comparable between wild type animals treated with IMR-1 or vehicle (Supplementary Fig. S10C, D). Together, these results demonstrate that aberrant NOTCH signaling impairs MCC differentiation in the CP that can be rescued by NOTCH inhibition, leading to reduced tumor growth.

Representative images of immunofluorescent staining for ARL13B (A, yellow; B, red) are shown in tumor cells at day E17.5 (A) and tumor cells isolated at day P7 (B) from Lcre;NICD1 (A, B) and Lcre;Ptchcko;NICD1 (A) animals treated with vehicle or IMR-1 from day E10.5 to day E16.5. Boxed region of ciliated cells is magnified in lower panel (A). DAPI staining (A, cyan; B, blue) labels nuclei. Scale bars, 5 µm (A), 10 µm (B). Results were obtained from at least three independent experiments. C Quantification total tumor cell numbers isolated at day P7 is shown in Lcre;NICD1 animals treated as described in A and B (n = 5 animals per treatment; mean ± s.e.m., two-tailed unpaired t-test, ***P < 0.001). D The expression of Ki-67 (red) in NICD1+/GFP+ tumor cells at day E17.5 is shown in Lcre;NICD1 animals treated with vehicle or IMR-1 from day E10.5 to day E16.5. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Quantification of Ki-67 expression in tumor cells is shown (right panel: n = 9 animals for vehicle, n = 8 animals for IMR-1; mean ± s.e.m., two-tailed unpaired t-test, **P < 0.01). Data are representative of three independent experiments.
Gmnc suppression by NOTCH mediates defective multiciliation in CP tumors
To understand the mechanisms of MCC regulation in tumor cells, we integrated RT-qPCR, RNAseq, and spatio-temporal gene expression data. Results from these assays consistently showed that both Foxj1 and Mcidas were expressed in tumor cells at lower levels than observed in wild type CP epithelium (Fig. 7A; Supplementary Fig. S11A–D) [34]. As this suggested that upstream regulators of the MCC program were impaired, we examined Gmnc expression. Although Gmnc exhibited ubiquitous expression in the CP epithelium, we consistently observed decreased levels of Gmnc and its downstream target TAp73 in CP tumors (Fig. 7A, B; Supplementary Fig. S11A, C, D). This was accompanied by a transient increase in the expression of Gmnn, a gene that is normally associated with proliferation and was shown to antagonize GMNC transcriptional functions (Fig. 7B) [15, 30]. These results demonstrate that the GMNC-MCIDAS program is profoundly repressed in NOTCH-driven CP tumors, and this can be modulated using NOTCH pathway inhibitors.

A Median FKPM (fragments per kilobase of exon per million reads mapped) values of genes in NOTCH-driven CP tumors and wild type CPs (n = 3 specimens per time point, mean ± s.e.m., two-tailed unpaired t-test, *P < 0.05; **P < 0.01). B RT-qPCR analysis of NOTCH-driven CPP and wild type CP (n = 3 animals per time point, mean ± s.e.m., two-tailed unpaired t-test, ***P < 0.001, ****P < 0.0001). Three independent experiments were conducted. C The expression of ARL13B (red) is shown in tumor cells infected with viruses expressing GMNC-myc, MCIDAS-myc, or control only. GMNC-myc (green), or MCIDAS-myc (green) labels infected cells. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Quantification of the percentage of MCCs in infected cells is shown on the right (n = 4 per treatment, mean ± s.e.m., one-way ANOVA, *P < 0.05). Results were obtained from three independent experiments. D The expression of Ki-67 (red) is shown in tumor cells from Lcre;NICD1 mice infected with viruses expressing GMNC-myc. GMNC-myc (green) labels infected cells. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Quantification of Ki-67 expression in tumor cells from Lcre;NICD1 mice infected with viruses expressing GMNC-myc or control vectors is shown in the lower panel (n = 5 per treatment, mean ± s.e.m., paired t-test, ****P < 0.0001). Data represent at least three independent experiments. E Representative images of Gmnc expression (green) by RNAscope are shown in tumor cells treated with vehicle or IMR-1. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Quantification of Gmnc transcript is shown (n = 7 per treatment; mean ± s.e.m., two-tailed unpaired t-test, ***P < 0.001). Three independent experiments were conducted. F The expression of ARL13B (red) is shown in Gmnc-deficient tumor cells treated with vehicle or IMR-1/IMR-1A. GFP (green) labels tumor cells. DAPI staining (blue) labels nuclei. Scale bar, 20 µm. Data represent five independent experiments. G RT-qPCR analysis of Foxj1 expression in tumor cells treated with vehicle or IMR-1 (tumors from Lcre;NICD1 mice: n = 6 per treatment; tumors from Lcre;NICD1;Gmncflox/− mice: n = 4 per treatment, mean ± s.e.m., paired t-test, *P < 0.05, NS, not significant). Results were obtained from three independent experiments. H Quantification of Ki-67 expression is shown in Gmnc-deficient tumor cells treated with vehicle or IMR-1 (n = 6 per treatment, mean ± s.e.m., two-tailed unpaired t-test, NS not significant). Three independent experiments were conducted.
To understand the role of the GMNC-MCIDAS program in defective multiciliation of CP tumors, myc-tagged GMNC or MCIDAS was expressed in tumor cells from Lcre;NICD1 mice using viral vectors. Enforced expression of Gmnc or Mcidas led to the formation of multiple cilia and reduced proliferation in infected tumor cells within 72 h (Fig. 7C, D; Supplementary Fig. S12A, B), phenocopying NOTCH inhibition with IMR-1 that significantly increased Gmnc levels in tumor cells (Fig. 7E). We subsequently eliminated Gmnc by crossing Gmncflox/− and Lcre;NICD1 animals. Tumor cells from Lcre;NICD1;Gmncflox/− mice became resistant to multiciliation, Foxj1 activation, and decreased proliferation induced by IMR-1/IMR-1A (Fig. 7F–H; Supplementary Fig. S12C, D). Conversely, overexpression of Gmnc increased Foxj1 expression in tumor cells (Supplementary Fig. S12E). Therefore, these results indicate that monociliation in tumor cells is maintained through NOTCH suppression of GMNC-MCIDAS signaling and suggest that GMNC loss prevents the rescue of multiciliation defects by NOTCH inhibition.
Similar to Rb1/Trp53-deficient CPC, despite the suppression of the GMNC-MCIDAS program by NOTCH, combined loss of Gmnc and Patched1 failed to induce CPC in Lcre;Ptchcko;Gmncflox/− mice (Supplementary Fig. S13; Supplementary Table 1), suggesting that loss of GMNC-driven multiciliation in the CP is insufficient to replace NOTCH or Rb1/Trp53 deletion in CPC. Together, these data indicate that GMNC-MCIDAS program deficiencies critically mediate cilia defects in CPC to modulate tumor growth.
Discussion
CPC clinical outcomes remain dismal, leaving patients vulnerable to devastating consequences [2, 3]. The gross genomic alterations in CP tumors have made the identification of driving events and actionable targets difficult [43, 44]. The GMNC-MCIDAS program promotes multiciliogenesis in different tissues, is required for MCC generation in mice, and mutations in both GMNC and MCIDAS have been identified in human ciliopathies [13, 14]. The observation that there is consistent disruption of multiciliogenesis program and prevalence of solitary cilia in CPC indicates that CPC has characteristics of a ciliopathy and that therapeutic strategies aimed at restoring multiciliogenesis may suppress CP tumors.
Our findings revealed the interaction of the multiciliogenesis program, NOTCH, and SHH pathways during CP differentiation and tumorigenesis. NOTCH suppressed multiciliation of roof plate progenitors, thereby preserving cilia-based signaling activated by SHH from postmitotic MCCs in CP epithelium [33]. Conversely, SHH signaling enhanced Hes1 and Hes5 expression in the roof plate in Lcre;Ptchcko mice and NOTCH-driven CP tumors. The expanded upper roof plate in Lcre;Ptchcko;NICD1 mice is consistent with the developmental origin and cilia defect of CPC being driven by NOTCH and SHH signaling. These animals represent an ideal therapeutic model for congenital or infantile CPC, a rare condition associated with high morbidity and mortality [50, 51]. Indeed, NOTCH inhibition by IMR-1 rescued the cilia deficit by inducing multiciliated tumor cells, whereas SHH pathway inhibitors suppressed tumor cell proliferation (Fig. 8) [34]. Thus, further study of the interactions between the SHH and NOTCH pathways in CP tumors is warranted to determine the therapeutic potential of activators of multiciliation and cilia-dependent signaling [52,53,54,55,56].

A Combined activation of NOTCH and SHH signaling, or loss of Rb1/Trp53 tumor suppressors drives CPC development. B GMNC-MCIDAS program mediates multiciliation in CP epithelium, and is repressed by NOTCH signaling in roof plate progenitors, whereas NOTCH inhibitor IMR-1 promotes GMNC-dependent multiciliation and suppresses tumor growth. GMNC-MCIDAS program suppression in Trp53-deficient CPC maintains monociliated tumor cells.
While the differentiation of MCCs requires NOTCH inhibition, it is unclear precisely how NOTCH impacts MCC fate during tumorigenesis [57,58,59,60,61]. Our data shows for the first time that NOTCH suppresses the expression of Gmnc and Mcidas to impair multiciliation during tumorigenesis. Gmnc is required for MCC differentiation following NOTCH inhibition, indicating that GMNC-MCIDAS signaling is required downstream of NOTCH regulation and represents a potent anti-tumor mechanism in CP tumors (Fig. 8).
Consistent with previous studies, we found that the GMNC-MCIDAS program was required for multiciliation in the CP epithelium. As progenitor cells exit the cell cycle to undergo multiciliogenesis, the expression of Gmnc, Mcidas, Foxj1, and TAp73 was upregulated, as has been observed in other multiciliated tissues [26]. Ectopic expression of either GMNC and MCIDAS stimulated Foxj1 and TAp73 expression, whereas Gmnc loss prevented the activation of Mcidas, TAp73, and Foxj1, in contrast to Mcidas-deficient MCCs that showed expression of both Foxj1 and TAp73 [14, 25]. Although both TAp73 and Foxj1 are sensitive to Gmnc status, loss of TAp73 failed to affect Foxj1 expression in the CP, as it does in other MCCs, indicating that TAp73 is not integrated into the Gmnc-Foxj1 axis in the CP [26]. Consistent with this, TAp73 expression varied greatly in Rb1/Trp53-deficient CPC in mice, as well as CP tumors in humans. These results highlight the need to further analyze the similarities and differences between different MCC types.
Overall, this study shows that the GMNC-MCIDAS program is required for MCC differentiation in the CP. The impairment of the program by oncogenic signals including Rb1/Trp53 defects or NOTCH activation prevents multiciliation and facilitates proliferation of CP tumor cells (Fig. 8). Therefore, activation of multiciliogenesis may serve as a potential therapeutic strategy in a subset of CP tumors. As the early events leading to the activation of the GMNC-MCIDAS program remain poorly characterized, a detailed understanding of its regulation and functions will be critical for developing strategies to target this pathway for the treatment of CPC.
Materials and methods
Animals
Gt(ROSA)26Sortm1.Notch1Dam/J (Rosa26-NICD1) mice, B6N.129-Ptch1tm1Hahn/J (Ptchflox/flox) mice, B6.129P2-Trp53tm1Brn/J (Trp53flox/flox) mice, Rb1tm2Brn/J (Rbflox/flox) mice, and C57BL/6 mice (all from Jackson Laboratory, Bar Harbor, ME, USA), Tg(Lmx1a-cre)1Kjmi (Lmx1a-Cre) mice, Gmnctm1Strc (Gemc1−/+) mice, and Gmnctm1.1Strc (Gemc1flox/+) mice were maintained by breeding with C57BL/6 mice (Supplementary Table 1). Animals were housed in the Animal Research Facility at New York Institute of Technology College of Osteopathic Medicine in accordance with NIH guidelines. All animal experimental procedures were approved by Institutional Animal Care and Use Committee (IACUC) and performed in compliance with national regulatory standards. Mcidas mutant mice were housed at the Biological Resource Center of the Agency for Science, Technology and Research (A*STAR) of Singapore, and experiments performed with these animals followed guidelines stipulated by the Singapore National Advisory Committee on Laboratory Animal Research. All experimental procedures at the Institute for Research in Biomedicine were conducted following European and National Regulation for the Protection of Vertebrate Animals used for experimental and other scientific purposes (directive 86/609), internationally established 3R principles, and guidelines established by the United Kingdom Coordinating Committee on Cancer Research.
The animal experiments were not randomized, and both male and female animals were used for experiments at different time points. For analysis of the Gmnc-Mcidas program in MCC differentiation in the CP, animals analyzed included: Gmnc−/− Mcidas−/−, and wild type animals (n = 3 for each at genotype at each time point). The investigators were blinded to group allocation during experiments and assessment of tumor development in wild type (n = 16), Lmx1a-Cre;p53flox/flox;Rbflox/flox (n = 23), Lmx1a-Cre;p53flox/flox;Rbflox/flox;Gmnc−/+ (n = 17), and Lmx1a-Cre;p53flox/flox;Rbflox/flox;Gmncflox/− (n = 36) mice. For NOTCH-driven CP tumors, animals analyzed used included: wild type (n = 11), Lmx1a-Cre;NICD1 (n = 4), Lmx1a-Cre;Ptchflox/flox (n = 3), and Lmx1a-Cre;Ptchflox/flox;NICD1 (n = 8) mice. For analysis of the role of Gmnc in NOTCH-driven CP tumors, 3 animals were used for wild type, Lmx1a-Cre;Ptchflox/+;Gmncflox/−, Lmx1a-Cre;Ptchflox/flox;Gmncflox/− mice. Experimental animals were administered 15 mg/kg IMR-1 (SML1812, Sigma-Aldrich, St. Louis, MO, USA) or vehicle by intraperitoneal injection of pregnant females. Animals used included: wild type (n = 3 for vehicle or IMR-1, respectively), Lmx1a-Cre;NICD1 (vehicle n = 14; IMR-1: n = 13), and Lmx1a-Cre;Ptchflox/flox;NICD1 (n = 5 for vehicle or IMR-1, respectively).
Human samples
CP specimens were procured with informed consent from patients following the requirements by institutional review boards at Shanghai East Hospital, Sanford Burnham Prebys Medical Discovery Institute, and University Medical Center Hamburg-Eppendorf. All CP specimens from Boston Children’s Hospital were obtained under an approved institutional review board protocol. All tissues were handled in accordance with guidelines and regulations for the research use of human brain tissue set forth by the NIH (http://osp.od.nih.gov/o_ce-clinical-research-and-bioethics-policy). Diagnoses of human CP specimens from Boston Children’s Hospital were reviewed by two neuropathologists (HGWL, S. Santagata) using standard WHO criteria [62].
Cell culture
Multiple sets of tissue specimens were collected from animals of appropriate genotype and maintained in culture. Gender information is not available for animals collected at day P7. Primary CP tumor cells were cultured as described previously [34]. Dissected specimens were dissociated and digested at 37 °C for 20 min with pronase (2 mg/ml, 537088, Calbiochem, San Diego, CA, USA) in Hank’s balanced salt solution (14170-112; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 2 mM glucose. Dissociated tumor cells were centrifuged at 100 g for 2 min at 4 °C. Cells were resuspended and cultured in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 Ham’s-Liquid Media (DMEM:F12, SH30261; HyClone Laboratories, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, R&D Systems, Inc., Minneapolis, MN, USA) and 100 IU/mL penicillin/streptomycin (Thermo Fisher Scientific). Cultured cells were treated with ShhN, with vehicle or IMR-1 (25 μM). HEK293 human embryonic kidney cells (ATCC, CRL-1573, Manassas, VA, USA), and AD-293 cell line (Agilent Technologies, Santa Clara, CA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; 5.5 mM glucose, Thermo Fisher Scientific) supplemented with 10% FBS (R&D Systems, Inc.), and 100 IU/ml penicillin/streptomycin (Thermo Fisher Scientific). Mouse inner medullary collecting duct cells (mIMCD3, ATCC, CRL-2123) were cultured in DMEM:F12 Medium (HyClone Laboratories) supplemented with 10% FBS (R&D Systems, Inc.), and 100 IU/mL penicillin/streptomycin (Thermo Fisher Scientific). These cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C in a cell culture incubator.
Analyses of gene expression, cell proliferation, and signal transduction were performed using CP tumor cells from Lmx1a-Cre;NICD1 (n = 13), Lmx1a-Cre;NICD1;Gmncflox/− (n = 8), and Lmx1a-Cre;p53flox/flox;Rbflox/flox;Gmncflox/− mice (n = 3). Results from these studies confirmed their identity (Figs. 3E, 5B–E, 7C–H; Supplementary Figs. S4B–D, S7C, D, S10A, B, S11D, S12A–E). HEK293, AD-293, and mIMCD3 cells were tested regularly for mycoplasma. Given the short time (<8–10 days) during which CP cells were maintained as primary cultures supplemented with antibiotics, we did not test for mycoplasma contamination.
Viruses
PmeI-linearized pShuttle-vectors carrying different cDNA fragments were introduced into the replication-deficient adenoviral vector pAdEasy-1 through homologous recombination in BJ5183 cells (Agilent Technologies). Successfully recombined adenoviral vector was verified by sequencing. The adenoviral plasmid was linearized by PacI digest and transfected into AD-293 cells (Agilent Technologies) to produce recombinant viral particles. All the procedures of production, purification, and use of adenoviruses were approved by Institutional Biosafety Committee.
Immunohistochemistry, immunofluorescence, and immunocytochemistry
Multiple sets of tissue specimens were collected from animals. Gender information is not available for animals collected during embryonic development and at days P0 and P7. Immunostaining was carried out as previously described [34]. Primary antibodies used and dilution ratios are: mouse monoclonal anti-Acetylated α-Tubulin (1:500, ab24610, clone 6-11B-1, abcam, Cambridge, MA, USA), mouse monoclonal anti-Acetylated α-Tubulin (1:500, T7451, clone 6-11B-1, Sigma-Aldrich), mouse monoclonal anti-ARL13B (1:500, clone N295B/66, NeuroMab, Davis, CA, USA), rabbit anti-ARL13B (1:500, 17711-1-AP, Proteintech, Chicago, IL, USA), mouse monoclonal anti-γ-Tubulin (1:10000, T6557, clone GTU-88, Sigma-Aldrich), chicken anti-GFP (1:1000, GFP-1010, Aves Lab, Tigard, OR, USA), rabbit monoclonal anti-Ki-67 (1:100, clone SP6, ab16667, abcam), mouse monoclonal anti-Aquaporin 1 (1:1000, clone 1/22, ab9566, abcam), rabbit anti-Aquaporin 1 (1:1000, AB2219, MilliporeSigma, Burlington, MA, USA), and rabbit anti-OTX2 (1:500, AB9566, MilliporeSigma), rabbit anti-Cytokeratins (1:100, Z0622, Dako, Carpinteria, CA, USA), mouse monoclonal anti-FOXJ1 (1:50, 14-9965, Clone 2A5, eBioscience, San Diego, CA, USA), and rabbit anti-TAp73 (1:200, ab40658, abcam), and sheep anti-Transthyretin (1:200, ab9015, abcam), and goat anti-Myc (1:1000, ab9132, abcam).
The investigators were blinded to group allocation in the following experiments. For analysis of TAp73 expression in human tissue samples, 9 CPPs, 8 CPCs, and 5 CPs were used. Four distinct tissue regions and 5 fields of view in each region were assessed to determine the average scores for each sample: percent positive fields of view were calculated by scoring each 40× field as “1” if there were TAp73+ cells present and “0” if there were none. For staining of primary cilia in CP tumors, the cilia pattern was assessed by analyzing five distinct tissue regions of each sample. Human tissues used included: 17 CPPs from 16 individuals, 6 CPCs from 6 individuals. Animals examined included: wild type and Mcidas−/− animals, Lmx1a-Cre;p53flox/flox;Rbflox/flox, Lmx1a-Cre;NICD1, Lmx1a-Cre;Ptchflox/flox, and Lmx1a-Cre;Ptchflox/flox;NICD1 mice, and wild type, Lmx1a-Cre;NICD1, and Lmx1a-Cre;Ptchflox/flox;NICD1 mice treated with vehicle or IMR-1. For analysis of the expression of Atoh1, SOX2, and PAX6 in embryonic cerebellum after treatment with vehicle or IMR-1, wild type animals were used.
For analysis of cell proliferation in tissues, the number of Ki-67+ cells in 200 cells was assessed from three distinct regions for each sample. The percentage of Ki-67+ cells was determined by calculating the number of Ki-67+ cells per 100 cells in each sample for each genotype or each treatment. For proliferation analysis of cultured cells, Ki-67 expression in 100–200 tumor cells or infected cells was assessed by analyzing three distinct fields. The percentage of Ki-67+ cells was determined by calculating the number of Ki-67+ cells per 100 tumor cells or infected cells in each sample for each treatment.
For analysis of multiciliation of cultured cells, primary cilia in 100–150 tumor cells or infected cells were assessed by analyzing ARL13B or γ-tubulin expression in three distinct fields. The percentage of MCCs was determined by calculating the number of MCCs per 100 tumor cells or infected cells of each sample for each treatment.
Immunoblotting
Immunoblotting was carried out as described previously [34]. Primary antibodies used included: mouse monoclonal anti-β-Actin (1:1000, clone AC-15, A5441, Sigma-Aldrich), and rabbit anti-TAp73 (1:1000, ab40658, abcam), and mouse monoclonal anti-FLAG (1:1000, clone M2, F3165, Sigma-Aldrich).
RT-qPCR, in situ hybridization and RNAscope
Multiple sets of tissue specimens were collected from animals. Gender information is not available for animals collected at days P0 and P7. Total RNA was extracted from tumor samples using Trizol (Thermo Fisher Scientific) and RNA Clean & Concentrator kits (Zymo Research, Irvine, CA, USA). Total RNA samples from normal human tissues were purchased (BioChain Institute, Inc., Newark, CA, USA). cDNA was synthesized using GoScript Reverse Transcription System (Promega, Madison, WI, USA). All reactions were run on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific). Gene-specific primers and probes were used (Gdf7, Mm.PT.58.12400445; GMNC, Hs.PT.58.15454001; MCIDAS, Hs.PT.58.22676071; FOXJ1, Hs.PT.58.40371261, Integrated DNA Technologies, Inc., Coralville, IA, USA) (Supplementary Table 2) [34]. Transcript levels were determined as the number of transcripts of genes of interest relative to those of Actb (mouse) or GAPDH (human) and normalized to the mean value of control samples. The results for each set of specimens were obtained by averaging transcript levels of technical triplicates and used for subsequent analyses. Exclusion was applied when one of the triplicates was a significant outlier, and the assay was repeated in independent experiments to validate the exclusion. For analysis of GMNC, MCIDAS, and FOXJ1 expression, human samples used included 10 CPPs, 8 CPCs, and 1 for brain, trachea, lung, testis, and epididymis. For analysis of Gmnc-deficient CP samples, animals included: 11 Lmx1a-Cre;Gmncflox/− and wild type animals, respectively. For CP tumor analysis, samples used included: wild type CP: n = 10; CPC: n = 11 from Lmx1a-Cre;p53flox/flox;Rbflox/flox mice, n = 10 for Lmx1a-Cre;p53flox/flox;Rbflox/flox;Gmncflox/− animals. For NOTCH-driven CP tumors, animals examined included: wild type, Lmx1a-Cre;NICD1 (n = 3 for each at genotype at each time point). For gene expression analysis of infected cells, 3 independent samples for infected and control cells were used, respectively.
In situ hybridization was performed as described at the In Situ Hybridization Core facility at Baylor College of Medicine [63]. Riboprobes for Gli1, Mycn, Shh, Hes1, Hes5, Gmnc, Mcidas, and Foxj1 were used. For RNAscope, Gmnc (510421), Mcidas (510401), Myb (510411), Ccno (546521), Foxj1 (317091), GMNC (566231), and FOXJ1 (430921) probes were used according to manufacturer’s instructions (Advanced Cell Diagnostics, Newark, CA, USA). For analysis of GMNC/FOXJ1 expression in human tissue samples, the investigator was blinded to group allocation. Human tissues used included: 31 CPPs from 31 individuals; 11 CPCs from 11 individuals. GEMC1 and FOXJ1 expression was assessed in five distinct tissue regions: the percentage of GEMC1- or FOXJ1-expressing cells was calculated by averaging the numbers of GEMC1+ or FOXJ1+ cells per 100 cells in five distinct tissue regions of each specimen. For RNAscope in cultured or infected tumor cells, 7 samples were used for each treatment. mRNA transcript copy number was assessed by counting the number of positive fluorescent spots in 50–100 tumor cells or infected cells in three distinct regions. The transcript levels were determined by averaging the transcript copy numbers of all cells for each treatment.
Electron microscopy and image acquisition
Transmission electron microscopy was performed as described previously [34]. The investigator was blinded to group allocation. A whole-mount bright field was obtained using a Nikon SMZ1000 Stereomicroscope. Light and fluorescent microscopic images were obtained by a Nikon Eclipse 90i microscope system, a Nikon confocal microscope system A1+ (Nikon Instruments, Melville, NY, USA), and a ZEISS LSM 980 with Airyscan 2 confocal microscope (Carl Zeiss Microscopy, LLC, White Plains, NY, USA).
Statistical analysis and reproducibility
Multiple specimens were collected from independent samples or animals for each treatment or genotype. Pilot studies were conducted, and results from these studies were used to determine the choice of sample size for the experiment. A group size of n = 10 (5 experimental, 5 control) will provide 90% power to detect a 22% change in assay results. No randomization was used to determine how samples were allocated to experimental groups. Both male and female animals were used for experiments. Experiments were repeated with similar results to eliminate the effects of gender and age on experimental findings. Information on experiment replication is provided in legends for figures and supplemental figures. Statistical analyses were performed with GraphPadPrism 9.0 (GraphPad Software Inc., La Jolla, CA, USA). All pooled data were expressed as the mean ± standard error of the mean (SEM). Variation within each group of data was examined based on the differences between each data point and the mean of the group. The Kolmogorov–Smirnov test was used to test the normal distribution of the data. Differences between two groups were compared using paired t-test or unpaired two-tailed t-test. Differences between multiple groups were analyzed with ANOVA followed by Tukey’s multiple comparisons test. Results were considered significant at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Accession numbers
Published data sets of human CP tumors (GSE14098, GSE60886) were downloaded from the GEO database. Hierarchical clustering was performed using Genesis (http://genome.tugraz.at/genesisclient/genesisclient_description.shtml). Pathway analysis using the GeneGoMetaCore Analytical Suite (http://genego.com; GeneGo) was used to score and rank pathways enriched in data sets by the proportion of pathway-associated genes with significant expression values. RNA-seq data (BioProject ID, PRJNA282889) were analyzed.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Fame RM, Lehtinen MK. Emergence and developmental roles of the cerebrospinal fluid system. Dev Cell. 2020;52:261–75.
Liddelow SA. Development of the choroid plexus and blood-CSF barrier. Front Neurosci. 2015;9:32.
Narita K, Takeda S. Cilia in the choroid plexus: their roles in hydrocephalus and beyond. Front Cell Neurosci. 2015;9:39.
Narita K, Kozuka-Hata H, Nonami Y, Ao-Kondo H, Suzuki T, Nakamura H, et al. Proteomic analysis of multiple primary cilia reveals a novel mode of ciliary development in mammals. Biol Open. 2012;1:815–25.
Nonami Y, Narita K, Nakamura H, Inoue T, Takeda S. Developmental changes in ciliary motility on choroid plexus epithelial cells during the perinatal period. Cytoskeleton. 2013;70:797–803.
Gozali AE, Britt B, Shane L, Gonzalez I, Gilles F, McComb JG, et al. Choroid plexus tumors; management, outcome, and association with the Li-Fraumeni syndrome: the Children’s Hospital Los Angeles (CHLA) experience, 1991-2010. Pediatr Blood Cancer. 2012;58:905–9.
Zaky W, Finlay JL. Pediatric choroid plexus carcinoma: Biologically and clinically in need of new perspectives. Pediatr Blood Cancer. 2018;65:e27031.
Shannon ML, Fame RM, Chau KF, Dani N, Calicchio ML, Geleoc GS, et al. Mice expressing Myc in neural precursors develop choroid plexus and ciliary body tumors. Am J Pathol. 2018;188:1334–44.
El Nagar S, Zindy F, Moens C, Martin L, Plassard D, Roussel MF, et al. A new genetically engineered mouse model of choroid plexus carcinoma. Biochem Biophys Res Commun. 2018;496:568–74.
Merve A, Zhang X, Pomella N, Acquati S, Hoeck JD, Dumas A, et al. c-MYC overexpression induces choroid plexus papillomas through a T-cell mediated inflammatory mechanism. Acta Neuropathol Commun. 2019;7:95.
Nassar M, Samaha H, Ghabriel M, Yehia M, Taha H, Salem S, et al. LC3A silencing hinders aggresome vimentin cage clearance in primary choroid plexus carcinoma. Sci Rep. 2017;7:8022.
Amer N, Taha H, Hesham D, Al-Shehaby N, Mosaab A, Soudy M, et al. Aggresomes predict poor outcomes and implicate proteostasis in the pathogenesis of pediatric choroid plexus tumors. J Neuro-Oncol. 2021;152:67–78.
Spassky N, Meunier A. The development and functions of multiciliated epithelia. Nat Rev Mol Cell Biol. 2017;18:423–36.
Lewis M, Stracker TH. Transcriptional regulation of multiciliated cell differentiation. Semin Cell Dev Biol. 2021;110:51–60.
Terre B, Piergiovanni G, Segura-Bayona S, Gil-Gomez G, Youssef SA, Attolini CS, et al. GEMC1 is a critical regulator of multiciliated cell differentiation. EMBO J. 2016;35:942–60.
Kyrousi C, Arbi M, Pilz GA, Pefani DE, Lalioti ME, Ninkovic J, et al. Mcidas and GemC1 are key regulators for the generation of multiciliated ependymal cells in the adult neurogenic niche. Development 2015;142:3661–74.
Lu H, Anujan P, Zhou F, Zhang Y, Chong YL, Bingle CD, et al. Mcidas mutant mice reveal a two-step process for the specification and differentiation of multiciliated cells in mammals. Development. 2019;146:dev172643.
Arbi M, Pefani DE, Kyrousi C, Lalioti ME, Kalogeropoulou A, Papanastasiou AD, et al. GemC1 controls multiciliogenesis in the airway epithelium. EMBO Rep. 2016;17:400–13.
Zhou F, Narasimhan V, Shboul M, Chong YL, Reversade B, Roy S. Gmnc is a master regulator of the multiciliated cell differentiation program. Curr Biol. 2015;25:3267–73.
Terre B, Lewis M, Gil-Gomez G, Han Z, Lu H, Aguilera M, et al. Defects in efferent duct multiciliogenesis underlie male infertility in GEMC1-, MCIDAS- or CCNO-deficient mice. Development. 2019;146:dev162628.
Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140:4277–86.
Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 2014;5:4418.
Ma L, Quigley I, Omran H, Kintner C. Multicilin drives centriole biogenesis via E2f proteins. Genes Dev. 2014;28:1461–71.
Wallmeier J, Al-Mutairi DA, Chen CT, Loges NT, Pennekamp P, Menchen T, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46:646–51.
Lalioti ME, Arbi M, Loukas I, Kaplani K, Kalogeropoulou A, Lokka G, et al. GemC1 governs multiciliogenesis through direct interaction with and transcriptional regulation of p73. J Cell Sci. 2019;67:2360–73.
Wildung M, Esser TU, Grausam KB, Wiedwald C, Volceanov-Hahn L, Riedel D, et al. Transcription factor TAp73 and microRNA-449 complement each other to support multiciliogenesis. Cell Death Differ. 2019;26:2740–57.
Nemajerova A, Kramer D, Siller SS, Herr C, Shomroni O, Pena T, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016;30:1300–12.
Marshall CB, Mays DJ, Beeler JS, Rosenbluth JM, Boyd KL, Santos Guasch GL, et al. p73 is required for multiciliogenesis and regulates the Foxj1-associated gene network. Cell Rep. 2016;14:2289–300.
Kyrousi C, Lalioti ME, Skavatsou E, Lygerou Z, Taraviras S. Mcidas and GemC1/Lynkeas specify embryonic radial glial cells. Neurogenesis. 2016;3:e1172747.
Ortiz-Alvarez G, Daclin M, Shihavuddin A, Lansade P, Fortoul A, Faucourt M, et al. Adult neural stem cells and multiciliated ependymal cells share a common lineage regulated by the geminin family members. Neuron. 2019;102:159–72 e7.
Lalioti ME, Kaplani K, Lokka G, Georgomanolis T, Kyrousi C, Dong W, et al. GemC1 is a critical switch for neural stem cell generation in the postnatal brain. Glia. 2019;67:2360–73.
Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci. 2015;16:445–57.
Beschorner R, Waidelich J, Trautmann K, Psaras T, Schittenhelm J. Notch receptors in human choroid plexus tumors. Histol Histopathol. 2013;28:1055–63.
Li L, Grausam KB, Wang J, Lun MP, Ohli J, Lidov HG, et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nat Cell Biol. 2016;18:418–30.
Eberhart C. Multiple cilia suppress tumour formation. Nat Cell Biol. 2016;18:368–9.
Huang X, Ketova T, Fleming JT, Wang H, Dey SK, Litingtung Y, et al. Sonic hedgehog signaling regulates a novel epithelial progenitor domain of the hindbrain choroid plexus. Development. 2009;136:2535–43.
Merino DM, Shlien A, Villani A, Pienkowska M, Mack S, Ramaswamy V, et al. Molecular characterization of choroid plexus tumors reveals novel clinically relevant subgroups. Clin Cancer Res. 2015;21:184–92.
Japp AS, Gessi M, Messing-Junger M, Denkhaus D, Zur Muhlen A, Wolff JE, et al. High-resolution genomic analysis does not qualify atypical plexus papilloma as a separate entity among choroid plexus tumors. J Neuropathol Exp Neurol. 2015;74:110–20.
Ruland V, Hartung S, Kordes U, Wolff JE, Paulus W, Hasselblatt M. Choroid plexus carcinomas are characterized by complex chromosomal alterations related to patient age and prognosis. Genes Chromosomes Cancer. 2014;53:373–80.
Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, et al. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature. 2013;504:456–9.
Chizhikov VV, Lindgren AG, Currle DS, Rose MF, Monuki ES, Millen KJ. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133:2793–804.
Tabori U, Shlien A, Baskin B, Levitt S, Ray P, Alon N, et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28:1995–2001.
Tong Y, Merino D, Nimmervoll B, Gupta K, Wang YD, Finkelstein D, et al. Cross-species genomics identifies TAF12, NFYC, and RAD54L as choroid plexus carcinoma oncogenes. Cancer Cell. 2015;27:712–27.
Wang J, Merino DM, Light N, Murphy BL, Wang YD, Guo X, et al. Myc and loss of p53 cooperate to drive formation of choroid plexus carcinoma. Cancer Res. 2019;79:2208–19.
Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004.
Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA. 2003;100:14920–5.
Uhmann A, Dittmann K, Nitzki F, Dressel R, Koleva M, Frommhold A, et al. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood 2007;110:1814–23.
Astudillo L, Da Silva TG, Wang Z, Han X, Jin K, VanWye J, et al. The small molecule IMR-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016;76:3593–603.
Chang AC, Fu Y, Garside VC, Niessen K, Chang L, Fuller M, et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell. 2011;21:288–300.
Crawford JR, Isaacs H Jr. Perinatal (fetal and neonatal) choroid plexus tumors: a review. Childs Nerv Syst. 2019;35:937–44.
Toescu SM, James G, Phipps K, Jeelani O, Thompson D, Hayward R, et al. Intracranial Neoplasms in the First Year of Life: Results of a Third Cohort of Patients From a Single Institution. Neurosurgery. 2019;84:636–46.
Wall DS, Mears AJ, McNeill B, Mazerolle C, Thurig S, Wang Y, et al. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J Cell Biol. 2009;184:101–12.
Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, et al. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PloS One. 2011;6:e14680.
Ingram WJ, McCue KI, Tran TH, Hallahan AR, Wainwright BJ. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene. 2008;27:1489–500.
Kong JH, Yang L, Dessaud E, Chuang K, Moore DM, Rohatgi R, et al. Notch activity modulates the responsiveness of neural progenitors to sonic hedgehog signaling. Dev Cell. 2015;33:373–87.
Stasiulewicz M, Gray SD, Mastromina I, Silva JC, Bjorklund M, Seymour PA, et al. A conserved role for Notch signaling in priming the cellular response to Shh through ciliary localisation of the key Shh transducer Smo. Development. 2015;142:2291–303.
Guseh JS, Bores SA, Stanger BZ, Zhou Q, Anderson WJ, Melton DA, et al. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development. 2009;136:1751–9.
Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–22.
Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 2009;136:2297–307.
Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci. 2010;123:213–24.
Deblandre GA, Wettstein DA, Koyano-Nakagawa N, Kintner C. A two-step mechanism generates the spacing pattern of the ciliated cells in the skin of Xenopus embryos. Development. 1999;126:4715–28.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer (IARC); 2016.
Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn. 2005;234:371–86.
Acknowledgements
We are grateful to Drs. Roger Packer, Huizhen Zhang, Brian Rood, William Weiss, Joanna Philips, Michael Taylor, James Loukides, and Sandro Santagata for providing tissue samples, and Monica Calicchio for assisting with tissue specimens from Boston Children’s Hospital (BCH). We thank Lars Udo-Bellner, Melanie Schweitzer, Amanda Chiang, Tamanna Sarowar, Brightlyn Kwa, Maheen Umer, Mariam Zahran, Navjot Guru, and Claire Evans for technical assistance. This project was supported by NIH T32 HL110852 and BCH Faculty Development Fellowship (RMF); NIH R01 NS088566 (MKL) and the New York Stem Cell Foundation (MKL). MKL is a New York Stem Cell Foundation—Robertson Investigator. US is supported by the Fördergemeinschaft Kinderkrebszentrum Hamburg. THS was supported by the Spanish Ministry of Science, Innovation and Universities (MCIU: PGC2018-095616-B-I00/GINDATA and FEDER), and is currently supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. SR is supported by funds from the A*STAR, Singapore. HZ is supported by New York Institute of Technology College of Osteopathic Medicine, New York Institute of Technology Imaging Center, Matthew Larson Foundation, NIH Institutional Development Awards (5P20GM103548 and 1P20GM103620-01A1) and NIH R01 CA220551. The RNA In Situ Hybridization Core facility at Baylor College of Medicine is supported by an NIH Shared Instrumentation grant (1S10OD016167).
Author information
Authors and Affiliations
Contributions
QL, ZH, ZL, and HZ conceived and designed the study; SR, ZL, THS, and HZ performed development of methodology and writing, review, editing, and revision of the manuscript; QL, ZH, NS, BT, RMF, MKL, HGWL, UA, TDP, JQV, and YZ provided acquisition, analysis and interpretation of data, and statistical analysis; TZ, AA, YH, PC, HL, SR, JW, RJW, KS, LW, US, and ZL provided technical and material support. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Tissue samples were obtained with informed consent from patients and approval by institutional review boards at Shanghai East Hospital, Sanford Burnham Prebys Medical Discovery Institute, and University Medical Center Hamburg-Eppendorf. All tissue specimens from Boston Children’s Hospital were obtained under an approved by institutional review board protocol. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by B. Dynlacht
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Han, Z., Singh, N. et al. Disruption of GMNC-MCIDAS multiciliogenesis program is critical in choroid plexus carcinoma development. Cell Death Differ 29, 1596–1610 (2022). https://doi.org/10.1038/s41418-022-00950-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-00950-z
- Springer Nature Limited
This article is cited by
-
Breakthroughs in choroid plexus and CSF biology from the first European Choroid plexus Scientific Forum (ECSF)
Fluids and Barriers of the CNS (2024)
-
RBL2 represses the transcriptional activity of Multicilin to inhibit multiciliogenesis
Cell Death & Disease (2024)
-
Multi-omics analyses of choroid plexus carcinoma cell lines reveal potential targetable pathways and alterations
Journal of Neuro-Oncology (2024)