
Abstract
During female mammal reproductive tract development, epithelial cells of the lower Müllerian duct are committed to become stratified squamous epithelium of the vagina and ectocervix, when the expression of ΔNp63 transcription factor is induced by mesenchymal cells. The absence of ΔNp63 expression leads to adenosis, the putative precursor of vaginal adenocarcinoma. Our previous studies with genetically engineered mouse models have established that fibroblast growth factor (FGF)/mitogen-activated protein kinase (MAPK), bone morphogenetic protein (BMP)/SMAD, and activin A/runt-related transcription factor 1 (RUNX1) signaling pathways are independently required for ΔNp63 expression in Müllerian duct epithelium (MDE). Here, we report that sine oculis homeobox homolog 1 (SIX1) plays a critical role in the activation of ΔNp63 locus in MDE as a downstream transcription factor of mesenchymal signals. In the developing mouse reproductive tract, SIX1 expression was restricted to MDE within the future cervix and vagina. SIX1 expression was totally absent in SMAD4 null MDE and was reduced in RUNX1 null and FGFR2 null MDE, indicating that SIX1 is under the control of vaginal mesenchymal factors: BMP4, activin A and FGF7/10. Furthermore, Six1, Runx1, and Smad4 gene-dose-dependently activated ΔNp63 expression in MDE within the vaginal fornix. Using a mouse model of diethylstilbestrol (DES)-associated vaginal adenosis, we found DES action through epithelial estrogen receptor α (ESR1) inhibits activation of ΔNp63 locus in MDE by transcriptionally repressing SIX1 and RUNX1 in the vaginal fornix.
Similar content being viewed by others


Introduction
In mammals, the majority of the female reproductive tract (FRT) develops from the Müllerian ducts (MDs) [1,2,3,4,5,6]. During embryogenesis, the MDs undergo a dynamic transformation from simple tubes consisting of homogeneous epithelium and mesenchyme into distinct organs, namely the oviduct, uterus, cervix, and vagina [7, 8]. Classic tissue recombination studies have established that organ-specific mesenchyme induces differentiation of MD epithelium (MDE) into epithelia with unique morphology and functions [9,10,11]. In the lower MD, epithelial cells are committed to become stratified squamous epithelium of ectocervix and vagina (together referred to as “vagina” hereafter), as the expression of ΔNp63 transcription factor is induced by vaginal mesenchyme [12,13,14]. In MDE of the developing vagina, the expression of ΔNp63 is activated by mesenchymal paracrine factors: bone morphogenetic protein (BMP) 4, activin A (ActA), and fibroblast growth factor (FGF) 7 or 10 [15, 16]. SMAD4 is essential for the activation of ΔNp63 in MDE, and this transcription factor binds on the 5′ sequence adjacent to the transcription start site (TSS) of ΔNp63 in future vaginal epithelium (VgE) [16]. This SMAD-dependent activation of the ΔNp63 locus requires runt-related transcription factor 1 (RUNX1). In MDE, the expression of RUNX1 is activated by ActA through a SMAD-independent mechanism [15]. In addition, activation of the mitogen-activated protein kinase (MAPK) pathway by FGF7/10-FGF receptor 2 IIIb (FGFR2IIIb) is essential for the activation of the ΔNp63 locus in MDE [15]. Once the ΔNp63 locus is activated in MDE, the transcriptional activity of the ΔNp63 locus is maintained by ΔNp63 protein itself independent of mesenchymal factors [11, 12, 16].
BMP4-SMADs, ActA-RUNX1, and FGF7/10-MAPK pathways are independently required for the vaginal cell fate commitment of MDE. Inactivation of Smad4, Runx1, or Fgfr2 in MDE results in uterine epithelial differentiation of MDE within the vagina, a congenital epithelial lesion called vaginal adenosis [15, 16]. Vaginal adenocarcinomas (VACs) are believed to arise from vaginal adenosis because of the presence of adenosis lesions at the primary site of VACs [17]. Hence, better understanding in etiology of vaginal adenosis is crucial in order to develop preventive and therapeutic approaches for VACs. The etiology of vaginal adenosis and VAC is commonly associated with intrauterine exposure to estrogenic compounds, including diethylstilbestrol (DES) [18]. Women who were exposed to DES in the womb of mothers (DES daughters) developed vaginal adenosis [19] and had a ~40 times higher risk of developing a specific type of VAC, vaginal clear cell adenocarcinoma [20]. Developmental exposures of rodents to estrogenic compounds also induce vaginal adenosis [21,22,23,24,25,26].
In this study, we investigated the role of sine oculis homeobox homolog 1 (SIX1) in the cell fate commitment of VgE. The vertebrate Six genes are homologs of Drosophila ‘sine oculis’ (so) [27]. The Six genes comprise an evolutionally conserved gene regulatory network with paired box (Pax) and eyes absent (Eya) [28, 29]. In mammals, the Six genes (Six1–6) cooperatively regulate the developmental process in multiple organs [30, 31]. Six1 null mutant mice die at birth, exhibiting craniofacial abnormalities and agenesis of thymus and kidney [32]. In humans, SIX1 mutations cause Branchiootic syndrome 3 (MIM#608389) [33], characterized by hearing loss, branchial cleft fistulas/cysts and renal dysplasia. SIX1 mutations also cause Deafness, autosomal dominant 23 (DFNA23, MIM#605192) [34], which is characterized by bilateral hearing impairment without renal malformations. In mouse FRTs, Six1 is enriched in the vagina compared with the uterus [16, 35]. However, its biological function in FRT remains unclear.
Our current mouse genetic study reveals that SIX1 cooperates with RUNX1 and SMAD4 in the activation of the ΔNp63 locus in MDE as a downstream transcription factor of BMP4, ActA, and FGF7/10. Here, we provide evidence that DES blocks the activation of ΔNp63 locus in future VgE by repressing SIX1 and RUNX1 through epithelial estrogen receptor α (ESR1). Such discoveries from our models may contribute to developing preventive and therapeutic treatments of VACs, the etiology of which is currently unknown.
Materials and methods
Mouse models
All animal procedures were approved by the Animal Care and Use Committee in the Ohio State University. The mouse strains carrying the following alleles were utilized: Six1flox [Six1tm2.1Mair] [36], Trp63flox [Trp63tm3.2Brd] [37], ΔNp63-EGFP knock-in (Trp63ΔNp63-EGFP-KI) [38], Runx1flox [Runx1tm1Tani] [39], Fgfr2flox [Fgfr2tm1Dor/J] [40], ROSAmT-mE [Gt(ROSA)26Sortm4(ACTB-tdTomato,–EGFP)Luo/J] [41], ROSAMAP2K1DD [Gt(ROSA)26Sortm8(Map2k1*,EGFP)Rsky/J] [42], Smad4flox (Smad4tm2.1Cxd/J) [43], Esr1flox [44], Pax2-Cre [Tg(Pax2-cre)1Akg] [45], Wnt7a-Cre [46], and ΔNp63-Cre [Trp63tm1.1(cre)Ssig/J] [47]. C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). MDE-specific conditional knockout (cKO) and conditional heterozygous (cHET) mice were generated by crossing lines carrying floxed alleles with Wnt7a-Cre mice, except for Trp63flox mice, which were crossed with Pax2-Cre because Trp63flox/flox; Wnt7a-Cre mice were embryonic lethal [16]. The day of birth was counted as postnatal day (PD) 1.
Neonatal DES treatment
The ~0.04 mg/mm DES filled tubing [16] was cut into 1 mm lengths and subcutaneously injected into newborn mice using a 19-gauge trocar.
Immunofluorescence (IF) and immunohistochemistry (IHC)
IF and IHC assays were performed as previously described [48]. The following primary antibodies were used at the indicated dilutions: anti-TRP63 (4A4) (1:200, 790–4509) from Ventana Medical Systems (Tucson, AZ); anti-ΔNp63 (1:2,000, PC373) from Millipore (Billerica, MA); anti-PGR (1:200, A0098) from Agilent Technologies (Santa Clara, CA); anti-RUNX1 (1:400, 2593-1) from Epitomics (Burlingame, CA); anti-phospho (p)-MAPK1/3 (p-T202/Y201, 1:30, #4370) and anti-pSMAD1/5/9 (1:50, #9511) from Cell Signaling Technology (Danvers, MA); anti-GFP (1:100, ab6673) from Abcam (Cambridge, MA); anti-SIX1 (1:800, HPA001893) from Millipore Sigma (St. Louis, MO); anti-ESR1 (1:100, RM-9101) from Lab Vision (Fremont, CA); anti-pan-Cytokeratin (AE1/AE3) (1:100, sc-81714) from Santa Cruz Biotechnology (Dallas, TX). For IF assay, Alexa-Fluor594 anti-mouse IgG (1:1,000, 715-586-150) and Alexa-Fluor488 anti-rabbit IgG (1:1,000, 711-546-152) (Jackson ImmunoResearch, West Grove, PA) were used for the secondary antibodies, and Hoechst 33258 (1:10,000, Sigma-Aldrich) was used for nuclear staining. For IHC with DAB (3,3′-diaminobenzidine, Sigma-Aldrich), biotinylated anti-rat goat IgG (1:800, 705-065-147) and streptavidin-horseradish peroxidase (1:400, 016-030-084) (Jackson ImmunoResearch) were used. Micrographs were captured using a BZ-9000 microscope (Keyence, Osaka, Japan) under identical conditions between samples for each antibody. The contrast of images was adjusted by applying identical parameters to the images for each antibody with the batch-process function of Adobe Photoshop CS6 (Adobe, San Jose, CA). To capture a wide area in a single image, tissue sections were scanned in multiple frames, and the images were automatically merged together utilizing the Image Stitching function of image analysis tool (Keyence).
Morphometric analysis
The methods for the quantitative analysis on the squamous transformation of MDE [16] and the IF signal [49] were previously described. We adapted these methods with some modifications. The length of epithelium at the basal lamina was measured in the outer wall of vaginal fornix in at least two TRP63-immunostained coronal-sections per animal. The proportion of epithelium with ΔNp63-positive basal cells was calculated by “length of epithelial basement membrane associated with TRP63-positive cells” ÷ “total epithelial basement membrane length” × 100, for each mouse. Basal cell density in the outer and inner fornix walls was calculated by the number of TRP63-positive pixels per epithelial basement membrane length. In tissue sections of vaginal fornices stained for TRP63, epithelial areas were manually selected, and the pixels positive for TRP63 signal within the epithelium were selected by adjusting the lower threshold for positivity to exclude background noise. Epithelial basement membrane was manually marked on the IF images, and the p63-positive area and the basement membrane length were measured utilizing Image J (NIH, Bethesda, MD). Analysis was performed on ≥4 fornices from ≥3 mice per group. The value in each fornix was considered as a single measurement. Statistical significance was analyzed by One-way ANOVA with post-hoc Tukey’s honestly significant difference test.
SIX1 IF analysis
Quantitative IF assay was performed as previously described with modifications [50]. Tissue sections were stained together, and images were captured at the same time under identical conditions. Images of ≥4 coronal tissue sections from n ≥ 3 independent animals were analyzed for each group. Epithelial areas were manually selected, and the signal intensity per pixel within the epithelial area was measured by Image J. In all experiments, approximately equivalent areas were analyzed in each sample. Since there was no significant intragroup difference in the average signal intensity, all samples in each group were plotted together, and the distributions of signals were compared between groups by the Mann–Whitney U test with continuity correction.
Immunoblot analysis
Ovaries, uteri and vaginae from PD2 mice (5–6 mice per blot) were homogenized in ice-cold lysis buffer containing protease (cOmplete Protease Inhibitor Cocktail, Roche, Basel, Schweiz) and phosphatase (phoSTOP, Roche) inhibitors and loaded onto NuPAGE 4–12% Bis-Tris precast SDS-PAGE gel (ThermoFisher, Waltham, MA). Proteins were transferred to a PVDF membrane (Millipore Sigma). The membrane was incubated with anti-RUNX1 (1:2,000, Epitomics), anti-SIX1 (1:1,000, Millipore Sigma) and anti-GAPDH (1:2,000, G8795, Millipore Sigma) antibodies in the OdysseyR Blocking buffer (LI-COR Biosciences, NE) overnight at 4 °C. After incubation with IRDye® 800CW and 680LT Donkey secondary antibodies, the signal was detected using Odyssey CLx Imaging System (LI-COR Biosciences). The analysis was repeated three times with independent samples.
Uterine organ culture
Uterine hanging drop organ culture was performed as previously described with minor modifications [15, 16]. Briefly, uteri were dissected from PD1 mice, cleaned by removing connective tissues, and cut into 3 pieces per uterine-horn in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F12 (DMEM/F12, 11039, ThermoFisher) containing 10 nM ICI 182,780 (Millipore Sigma). The uterine pieces were then placed in autoclaved PCR tube caps (AXYGEN, Union City, CA) with basal medium (10 nM ICI 182,780 DMEM/F12 with Insulin-Transferrin-Selenium and Antibiotic-Antimycotic) with/without 20 ng/ml human recombinant BMP4, ActA and/or FGF10 (ThermoFisher), inverted, and incubated. Uterine pieces were cultured up to 3 days with daily medium change, fixed with Modified Davidson’s fixative, and processed for histological analysis.
Quantitative real-time PCR (qRT-PCR)
Newborn female mice were implanted with a vehicle (Veh, empty pellet) or DES pellet within 12 h after birth (n ≥ 3). At 24 and 48 h after pellet implantation, uterine horns and the upper half of vaginae were collected, immediately snap-frozen and stored at −80 °C until use. Total RNA was extracted from vagina and uteri of each mouse separately utilizing RNeasy Plus Mini or Micro Kits (Qiagen, Hilden, Germany). cDNA was synthesized using SuperScript II (ThermoFisher) with oligo(dT) primer, and qRT-PCR was performed on a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA) using KAPA SYBR FAST qPCR kits (Kapa Biosystems, Inc., Wilmington, MA). Primer sequences in this study are available upon request. The relative expression values of target transcripts were calculated by normalizing the threshold cycle (CT) value to that of Cdh1 for epithelial genes (Six1, Runx1, Fgfr2IIIb, and ΔNp63), and Vim for mesenchymal genes (Bmp4, Fgf7, Fgf10, and Inhba). The average values of Veh-treated vaginal or uterine samples at 24 h were set as 1.0. The qRT-PCR data were statistically analyzed by F-test followed by a Student’s t test or Welch’s t test between Veh-treated and DES-treated tissues in each time point.
Results
Expression patterns of SIX1 in neonatal FRTs
ΔNp63α is the dominant isoform of TRP63/TP63 in mouse/human VgE [14, 16]. To identify molecules that control epithelial cell fate in the lower FRT, we conducted microarray analysis of postnatal day 2 (PD2) vagina and uterus from MDE-specific cKO and conditional heterozygous (cHET, control) mice of Trp63 with Pax2-Cre [16]. Differential gene expression between cHET vagina and cHET uterus (Fig. 1a) reflects both the upstream and downstream signaling of TRP63 [16], whereas the comparison of Trp63 cKO and cHET vaginae identifies the downstream targets of TRP63. In the analysis, Six1 was more enriched in vaginae than uteri (2.03-fold-change, p = 0.0013) (Fig. 1a). The level of Six1 in the neonatal vagina was not significantly different between Trp63 cKO and cHET mice (Log2 cHET/cKO = −0.176, p = 0.23) [16], indicating that Six1 is not the target of TRP63. Immunoblotting confirmed the result: SIX1 protein was detected in PD2 vaginae but not in uteri (Fig. 1b).

a Volcano plot displaying differential expressed genes in mouse PD2 uterus and vagina. Genes significantly enriched in vagina and uterus in microarray analysis [16] are marked in red and green, respectively. b Immunoblot analysis of SIX1 and RUNX1 expression in PD2 mouse FRT and ovary. The vagina was divided into upper and lower half. Immunofluorescence assay for RUNX1, SIX1 (green) and TRP63 (red) in the lower FRT at PD1 (c) and PD2 (d, e). Outer wall of fornix is marker with dotted line (d). In the vaginal fornix (e), RUNX1 is downregulated in MDE upon expression of TRP63 (white arrows), whereas TRP63 and SIX1 are co-expressed (yellow arrows). Bar = 100 µm (c, d), =50 µm (e).
SIX1 expression progressed from posterior to anterior in developing vagina. At birth, SIX1 was expressed in the MDE of the lower vagina but not in the upper vagina and cervix, where RUNX1 was already expressed (Fig. 1c). By PD2, SIX1 expression extended to the cervix (Fig. 1d). However, there were substantial differences in the expression patterns of RUNX1 and SIX1. RUNX1 expression was reduced in the posterior portion from the outer wall of the fornix (Fig. 1d, outer wall of fornix is marked with white dotted line), whereas SIX1 was expressed at similar levels in both inner and outer walls of the fornix (Fig. 1d). In addition, RUNX1 was downregulated upon expression of ΔNp63 (Fig. 1e, white arrow) [16], whereas SIX1 expression persisted in ΔNp63-positive cells (Fig. 1e, yellow arrow).
Regulation of SIX1 expression in MDE
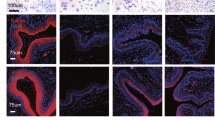
SIX1 was expressed in the fornices of ΔNp63 cKO and cHET mice [16] at PD14, confirming that expression of SIX1 is independent of ΔNp63 (Fig. 2a). In contrast, PD2 Smad4 cKO mice with Wnt7a-Cre [16] completely lacked the expression of SIX1 in the entire MDE (n = 5) (Fig. 2b). In PD2 Runx1 cKO mice with Wnt7a-Cre [16], SIX1 was expressed throughout the VgE, however the expression of SIX1 in the fornix was reduced compared with Runx1 cHET mice (Figs. 2c and 3a, b; p value < 2.2 × 10–16). This was not due to delayed development, as SIX1 expression in the vaginal fornix remained low in PD4 Runx1 cKO mice (Fig. 3a). Thus, the expression level of SIX1 in MDE is positively regulated by ActA-RUNX1 signaling activity. Similarly, SIX1 expression was slightly reduced in the fornix of Fgfr2 cKO mice with Wnt7a-Cre, in which vaginal MDE undergoes uterine differentiation [15] (Fig. 3c). However, SIX1 expression in the fornix was upregulated when the vaginal defect of Fgfr2 cKO MDE was corrected with the expression of a constitutively active MAP2K1 (MAP2K1DD) [15] (Fig. 3c, d, cHET v.s. cKO; p value < 2 × 10–16, cKO v.s. cKO + MK; p value < 2 × 10–16, cHET v.s cKO + MK; p value < 2 × 10–16), suggesting that MAPK activity modulates the expression level of SIX1 in the vaginal fornix. Accordingly, we tested the effect of BMP4, ActA and FGF10, three vaginal mesenchymal factors that induce ΔNp63 in MDE, on SIX1 expression in uterine organ culture assay. ActA and FGF10 had no effect on SIX1 expression in uterine explants and only BMP4 slightly increased SIX1 in UtE, but nuclear expression was mostly absent (Fig. 3e). When all three factors were combined, nuclear SIX1 expression was detected in a portion of UtE. The areas showing nuclear SIX1 expression also contained ΔNp63-positive cells, suggesting that SIX1 is involved in the signaling pathway that activates ΔNp63 in MDE. In uterine organ culture, diffusion of FGF10 within connective tissues is limited, due to its high affinity to heparan sulfate [51]. Accordingly, we replaced FGF10 with the epithelial expression of MAP2K1DD, which by itself did not induce expression of ΔNp63 [15]. ActA and BMP4 efficiently induced SIX1 as well as ΔNp63 in Map2k1DD transgenic UtE (Fig. 3e, f), suggesting that SIX1 is the downstream transcription factor of vaginal mesenchymal factors.

In all figures, outer wall of fornix is shown on the right side. a SIX1 expression is maintained in the vaginal fornix of ΔNp63 cKO mice (PD14) (n ≥ 4). Bar = 50 µm. b SIX1 expression in MDE is SMAD4 dependent. At PD2, SIX1 is totally absent in the MDE of Smad4 cKO mice, which normally express RUNX1 in MDE. c Expression of RUNX1 and SIX1 in the lower FRT of PD2 Runx1 cHET and cKO mice. RUNX1 null vaginal/cervical epithelium is outlined by doted lines. Nuclear expression of SIX1 expression is reduced in the fornices of Runx1 cKO mice. Bars = 100 µm.

a SIX1 expression patterns in the vaginal fornices of Runx1 cHET and cKO mice at PD2 and PD4. In the fornix of Runx1 cKO mice, nuclear expression of SIX1 increases from PD2 to PD4, but the overall expression level of SIX1 in MDE remains low and uneven. b Violin plot of SIX1 IF signal distribution in the fornix of PD2 Runx1 cHET and cKO mice (n ≥ 4 per group). The signal distributions of two groups are significantly different (p < 2 × 10–16). c Expression of SIX1 in the vaginal fornix of Fgfr2 mutant mice. SIX1 is reduced in the fornix of Fgfr2 cKO mice, but the SIX1 expression level is restored by expression of MAP2K1DD. d Violin plot of SIX1 IF signal distribution in the fornix of PD2 Fgfr2 cHET, Fgfr2 cKO, and Fgfr2 cKO with MAP2K1DD (cKO + MK) mice (n = 4 per group). The signal distributions are significantly different among three groups (p < 2 × 10–16). e Regulation of SIX1 in cultured uterine explants. 20 ng/ml BMP4 has a weak effect on the expression of SIX1 in UtE. The combination of BMP4 (b), ActA (a), and FGF10 (f) (20 ng/ml each) induced nuclear expression of SIX1 and ΔNp63 in restricted regions of UtE. Replacement of FGF10 with Map2k1DD transgene (MK) efficiently induced SIX1 and ΔNp63 in UtE. f Violin plot of SIX1 IF signal distribution in the UtE of cultured uterine explants (n ≥ 4 per group). The signal distributions are significantly different among groups (p < 2 × 10–16). Bars = 100 µm.
Six1 and Runx1 dose-dependently promote ΔNp63 expression in MDE
Since Six1 null mice die before vaginal epithelial differentiation occurs [52], the role of SIX1 in VgE differentiation was assessed by genetically inactivating Six1 in MDE by Wnt7a-Cre [46]. The loss of SIX1 in MDE affected the formation of the ΔNp63-positive basal epithelial layer in the vaginal fornix (Fig. 4a). Thus, SIX1 is one of several key transcription factors in the vaginal cell fate commitment of MDE. However, the vaginal defect of Six1 cKO mice was relatively minor compared with Smad4, Runx1, and Fgfr2 cKO mice: while Smad4, Runx1, and Fgfr2 cKO mice lost ΔNp63 expression in a significant portion of vagina, the defect of Six1 cKO mice was restricted to the epithelium on the outer wall of vaginal fornix, where the expression of RUNX1 is reduced (Fig. 1d). Meanwhile, RUNX1 expression in the vaginal fornix was not affected in Six1 cKO mice (Fig. 4a). Hence, we generated compound conditional mutant mice of Six1 and Runx1 with Wnt7a-Cre to assess if SIX1 and RUNX1 collaborate in the ΔNp63 expression of MDE in the outer wall of the vaginal fornix. Monoallelic loss of Runx1 in MDE exaggerated the effect of Six1 allelic loss on ΔNp63 expression: While monoallelic loss (cHET) of Six1 or Runx1 alone had no evident effect on the formation of ΔNp63-positive basal layer, the Six1;Runx1 double cHET mice showed gaps in the ΔNp63-positive basal cells in the outer wall of the fornix (Fig. 4b, c). The ΔNp63-negative epithelial area expanded further to the inner wall of the fornix when biallelic loss (cKO) of Six1 was combined with monoallelic loss of Runx1 (Fig. 4b, c). Moreover, the ΔNp63-negative MDE within the fornix expressed progesterone receptor (PGR), indicating the uterine cell fate commitment [53] (Fig. 4d).

a Six1 cKO mice showed minor defects in ΔNp63 expression in the outer wall (ow) of vaginal fornix. The ΔNp63-negative epithelial regions are indicated by arrows. b–d Gene-does effect of Six1 and Runx1 on vaginal cell fate commitment of MDE in the vaginal fornix. The outer fornix wall is on the right side. b Expression of ΔNp63 (red) and RUNX1 (green). c Proportion of MDE lined with ΔNp63-psotive basal layer on the outer wall of vaginal fornix. d Expression of uterine epithelial marker (PGR, green). The epithelium is highlighted with cytokeratin (red). Bars = 100 µm.
Gene-dose-dependent function of Six1, Runx1, and Smad4 in activation of ΔNp63 locus in MDE
The distinctive vaginal phenotypes of Six1 cKO and Smad4 cKO mice indicate that SMAD4 works independent of SIX1 in vaginal cell fate commitment of MDE. Accordingly, we assessed if the efficacy of SIX1 and RUNX1 in the activation of ΔNp63 expression in MDE is affected by monoallelic loss of Smad4 gene, which alone does not affect the formation of ΔNp63-positive basal layer in VgE [16]. Six1;Smad4 double cHET mice expressed ΔNp63 throughout the vagina at PD4. However, the density of basal cells on the outer wall of the fornix was reduced (Fig. 5a). The combinatorial effect of Six1 and Smad4 alleles became more prominent when an additional Six1 allele was inactivated (Fig. 5a). Similarly, monoallelic loss of Smad4 and Runx1 does-dependently affected the density of ΔNp63 in the fornix (Fig. 5a). Accordingly, Six1;Smad4;Runx1 triple cHET mice demonstrated gaps in the ΔNp63-positive basal layer throughout the vaginal fornix (Fig. 5a). The effect of monoallelic Smad4 loss on the density of TRP63-positive cells was statistically significant in mice with certain genotypes (Fig. 5b, c, Table S1). For instance, the TRP63-positive cell density in Six1 cHET mice became significantly lower than WT mice with the monoallelic loss of Smad4 (Six1 cHET; Smad4 cHET) (Fig. 5b, c).

a Monoallelic loss of Smad4 exaggerates effects of Six1 and Runx1 null alleles on ΔNp63 expression (green) in MDE. The outer fornix wall is shown on the right side. Breaks in the ΔNp63-positive basal layer in the inner fornix wall are marked by arrowheads. Bar = 50 µm. b, c Basal cell density (TRP63-positive nuclear area per epithelial basement membrane length) in the outer and inner fornix walls of Six1, Runx1, and Smad4 compound mutant mice. The sample number in each group is marker on the bars. The result is demonstrated by average means ± SD. The comparisons that become significantly different by monoallelic Smad4 loss are marked by lines with p value.
Regulatory elements of ΔNp63
The gene-dose-dependent effect of Six1, Runx1, and Smad4 on ΔNp63 activation suggests collaboration among these transcription factors. The analysis of evolutionally conserved regions by ECR browser [54] identified numerous numbers of putative enhancer elements within TP63/Trp63 locus. Many of these conserved sequences near ΔNp63 TSS contained binding sites for SMADs, RUNX1 and SIX1 (Fig. S1). The 5′ sequence proximal to ΔNp63 TSS, to which SMAD4 binds in VgE [16], also contained binding sites of RUNX1 and SIX1 (Fig. S2A). Thus, we generated transgenic mice to test if the putative 5′ proximal enhancer and the promoter are sufficient to replicate the expression patterns of ΔNp63 (Fig. S2). However, the transgene (Cre-ires-EGFP) was not expressed in any tissues of five founders and their progenies, indicating the insufficiency of the sequence to replicate ΔNp63 expression. Furthermore, when ΔNp63-Cre knock-in mice, in which the coding sequence in the first exon of ΔNp63 was replaced with Cre [47] were crossed with ROSAmT-mE reporter mice [41], VgE was mostly negative for mEGFP (n = 3, Fig. S2C). ConTra v3 analysis [55] identified conserved binding sites of SMAD1, SMAD4 and RUNX1 within the sequence deleted in ΔNp63-Cre knock-in mice (Fig. S1E). Thus, the efficient activation of ΔNp63 locus in MDE appears to require cooperation of multiple regulatory elements including the protein coding sequence within exon 1.
Developmental exposure to diethylstilbestrol (DES) downregulates Runx1 and Six1 in the vaginal fornix
Previously, we demonstrated that downregulation of RUNX1 is involved in the pathogenesis of DES-associated vaginal adenosis [16] (Fig. 6a). However, the effect of 24-h DES treatment was more prominent on SIX1 than RUNX1: Nuclear expression of SIX1 disappeared from the MDE in the vaginal fornix and the cervix of DES-treated mice (Fig. 6b, c). Meanwhile, the downregulation of SIX1 was not likely due to the repression of BMP4-SMAD or FGF7/10-MAPK activities because DES treatment increased pMAPK1/3 in VgE and mesenchyme while it had no evident effect on the levels of pSMAD1/5/9 (Fig. 6c).

IF assay of RUNX1, SIX1 (a), pSMAD1/5/9 and pMAPK1/3 (b) Violin plot presentation of SIX1 IF signals in the outer and inner fornix walls of control and DES-treated PD2 mice (n = 4 each). SIX1 IF signals in MDE were significantly higher (***p < 2 × 10–16) in control than DES-treated mice in both outer and inner fornix walls. c for DES effects in MDE. mes; mesenchyme. FRTs are collected from PD2 female mice with/without DES treatment (24 h after DES-pellet injection). d, e Effect of DES on transcript levels of essential factors for ΔNp63 expression in the upper part of vagina. Epithelial genes (d) are normalized by Cdh1. Mesenchymal genes (e) are normalized by Vim. V, Vehicle; D, DES. f Effect of DES on the SIX1 (green) and TRP63 (red) expression in the fornix of Map2k1DD transgene mice. Bars = 100 µm.
We tested the effect of DES on mRNA levels of factors essential for ΔNp63 expression. Consistent to IF analysis, DES reduced the transcript level of Six1 in the upper vagina by 76% at 24 h and 83% at 48 h compared with the vehicle (Veh) control (Fig. 6d). Runx1 mRNA was also downregulated by ~50% in DES-treated group (Fig. 6d). In contrast, DES significantly upregulated transcripts for Fgf7 and Bmp4 in the upper vagina at 24 and 48 h (Fig. 6e). Inhba, encoding subunit of ActA, was also upregulated, but it did not reach statistically significant levels (Fig. 6e). Among four vaginal mesenchymal factors we measured, only Fgf10 was downregulated in the DES group at 24 h. Nevertheless, Fgf10 mRNA levels became comparable between DES and control groups by 48 h (Fig. 6e). Unexpectedly, DES reduced the expression of Fgfr2IIIb by more than 80% (Fig. 6e), suggesting a possibility that DES alters the epithelial cell fate in the upper vagina by blocking FGF7/10-MAPK pathway via downregulation of Fgfr2IIIb. However, DES increased the phosphorylation of MAPK1/3 in MDE within the fornix (Fig. 6c). Furthermore, nuclear staining of SIX1, which was evident in the MDE of Fgfr2 cKO mice (Fig. 3c), disappeared from the outer fornix of DES-treated mice (Fig. 6a). These observations do not support the hypothesis that DES downregulates SIX1 by blocking the FGF-MAPK pathway. Thus, we tested if the activation of MAPK1/3 by Map2k1DD counteracts DES effects on ΔNp63 and SIX1 expression in MDE. DES repressed the expression of SIX1 and ΔNp63 in Map2k1DD conditional transgenic mice with Wnt7a-Cre (Fig. 6f), rejecting the hypothesis.
Interestingly, DES upregulated mRNAs for vaginal mesenchymal factors and downstream transcription factors in the uterus (Fig. S3A). In the mouse uterus, ESR1 is expressed in the mesenchyme but not in the epithelium at birth [7, 56, 57], suggesting that DES induces vaginal mesenchymal factors in the uterus via mesenchymal ESR1.
DES inhibits activation of ΔNp63 locus in MDE through downregulation of RUNX1 and SIX1 via epithelial ESR1
Our previous tissue recombination study has established that DES blocks expression of ΔNp63 in MDE through ESR1 within the epithelial cells [12, 58]. In the experiment, expression of ΔNp63 in ESR1 null MDE was not inhibited by direct contact with ESR1-positive MDE, reasonably excluding the involvement of a juxtacrine or paracrine mechanism among epithelial cells [58]. However, the fornix-specific effect of DES was not assessed in these studies, as the anatomical structure of vagina was lost by tissue recombination. We generated MDE-specific Esr1 cKO mice with Wnt7a-Cre. The deletion of Esr1 in MDE did not change the expression patterns of SIX1, RUNX1 and ΔNp63 in the fornix (Fig. 7a). DES did not block the induction of ΔNp63 in the VgE of Esr1 cKO mice (Fig. 7b). Instead, DES exposure promoted the expression of ΔNp63 in VgE in Esr1 cKO mice (Fig. 7b), forming a continuous layer of ΔNp63-positive cells by PD3, ≥1 day earlier than normal development. This is likely due to the up-regulation of BMP4 and ActA (Fig. 6e). DES induced RUNX1 and SIX1 in the UtE of Esr1 cKO mice (Fig. S3B), further demonstrating that DES action via mesenchymal ESR1 promotes vaginal epithelial cell fate in MDE. The expression of SIX1 and RUNX1 was maintained in the vaginal fornices of DES-treated Esr1 cKO mice (Fig. 7c), suggesting that DES-ESR1 attenuates the expression of SIX1 and RUNX1 in MDE cell autonomously.

a Expression patterns of ESR1 (green) and TRP63 (red) in Esr1 cKO mice (PD3). RUNX1 and SIX1 in Esr1 cKO mice are indistinguishable from wild type mice. Effect of DES on the FRT of Esr1 cHET and cKO mice (PD3): (b) IF assay of ESR1 (green) and TRP63 (red), (c) IF assay of SIX1 and RUNX1. d Effect of DES (or control) on the phosphorylation of MAPK1/3 (green). Bars = 100 µm.
Interestingly, DES increased MAPK1/3 activities in the entire vagina of Esr1 cKO mice (Fig. 7d). This suggests that DES activates the MAPK1/3 pathway in MDE and vaginal mesenchymal via ESR1 in the mesenchyme.
Discussion
It has long been known that the differentiation of MDE into distinctive epithelia of uterus and vagina is under the control of organ-specific mesenchyme [9]. Our group has established that ΔNp63 is the master regulator of vaginal epithelial differentiation in MDE [12], and that the expression of ΔNp63 is induced by mesenchymal paracrine factors, BMP4, ActA, and FGF7/10 [15, 16]. Within MDE, the signals from underlying mesenchyme are transduced by BMP4-SMADs, ActA-RUNX1, and FGFs-MAPKs. Since mouse vaginal mesenchyme can induce ΔNp63 and squamous differentiation in human MDE, the molecules that mediate communication between mesenchyme and epithelium in the commitment of MDE to vaginal cell fate must be common between these two species [59].
In this study, we identified SIX1 as one of several key transcription factors that mediate the mesenchymal signals in the activation of ΔNp63 locus during vaginal cell fate commitment of MDE. Subsequently, we propose that vaginal mesenchymal factors induce MDE to commit to vaginal epithelial cell fate by activating the ΔNp63 locus through cooperation of multiple enhancer elements, which are activated by SMADs, RUNX1, and/or SIX1 (Fig. 8). An enhancer is a short genomic region that contains clustered binding sites for multiple transcription factors. Although many transcription factors cannot bind their target site in the context of nucleosomal DNA, enhancer-mediated simultaneous binding of multiple transcription factors can overcome the nucleosome barrier [60]. Thus, enhancers integrate multiple signaling pathways through binding of downstream effectors, and regulate gene expression by organizing accessible chromatin in cooperation with promoters [61, 62]. In cell fate commitment of MDE to VgE, BMP, ActA, and FGF pathways are integrated to prime MDE for VgE-specific gene expression programs through the simultaneous binding of SMADs, RUNX1, and SIX1 to ΔNp63 enhancers (Fig. 8). The enhancers that regulate ΔNp63 expression in MDE must be distinctive from those in the skin because Six1 null [52] and Runx1 null [63] mice do not exhibit the deformation of skin and appendages observed in ΔNp63 mutant mice [38]. The identification of key regulator elements of ΔNp63 in MDE is imperative to fully appreciate the pathogenesis of vaginal adenosis. The usage of ΔNp63 enhancers must be unique between different regions of MDE as demonstrated by the difference in the requirement of SMAD4, RUNX1, and SIX1 for ΔNp63 expression in mouse genetic studies. Our particular interest is in the ΔNp63 enhancers utilized by MDE in the outer wall of the vaginal fornix, the primary site of vaginal adenosis development. Given the heterogeneity of the cell population (only a subpopulation of epithelial cells in the vaginal fornix express ΔNp63), the narrow developmental time window (ΔNp63 expression in the upper vagina gradually progresses from caudal to cranial between PD1 and PD4), and the small tissue amount of MDE within the fornix of neonatal mice, the identification of ΔNp63 regulatory elements in MDE by current technologies is challenging.

Signals of vaginal mesenchymal factors are transduced to downstream transcription factors, and the transcription factors dose-dependently activate enhancers of ΔNp63 in MDE. Upon differentiation of VgE, ΔNp63 itself maintains the transcriptional activity of ΔNp63 locus in VgE fate independently of vaginal mesenchymal factors. DES-ESR1 activity within MDE causes vaginal adenosis by blocking the vaginal cell fate commitment of MDE interfering the signal transduction.
Since the expression patterns of ΔNp63 and RUNX1 as well as the effect of DES on the expression of these transcription factors are identical between human and mouse MDE [1, 14, 59, 64], the molecular model established in mice (Fig. 8) should explain the etiology of vaginal adenosis in DES daughters. However, VACs also occur in women who have no history of DES exposure [18, 65]. In human fetuses, the expression of ΔNp63 in the lower MD occurs during the first trimester [14, 64]. Hence, the pathogenesis of non-DES-associated VACs should involve an in utero event that disturbs cell fate commitment in MDE. In this regard, exposure to a compound that inhibits any pathways/molecules described in Fig. 8 can lead to vaginal adenosis. Some studies suggest the de novo formation of adenosis in the vagina of adult women following intravaginal applications of 5-fluorouracil cream [66,67,68], challenging our hypothesis. However, given the low detection sensitivity of routine cytology screenings for adenosis [69,70,71], adenosis cases that reported to be de novo are probably due to an increased visibility of previously imperceptible adenosis lesions enlarged by a reactive change to medical treatments.
In addition to vaginal adenosis, perinatal DES exposure of female mice induces uterine squamous metaplasia [72], a formation of squamous epithelium within the UtE. The gene expression pattern suggests that uterine squamous metaplasia results from vaginal cell fate commitment of MDE within the uterus [12, 58]. This intriguing dual-effect of DES is explained by the opposite functions of epithelial versus mesenchymal ESR1: DES action through epithelial ESR1 interferes the activation of ΔNp63 locus, whereas DES action through mesenchymal ESR1 promotes ΔNp63 expression (Fig. S4). When ESR1 is expressed in both epithelium and mesenchyme, DES effects via epithelial ESR1 are dominant. The molecular mechanisms through which DES-ESR1 represses RUNX1 and SIX1 in MDE remain unclear. Further study to elucidate the underlying molecular pathogenesis of DES-associated adenosis is essential to identify etiology of non-DES-associated vaginal adenosis and VAC.
References
Robboy SJ, Kurita T, Baskin L, Cunha GR. New insights into human female reproductive tract development. Differentiation. 2017;97:9–22.
Roly ZY, Backhouse B, Cutting A, Tan TY, Sinclair AH, Ayers KL, et al. The cell biology and molecular genetics of Mullerian duct development. Wiley Interdiscip Rev Dev Biol. 2018;7:e310.
Kobayashi A, Behringer RR. Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet. 2003;4:969–80.
Ma L. Endocrine disruptors in female reproductive tract development and carcinogenesis. Trends Endocrinol Metab. 2009;20:357–63.
Kurita T. Developmental origin of vaginal epithelium. Differentiation. 2010;80:99–105.
Narang K, Cope ZS, Teixeira JM. Chapter 6—developmental genetics of the female reproductive tract. In: Leung PCK, Qiao J, editors. Human reproductive and prenatal genetics. San Diego: Academic Press; 2019. p. 129–53.
Kurita T, Nakamura H. Embryology of the uterus. In: Aplin JD, Fazleabas AT, Glasser SR, Giudice LC, editors. Endometrium, 2 ed. London: Informa UK Ltd; 2008. p. 1–18.
Kurita T, Terakawa J. Endometrial development and its fine structure. Endometrial gene expression: an emerging paradigm for reproductive disorders. Springer International Publishing; 2020, p. 1–32.
Cunha GR. Stromal induction and specification of morphogenesis and cytodifferentiation of the epithelia of the Müllerian ducts and urogenital sinus during development of the uterus and vagina in mice. J Exp Zool. 1976;196:361–70.
Boutin E, Sanderson R, Bernfield M, Cunha GR. Expression of syndecan, a cell surface proteoglycan, correlates with induced changes in cellular organization. J Cell Biol. 1989;107:605a.
Kurita T, Cooke PS, Cunha GR. Epithelial-stromal tissue interaction in paramesonephric (Müllerian) epithelial differentiation. Dev Biol. 2001;240:194–211.
Kurita T, Mills AA, Cunha GR. Roles of p63 in the diethylstilbestrol-induced cervicovaginal adenosis. Development. 2004;131:1639–49.
Kurita T, Cunha GR. Roles of p63 in differentiation of Müllerian duct epithelial cells. Ann NY Acad Sci. 2001;948:9–12.
Kurita T, Cunha GR, Robboy SJ, Mills AA, Medina RT. Differential expression of p63 isoforms in female reproductive organs. Mech Dev. 2005;122:1043–55.
Terakawa J, Rocchi A, Serna VA, Bottinger EP, Graff JM, Kurita T. FGFR2IIIb-MAPK activity is required for epithelial cell fate decision in the lower Müllerian duct. Mol Endocrinol. 2016;30:783–95.
Laronda MM, Unno K, Ishi K, Serna VA, Butler LM, Mills AA, et al. Diethylstilbestrol induces vaginal adenosis by disrupting SMAD/RUNX1-mediated cell fate decision in the Müllerian duct epithelium. Dev Biol. 2013;381:5–16.
Robboy SJ, Young RH, Welch WR, Truslow GY, Prat J, Herbst AL, et al. Atypical vaginal adenosis and cervical ectropion. Association with clear cell adenocarcinoma in diethylstilbestrol-exposed offspring. Cancer. 1984;54:869–75.
Laronda MM, Unno K, Butler LM, Kurita T. The development of cervical and vaginal adenosis as a result of diethylstilbestrol exposure in utero. Differentiation. 2012;84:252–60.
Robboy SJ, Szyfelbein WM, Goellner JR, Kaufman RH, Taft PD, Richard RM, et al. Dysplasia and cytologic findings in 4,589 young women enrolled in diethylstilbestrol-adenosis (DESAD) project. Am J Obstet Gynecol. 1981;140:579–86.
Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011;365:1304–14.
Forsberg JG, Lannerstad B. Abnormalities in the adult mouse vagina after neonatal estradiol treatment. Biol Neonat. 1968;12:175–9.
Takasugi N. Morphogenesis of estrogen-independent proliferation and cornification of the vaginal epithelium in neonatally estrogenized mice. Proc Jpn Acad. 1971;47:193–8.
Forsberg JG. Estrogen, vaginal cancer, and vaginal development. Am J Obstet Gynecol. 1972;113:83–7.
Plapinger L, Bern HA. Adenosis-like lesions and other cervicovaginal abnormalities in mice treated perinatally with estrogen. J Natl Cancer Inst. 1979;63:507–18.
Forsberg JG, Kalland T. Neonatal estrogen treatment and epithelial abnormalities in the cervicovaginal epithelium of adult mice. Cancer Res. 1981;41:721–34.
Newbold RR, McLachlan JA. Vaginal adenosis and adenocarcinoma in mice exposed prenatally or neonatally to diethylstilbestrol. Cancer Res. 1982;42:2003–11.
Cheyette BN, Green PJ, Martin K, Garren H, Hartenstein V, Zipursky SL. The Drosophila sine oculis locus encodes a homeodomain-containing protein required for the development of the entire visual system. Neuron. 1994;12:977–96.
Fortunato SA, Leininger S, Adamska M. Evolution of the Pax-Six-Eya-Dach network: the calcisponge case study. Evodevo. 2014;5:23.
Byrne M, Koop D, Morris VB, Chui J, Wray GA, Cisternas P. Expression of genes and proteins of the pax-six-eya-dach network in the metamorphic sea urchin: insights into development of the enigmatic echinoderm body plan and sensory structures. Dev Dyn. 2018;247:239–49.
Kawakami K, Sato S, Ozaki H, Ikeda K. Six family genes–structure and function as transcription factors and their roles in development. Bioessays. 2000;22:616–26.
Kochhar A, Fischer SM, Kimberling WJ, Smith RJ. Branchio-oto-renal syndrome. Am J Med Genet A. 2007;143A:1671–8.
Laclef C, Souil E, Demignon J, Maire P. Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech Dev. 2003;120:669–79.
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA. 2004;101:8090–5.
Salam AA, Hafner FM, Linder TE, Spillmann T, Schinzel AA, Leal SM. A novel locus (DFNA23) for prelingual autosomal dominant nonsyndromic hearing loss maps to 14q21-q22 in a Swiss German kindred. Am J Hum Genet. 2000;66:1984–8.
Jefferson WN, Chevalier DM, Phelps JY, Cantor AM, Padilla-Banks E, Newbold RR, et al. Persistently altered epigenetic marks in the mouse uterus after neonatal estrogen exposure. Mol Endocrinol. 2013;27:1666–77.
Le Grand F, Grifone R, Mourikis P, Houbron C, Gigaud C, Pujol J, et al. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J Cell Biol. 2012;198:815–32.
Mills AA, Qi Y, Bradley A. Conditional inactivation of p63 by Cre-mediated excision. Genesis. 2002;32:138–41.
Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. ΔNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–82.
Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–33.
Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–74.
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605.
Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–86.
Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81.
Singh SP, Wolfe A, Ng Y, DiVall SA, Buggs C, Levine JE, et al. Impaired estrogen feedback and infertility in female mice with pituitary-specific deletion of estrogen receptor alpha (ESR1). Biol Reprod. 2009;81:488–96.
Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–9.
Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci USA. 2010;107:19272–7.
Pignon JC, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S. p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci USA. 2013;110:8105–10.
Kim SY, Cordeiro MH, Serna VA, Ebbert K, Butler LM, Sinha S, et al. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell Death Differ. 2013;20:987–97.
Terakawa J, Serna VA, Taketo MM, Daikoku T, Suarez A, Kurita T. Ovarian-insufficiency and CTNNB1 mutations drive malignant transformation of endometrial hyperplasia with altered PTEN/PI3K activities. Proc Natl Acad Sci USA. 2019;116:4528–37.
Serna VA, Wu X, Qiang W, Thomas J, Blumenfeld ML, Kurita T. Cellular kinetics of MED12-mutant uterine leiomyoma growth and regression in vivo. Endocr Relat Cancer. 2018;25:747–59.
Izvolsky KI, Shoykhet D, Yang Y, Yu Q, Nugent MA, Cardoso WV. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol. 2003;258:185–200.
Laclef C, Hamard G, Demignon J, Souil E, Houbron C, Maire P. Altered myogenesis in Six1-deficient mice. Development. 2003;130:2239–52.
Kurita T, Lee KJ, Cooke PS, Taylor JA, Lubahn DB, Cunha GR. Paracrine regulation of epithelial progesterone receptor by estradiol in the mouse female reproductive tract. Biol Reprod. 2000;62:821–30.
Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–286.
Kreft L, Soete A, Hulpiau P, Botzki A, Saeys Y, De Bleser P. ConTra v3: a tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017;45(W1):W490–W494.
Bigsby RM, Li AX, Luo K, Cunha GR. Strain differences in the ontogeny of estrogen receptors in murine uterine epithelium. Endocrinology. 1990;126:2592–6.
Bigsby RM, Cunha GR. Estrogen stimulation of deoxyribonucleic acid synthesis in uterine epithelial cells which lack estrogen receptors. Endocrinology. 1986;119:390–6.
Kurita T. Normal and abnormal epithelial differentiation in the female reproductive tract. Differentiation. 2011;82:117–26.
Cunha GR, Kurita T, Cao M, Shen J, Cooke PS, Robboy SJ, et al. Tissue interactions and estrogenic response during human female fetal reproductive tract development. Differentiation. 2018;101:39–45.
Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–41.
Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, Rohs R. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014;39:381–99.
Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 2012;13:59–69.
Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA. 1996;93:3444–9.
Cunha GR, Kurita T, Cao M, Shen J, Robboy S, Baskin L. Molecular mechanisms of development of the human fetal female reproductive tract. Differentiation. 2017;97:54–72.
Smith EK, White MC, Weir HK, Peipins LA, Thompson TD. Higher incidence of clear cell adenocarcinoma of the cervix and vagina among women born between 1947 and 1971 in the United States. Cancer Causes Control. 2012;23:207–11.
Goodman A, Zukerberg LR, Nikrui N, Scully RE. Vaginal adenosis and clear cell carcinoma after 5-fluorouracil treatment for condylomas. Cancer. 1991;68:1628–32.
Dungar CF, Wilkinson EJ. Vaginal columnar cell metaplasia. An acquired adenosis associated with topical 5-fluorouracil therapy. J Reprod Med. 1995;40:361–6.
Bornstein J, Sova Y, Atad J, Lurie M, Abramovici H. Development of vaginal adenosis following combined 5-fluorouracil and carbon dioxide laser treatments for diffuse vaginal condylomatosis. Obstet Gynecol. 1993;81(5 (Pt 2)):896–8.
Robboy SJ, Friedlander LM, Welch WR, Keh PC, Taft PD, Barnes AB, et al. Cytology of 575 young women with prenatal exposure to diethylstilbestrol. Obstet Gynecol. 1976;48:511–5.
Stafl A, Mattingly RF, Foley DV, Fetherston WC. Clinical diagnosis of vaginal adenosis. Obstet Gynecol. 1974;43:118–28.
Gunning JE, Ostegard DR. Value of screening procedures for the detection of vaginal adenosis. Obstet Gynecol. 1976;47:268–71.
McLachlan JA, Newbold RR, Bullock BC. Long-term effects on the female mouse genital tract associated with prenatal exposure to diethylstilbestrol. Cancer Res. 1980;40:3988–99.
Acknowledgements
The authors thank Dr. Altea Rocci, Shayna Wallace and Justin Thomas for technical help. This work was supported by the National Institutes of Health [R01CA154358, R01HD064402, P30CA016058 to TK] and The Ohio State University Comprehensive Cancer Center [Pelotonia Idea Grant to TK].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Terakawa, J., Serna, V.A., Nair, D.M. et al. SIX1 cooperates with RUNX1 and SMAD4 in cell fate commitment of Müllerian duct epithelium. Cell Death Differ 27, 3307–3320 (2020). https://doi.org/10.1038/s41418-020-0579-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0579-z
- Springer Nature Limited