
Abstract
The primary function of the immune system is to protect the host from invading pathogens. In response, microbial pathogens have developed various strategies to evade detection and destruction by the immune system. This tug-of-war between the host and the pathogen is a powerful force that shapes organismal evolution. Regulated cell death (RCD) is a host response that limits the reservoir for intracellular pathogens such as viruses. Since pathogen-specific T cell and B cell responses typically take several days and is therefore slow-developing, RCD of infected cells during the first few days of the infection is critical for organismal survival. This innate immune response not only restricts viral replication, but also serves to promote anti-viral inflammation through cell death-associated release of damage-associated molecular patterns (DAMPs). In recent years, necroptosis has been recognized as an important response against many viruses. The central adaptor for necroptosis, RIPK3, also exerts anti-viral effects through cell death-independent activities such as promoting cytokine gene expression. Here, we will discuss recent advances on how viruses counteract this host defense mechanism and the effect of necroptosis on the anti-viral inflammatory reaction.
Similar content being viewed by others


Facts
-
Necroptosis is mediated through cellular RHIM domain-containing proteins, RIPK1, RIPK3, ZBP1, and TRIF.
-
Virus infections activate RHIM-dependent necroptosis in host, which leads to anti-viral inflammation.
-
Viruses modulate necroptosis pathway through various inhibitors competing for RHIM binding.
-
RIPK3 activates the NLRP3 inflammasome in certain RNA virus infection independent of its necroptotic function.
Open questions
-
Is necroptosis a primary driver or secondary form of cell death upon virus infection?
-
Do lytic viruses like IAV activate necroptosis contribute to pathogenesis?
-
Is ZBP1 sensing of viral RNA a common phenomenon among different virus infections?
-
Does RIPK3 have roles beyond necroptosis in virus infections?
-
Can targeting cellular RHIM proteins be an effective strategy as anti-viral therapeutics?
Introduction
Regulated cell death (RCD) constitutes an important aspect of organismal development. RCD drives crucial cellular responses and is especially important for immune homeostasis. Until recently, apoptosis was synonymous with programmed cell death. Apoptosis is characterized by cell shrinking, chromatin condensation, nuclear fragmentation (karyorrhexis) and plasma membrane blebbing. Apoptotic cells also expose the “eat-me” signal phosphatidyl serine on the cell surface to promote engulfment by resident phagocytes. Thus, apoptosis can be considered a “contained” and immunologically silent form of cell death. This is in contrast to cell death by necrosis, which is marked by cell lysis and the release of inflammatory mediators. Although non-apoptotic cell death pathways are sometimes considered as secondary responses to apoptosis, there is growing evidence that they can also be “programmed” and controlled by a dedicated molecular circuitry. Moreover, these pathways can be activated independently or as a consequence of apoptosis inhibition [1].
Many forms of non-apoptotic RCD have been described in recent years. These include necroptosis, parthanatos, ferroptosis, mitochondrial permeability transition (MPT)-dependent necrosis, pyroptosis and pyronecrosis, and NETosis or ETosis [1]. The molecular mechanisms and biological roles for some of these cell death modes are still poorly defined. By contrast, necroptosis has emerged as a pivotal cell death response in microbial infections. Necroptosis is initiated in response to toll-like receptor 3 (TLR3), TLR4, and death receptors in the tumor necrosis factor receptor (TNFR) superfamily. Necroptosis is marked by rupture of the plasma membrane and release of pro-inflammatory damage-associated molecular patterns (DAMPs) (Box 1). Hence, necroptosis is distinct from apoptosis in both morphology and biological consequences [2]. Receptor interacting protein kinase 3 (RIPK3) is a serine/threonine kinase and key adaptor of necroptosis. In addition to the kinase domain, RIPK3 also contains a “RIP homotypic interaction motif (RHIM)” at the C-terminus. As its name implies, the RHIM of RIPK3 mediates homotypic interaction with other mammalian RHIM-containing signal adaptors [3]. Consistent with the pro-inflammatory role of necroptosis, RIPK3 has critical functions in microbial infections [4]. Here, we discuss how viruses target RIPK3 and other components of the necroptosis pathway to manipulate host-defense and anti-viral inflammation (Table 1).
Necroptosis upon poxvirus infection
RIPK1, the upstream activator of RIPK3 in the TNF receptor pathway, is a cleavage substrate of caspase 8. Caspase 8 cleaves RIPK1 at D324, the boundary between the N-terminal kinase domain and the C-terminal RHIM and death domain [5]. The C-terminal cleavage product, which contains the death domain, was reported to enhance apoptosis by promoting TRADD and FADD interaction [5]. However, since this cleavage event results in the release of the kinase domain from the signaling complex, necroptosis is inhibited. This explains why inhibition of caspase 8 is a key priming signal for necroptosis. Although the supporting biochemical evidence is still lacking, similar cleavage of RIPK3 by caspase 8 has also been implicated [6, 7]. Large DNA viruses often target caspases for inhibition, a strategy that promotes viral persistence and immune evasion within the host. For example, poxviruses encode serpins that inhibit caspase 1 and caspase 8 [8]. Poxviral inhibition of caspase 8 may therefore provide a natural priming signal for necroptosis. Indeed, cells infected with vaccinia virus (VV), the poxvirus strain that is used to vaccinate the world population against smallpox, are highly sensitive to TNF-induced necroptosis [9]. By contrast, VV-infected cells deficient in RIPK1 or RIPK3 were resistant to TNF-induced necroptosis [10]. As a consequence, Ripk3−/− mice and mice expressing the kinase-inactive RIPK1 mutant D138N both failed to control VV replication in vivo [10, 11]. The phenotypes of Ripk3−/− mice were reminiscent of mice lacking TNF or TNF receptor expression [9]. These results indicate that TNF, RIPK1, and RIPK3-dependent necroptosis is a key innate immune host defense mechanism against VV.
Interferon (IFN) response is a powerful host response against pathogens. VV encodes many immune evasion genes including those that neutralize the inflammatory cytokines TNF and IFNs and other innate immune signaling pathways. One of the VV-encoded immune modulators is E3L, which contains a z-DNA binding domain that is also found in the mammalian RHIM adaptor z-DNA binding protein 1 (ZBP1, aka DAI for DNA activator of interferon). Infection with VV expressing a truncated E3L lacking this z-DNA binding domain led to increased ZBP1 expression and excessive Type I IFN-induced RIPK3/MLKL-dependent necroptosis. Moreover, the E3L mutant VV failed to cause disease in wild type mice [12]. Strikingly, pathogenicity was restored when the E3L mutant VV was used to infect Ripk3−/− and Zbp1−/− mice [12]. These results indicate that while TNF-induced, RIPK1/RIPK3/MLKL-mediated necroptosis confers protection to the host, E3L-mediated inhibition of ZBP1 and RIPK3 enables VV to evade the anti-viral effects of IFNs (Fig. 1). The opposing effects of the two RIPK3-driven responses suggest that the timing and context of RIPK3-mediated necroptosis may impact host response as well as viral growth and dissemination in the host.

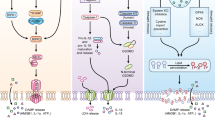
Necroptosis in poxvirus infection. Control of vaccinia virus (VV) infection requires TNF signaling [9]. TNF signaling through its trimeric receptor (TNFR-1) triggers three distinct signaling responses: NF-κB activation, apoptosis or necroptosis. Vaccinia virus encodes the caspase inhibitor B13R/Spi2, which inhibits caspase 1 and caspase 8 and blocks apoptosis. Thus, in VV infection, TNF stimulation favors necroptosis. This leads to assembly of the RHIM–RHIM interaction between RIPK1–RIPK3 and necrosome formation. Certain viral FLIPs such as MC159 from Molluscum contagious virus (MCV) inhibit both apoptosis and necroptosis, although the mechanisms are not fully elucidated. The VV-encoded protein E3L contains a z-DNA binding domain that interacts with ZBP1 and senses viral RNA. E3L binding sequesters ZBP1 from RIPK3 and therefore prevents necroptosis. However, E3L does not interfere with TNF-induced necroptosis. VV Vaccinia virus, TRADD TNFRSF1A associated death domain, TRAF2 TNF receptor associated factor 2, cIAP cellular inhibitor of apoptosis, CYLD cylindromatosis, FADD Fas associated via death domain, IKK inhibitor of nuclear factor kappa B kinase, cFLIPL CASP8 and FADD like apoptosis regulator long isoform, MLKL mixed lineage kinase domain-like, RHIM RIP homotypic interaction motif, Zα1, Zα2 zDNA binding domain
Necroptosis upon herpesvirus infection
Cytomegalovirus
Herpesvirus is another large DNA virus family that exploits the necroptosis pathway. In a forward genetic screen using transposon mutagenesis, Brune and colleagues identified the murine cytomegalovirus (MCMV) encoded M45 gene as a factor that promotes viral growth and endothelial cell survival [13]. M45 encodes a large polypeptide with homology to the large subunit of ribonucleotide reductase (R1) at the C-terminus. However, unlike other viral R1 subunits, M45 lacks enzymatic activity [14]. In addition to the R1-like domain, M45 contains a RHIM at the N-terminus that mediates binding to mammalian RHIM-containing adaptors such as RIPK1 and RIPK3 [15, 16]. In over-expression studies, M45 inhibited both TNF- and TLR3-driven NF-κB and MAPK activation [15]. More recent studies showed that M45 also inhibits necroptosis during MCMV infection. For example, mutant MCMV expressing a M45 RHIM mutant failed to replicate in cells due to excessive activation of RIPK3-dependent necroptosis [17]. However, M45 was only required for optimal viral replication in certain cell types in tissue culture [18]. Mutant virus replication was also compromised in the lungs, spleen, and salivary glands of wild type mice. Thus, M45 is the first viral inhibitor of RIP activation (vIRA) identified. Strikingly, replication of the M45 RHIM mutant virus was restored in Ripk3−/− or Zbp1−/− cells and mice [19], indicating that M45 prevents premature cell death by inhibiting ZBP1/RIPK3-induced necroptosis. In contrast to TNF-induced necroptosis in VV-infected cells, RIPK1 is dispensable for necroptosis induced by the M45 RHIM mutant MCMV [17]. These results establish necroptosis as a bona fide host defense mechanism in MCMV. They also provide the first examples in which mammalian RHIM-containing signal adaptors, other than RIPK1, activate RIPK3 and necroptosis.
Recent studies suggest that MCMV transcription is required for ZBP1-dependent necroptosis. When MCMV transcription is inhibited at an early time point with chemical inhibitors, M45 RHIM mutant virus-infected cells become resistant to necroptosis. The viral immediate early protein 3 (IE3), a crucial transcriptional activator of MCMV, was required for ZBP1 activation. Moreover, mutation of key amino acid residues in the z-DNA binding domain of ZBP1 abolished this response, suggesting that ZBP1 activates necroptosis upon sensing viral nucleic acid [20]. Hence, viral RNA, but not viral DNA, appears to be the ligand that activates ZBP1 in response to MCMV infection [21].
ZBP1 was originally identified as a sensor of viral DNA that regulates virus-induced NF-κB activation and interferon expression [22, 23]. However, DNA-induced IFN expression was normal in Zbp1−/− macrophages, dendritic cells and embryonic fibroblasts [24]. Hence, it is noteworthy that viral RNA sensing appears to be the key mechanism by which ZBP1 becomes activated in response to infection by mutant MCMV expressing a RHIM-mutated M45/vIRA. As we shall see below, viral RNA sensing by ZBP1 is also found in other viruses (Fig. 2). Hence, unlike the TNF/RIPK1/RIPK3 necroptosis pathway, which has a more prominent role in tissue homeostasis, ZBP1-mediated necroptosis functions predominantly as a critical host defense mechanism. These observations also argue strongly that the ZBP1/RIPK3 necroptosis pathway may be the product of host-pathogen co-evolution.

Necroptosis in herpesvirus and Influenza virus infection. Herpesviruses exploit the necroptotic pathway via various RHIM adaptors such as M45 from MCMV, ICP6 from HSV-1 and ICP10 from HSV-2. These viral inhibitors sequester ZBP1 and RIPK3 to prevent their interaction and activation. When viral RHIM inhibitors are absent, ZBP1 senses viral RNA (vRNA) to trigger RIPK3 binding and activation. The necroptosis modulating function of these viral RHIM adaptors varies with host species. For example, ICP6 and ICP10 prevent necroptosis in human cells, but stimulate RIPK3-dependent necroptosis in mouse cells. Though the molecular basis for the differential effects of ICP6 or ICP10 in different species is unknown, it may account for the species tropism of these viruses. In contrast to herpesvirus, IAV uses this mechanism to cause ZBP1/RIPK3-dependent necroptosis. MCMV murine cytomegalovirus, HSV herpes simplex virus, IAV Influenza A virus, vRNA viral RNA
Besides M45, MCMV encodes another viral inhibitor called vICA (for viral inhibitor of caspase 8-induced apoptosis). vICA is encoded by the M36 gene. A recent study indicates that M36 and M45 cooperate with each other to suppress cell death-associated inflammation [18]. Infection of cells with mutant MCMV that lacks M36 and M45 led to caspase 8 and RIPK3/MLKL activation and severely reduced viral replication. The activation of RIPK3 and MLKL in the presence of intact caspase 8 activation is interesting and significant, since it challenges the paradigm that necroptosis induction requires caspase 8 inhibition. However, it is consistent with the involvement of ZBP1 rather than RIPK1 as the upstream activator of RIPK3 [19]. When compared to MCMV with single mutation in either M36 or M45, M36/M45 double mutant virus elicited stronger expression of many pro-inflammatory cytokines and early CD8+ T cell activation [18]. These results highlight the cooperative nature of apoptosis and necroptosis in driving robust host anti-viral inflammation.
Similar to MCMV, human cytomegalovirus (HCMV) also suppresses necroptosis induced by TNF, Smac mimetics and caspase inhibitors. However, in contrast to MCMV, HCMV does not inhibit RIPK3 phosphorylation, and MLKL phosphorylation was readily detected [25]. These results indicate that HCMV interferes with necroptosis at a step after MLKL activation. Consistent with the notion that HCMV does not target ZBP1 or RIPK3, super-infection with M45 RHIM mutant MCMV in HCMV-infected cells no longer sensitize the cells to necroptosis. While the precise mechanism by which HCMV inhibits necroptosis is still unknown, viral gene products under the control of the regulatory protein IE1 appear to be important, since infection with mutant HCMV lacking IE1 no longer conferred protection against TNF, Smac mimetic and caspase inhibitor-induced necroptosis [25] (Fig. 2). It will be interesting to determine if the differential mechanisms of necroptosis inhibition by MCMV and HCMV is the driver or consequence of viral evolution. It will also be important to evaluate how this switch in cell death inhibitory mechanism may influence anti-viral inflammation.
Other herpesviruses
Similar to cytomegaloviruses, the alpha herpesviruses human herpes simplex virus 1 (HSV-1) and HSV-2 also modulate necroptosis through the large subunit of the ribonucleotide reductase (RR), ICP6 for HSV-1 and ICP10 for HSV-2. ICP6 and ICP10 share similar domain organization with M45, with an N-terminal RHIM and a C-terminal RR. However, in contrast to M45, ICP6 and ICP10 retain intact ribonucleotide reductase function. In addition, ICP6 and ICP10 interact with and inhibit caspase 8 [26]. Thus, they are dual inhibitors of apoptosis and necroptosis. The RHIM of ICP6 and ICP10 mediates RIPK1 and RIPK3 interaction [27,28,29]. HSV-1 triggered RIPK3/MLKL activation and necroptosis in mouse cells and increased viral titers and mortality in mice [28]. By contrast, HSV-1 or expression of ICP6 or ICP10 inhibits necroptosis in human cells. The differential effects of ICP6 and ICP10 in human versus mouse cells distinguish them from M45, which potently inhibits necroptosis in both human and mouse cells. Since HSV-1 and HSV-2 are human pathogens, the differential activity of ICP6 and ICP10 may account for their species tropism.
Epstein–Barr virus (EBV) is a gamma herpesvirus that infects the majority of human adults. A recent report suggests that the latent membrane protein 1 (LMP1) of EBV interacts with RIPK1 and RIPK3, modulates their ubiquitination status, and inhibits TNF, Smac mimetic and caspase inhibitor-induced necroptosis [30]. Interestingly, a mapping study using truncation mutants suggests that the interaction between LMP1 and RIPK1 or RIPK3 is independent of the RHIM. Further studies are required to confirm this observation and to examine whether this interaction plays any role in anti-viral immune responses and disease pathology.
Viral FLIPs and necroptosis
Like caspase 8, cellular FLICE (caspase 8)-like inhibitor protein (cFLIP) contains two tandem death effector domains at the N-terminus. However, the long form of cFLIP (cFLIPL) harbors mutations at the C-terminal caspase domain, thus rendering it enzymatically inactive. By contrast, the short form of cFLIP, cFLIPs, lacks the C-terminal caspase domain. While cFLIPL promotes caspase 8 activation through heterodimerization, cFLIPs inhibits caspase 8 activity [31]. Interestingly, a class of viral FLIP proteins (vFLIPs) has been identified in herpesviruses and the human poxvirus Molluscum contagiosum virus (MCV) [32, 33]. vFLIPs lack the C-terminal caspase domain and thus resemble cFLIPs in both structure and function [34, 35]. Surprisingly, many vFLIPs also exhibit necroptosis inhibitory activity [9]. vFLIPs have been reported to interact with RIPK1 [36]. However, we were not able to detect such interaction (unpublished observation). Hence, the mechanism by which vFLIPs inhibit necroptosis remains elusive.
Besides apoptosis and necroptosis, vFLIPs also interfere with NF-κB activation. Several reports showed that transgenic expression of MC159 inhibited NF-κB activation and triggered a loss of memory CD8+ T cells in mice [37, 38]. However, NF-κB inhibition requires high expression of MC159. At low level of expression, MC159 actually stimulates NF-κB activation [39]. In contrast to transgenic mice expressing high level of MC159, mice with low MC159 expression exhibited enhanced NF-κB activation, chemokine expression and viral clearance in response to VV infection [39]. The functional relevance of MC159 in virus infection was further characterized by Hüttmann and coworkers. When expressed in recombinant MCMV lacking either M36 or M45, MC159 blocked TNF-induced apoptosis. However, necroptosis in human, but not mouse cells, was inhibited by recombinant MCMV expressing MC159 [40]. These discrepant results highlight a potential caveat in using over-expression to study viral necroptosis inhibitors suggesting that their behavior can vary depending on the expression level and the cell type studied.
Necroptosis in RNA virus infection
In a survey for additional viruses that might interfere with necroptosis, Schock and colleagues found that Sendai virus induced necroptosis in the presence of the pan-caspase inhibitor zVAD-fmk [41]. Sendai virus drives necroptosis via the RNA sensor RIG-I [41]. In Ripk3−/− mice, Sendai virus infection resulted in increased inflammation. Although more work is required to fully elucidate the mechanism by which Sendai virus triggers necroptosis and inhibits inflammation, these results do highlight the fact that RIPK3 does not always facilitate detrimental inflammation. Rather, in certain situations such as that in Sendai virus infection, RIPK3 can play an anti-inflammatory and protective role.
Recently, necroptosis has emerged as a critical pathway in influenza virus-induced cell death and pathology. For instance, Rodrigue-Gervais and coworkers found that Birc3−/− mice, which are deficient in the anti-apoptotic factor cIAP2, were hypersensitive to influenza A virus (IAV) infection and developed severe loss of lung epithelium [42]. Importantly, pharmacologic inhibition of RIPK1 kinase activity or genetic deletion of Ripk3 rescued Birc3−/− mice from IAV-induced epithelial necrosis, tissue damage and lethality. These results are consistent with the known function of cIAP1 and cIAP2 as apoptosis and necroptosis inhibitors.
The study in Birc3−/− mice suggests a role for RIPK1 and RIPK3 in IAV pathogenesis. Indeed, several recent studies indicate that RIPK3-dependent necroptosis plays a key role in IAV-induced cell death. Similar to infection with MCMV, ZBP1 functions as the upstream sensor of IAV RNA to activate RIPK3 [43, 44]. ZBP1 was reported to sense IAV nucleoprotein (NP) and polymerase subunit (PB1) to induce necroptosis [43]. Interestingly, NP was also reported to induce apoptosis in IAV-infected cells [45, 46]. In contrast to Birc3−/− mice, RIPK1 was mostly dispensable for IAV-induced cell death in wild type cells or mice [47]. Ripk3−/− and Zbp1−/− mice failed to control viral replication and exhibited increased mortality in response to IAV infection [44, 48]. However, increased mortality was not observed in another report [43].
Mechanistically, IAV-induced expression of cytokines such as type I IFNs via RIPK3 and ZBP1. As such, IAV-induced IFNβ expression was impaired in Ripk3−/− and Zbp1−/− cells [43]. While reduced IFNβ expression might be due to decreased necroptosis and necroptosis-driven inflammation, it could also be caused by direct effects on gene transcription [49,50,51,52,53]. For example, in West Nile virus infection, chemokine expression in the central nervous system was driven by RIPK3 but independent of cell death [52]. In macrophages, IAV induced binding between RIPK3 and the mitochondrial associated RNA sensor MAVS, which stimulated TBK1 and IRF1/IRF3 activation to promote IFNβ mRNA stability [48, 54]. This response is not only critical for virus-induced IFN expression, but also for the expression of the necroptosis genes Zbp1 and Mlkl. The relative contribution of cell death-dependent and independent activities of RIPK3 in influenza virus infection will require further investigation.
Further evidence supporting a role for RIPK3-dependent necroptosis in shaping influenza virus-induced pathogenesis comes from a recent study comparing the host response to seasonal versus pandemic strains of IAV. Remarkably, virus-induced necroptosis in dendritic cells (DCs) was readily detected by seasonal IAV strains, but not by pandemic IAV strains [55]. In co-culture experiments, seasonal IAV-infected DCs underwent necroptosis and induced bystander DC maturation and virus-specific T cell proliferation. By contrast, DC maturation and T cell proliferation were significantly impaired in pandemic IAV-infected DC cultures. At present, the molecular mechanism by which seasonal and pandemic IAV strains induces differential necroptosis in DCs is unknown. However, differences in the viral hemagglutinin gene segment between the two groups of IAVs have been implicated [55]. These results highlight the interesting scenario that necroptosis may have broad functions in shaping viral evolution and pathogenicity.
To further complicate matters, recent studies have clearly demonstrated a necroptosis-independent role for RIPK3 inNLRP3 inflammasome activation [56]. Inflammasome activation results in two major biological outcomes: processing and secretion of pro-inflammatory cytokines IL-1β and IL-18, and induction of pyroptosis, another lytic form of cell death [57]. RIPK3-mediated inflammasome activity has been reported to promote IL-1β secretion in response to IAV, Sendai virus and Vesicular stomatitis virus (VSV). Interestingly, the mitochondrial fission protein DRP1 was implicated to signal downstream of RIPK3 to promote RNA virus-induced inflammasome activity [58]. However, these results were challenged by another study, which showed that VSV-induced IL-1β secretion was normal in Ripk3−/−macrophages and macrophages expressing the kinase inactive RIPK3 mutant K51A [59]. Nonetheless, the artificial ligand for TLR3 and RIG-I poly(I:C), which mimics viral double-stranded RNA, could clearly activate RIPK3 signaling, especially in the presence of caspase inhibition [60,61,62]. Thus, we cannot rule out the possibility that RIPK3 can indeed stimulate inflammasome activation in certain situations. In this light, it is noteworthy that RIPK3-dependent inflammasome activity was particularly prominent in human monocytes, mouse dendritic cells and macrophages [49, 63, 64]. Thus, the anti-viral mechanism employed by RIPK3 in response to different pathogens will likely vary depending on the cell type under investigation.
Conclusion
Research in the last decade has clearly established necroptosis as a crucial host defense response against many pathogens. In response, viruses have developed different strategies to neutralize the host necroptosis machinery. In addition to viral pathogens, an increasing number of non-viral pathogens have also been found to target the necroptosis machinery. For example, atypical proteases from certain pathogenic bacteria specifically target mammalian RHIM-containing signal adaptors for cleavage and inactivation [65]. An important lesson we learned from studies with different pathogens is that while apoptosis and necroptosis are often mutually exclusive responses in tissue culture, this is not the case in pathogen infections. For instance, IAV infection elicits concomitant activation of caspase-dependent apoptosis and RIP kinase-mediated necroptosis [66]. The cooperative nature of apoptosis and necroptosis in anti-microbial defense is further illustrated by the fact that caspase 8, its adaptor FADD, RIPK1 and RIPK3 are often found in the same signaling complex regardless of whether apoptosis or necroptosis is the dominant response. This concept is further bolstered by the many examples in which pathogens often target both pathways of cell death. The knowledge gained from studying how various pathogens counteract the host cell death machinery may lead us to better therapeutic strategies against viral and sterile inflammatory diseases.
References
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–30.
Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–11.
Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106.
Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–26.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6.
Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–67.
Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem. 1997;272:7797–800.
Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–21.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193:1539–43.
Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci USA. 2017;114:11506–11.
Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 2001;291:303–5.
Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, et al. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol. 2004;78:4278–88.
Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA. 2008;105:3094–9.
Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008;283:16966–70.
Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–13.
Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, et al. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci USA. 2017;114:E2786–e95.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–7.
Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017;36:2529–43.
Sridharan H, Ragan KB, Guo H, Gilley RP, Landsteiner VJ, Kaiser WJ, et al. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep. 2017;18:1429–41.
Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–5.
Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–34.
Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–9.
Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem. 2015;290:11635–48.
Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-inducedapoptosis by interacting with caspase-8. Apoptosis Int J Program Cell death. 2011;16:256–71.
Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–42.
Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA. 2014;111:15438–43.
Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–51.
Liu X, Li Y, Peng S, Yu X, Li W, Shi F, et al. Epstein–Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018;9:53.
Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–49.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21.
Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:1172–6.
Garvey T, Bertin J, Siegel R, Lenardo M, Cohen J. The death effector domains (DEDs) of the molluscum contagiosum virus MC159 v-FLIP protein are not functionally interchangeable with each other or with the DEDs of caspase-8. Virology. 2002;300:217–25.
Garvey TL, Bertin J, Siegel RM, Wang GH, Lenardo MJ, Cohen JI. Binding of FADD and caspase-8 to molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J Virol. 2002;76:697–706.
Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene. 1999;18:5738–46.
Wu Z, Roberts M, Porter M, Walker F, Wherry EJ, Kelly J, et al. Viral FLIP impairs survival of activated T cells and generation of CD8+ T cell memory. J Immunol. 2004;172:6313–23.
Randall CM, Jokela JA, Shisler JL. The MC159 protein from the molluscum contagiosum poxvirus inhibits NF-kappaB activation by interacting with the IkappaB kinase complex. J Immunol. 2012;188:2371–9.
Challa S, Woelfel M, Guildford M, Moquin D, Chan FK. Viral cell death inhibitor MC159 enhances innate immunity against vaccinia virus infection. J Virol. 2010;84:10467–76.
Huttmann J, Krause E, Schommartz T, Brune W. Functional comparison of molluscum contagiosum virus vFLIP MC159 with murine cytomegalovirus M36/vICA and M45/vIRA proteins. J Virol. 2015;90:2895–905.
Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017;24:615–25.
Rodrigue-Gervais IG, Labbe K, Dagenais M, Dupaul-Chicoine J, Champagne C, Morizot A, et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe. 2014;15:23–35.
Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045.
Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses Influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. 2016;20:674–81.
Nailwal H, Sharma S, Mayank AK, Lal SK. The nucleoprotein of influenza A virus induces p53 signaling and apoptosis via attenuation of host ubiquitin ligase RNF43. Cell Death Dis. 2015;6:e1768.
Mayank AK, Sharma S, Nailwal H, Lal SK. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015;6:e2018.
Nogusa S, Slifker MJ, Ingram JP, Thapa RJ, Balachandran S. RIPK3 is largely dispensable for RIG-I-like receptor- and Type I interferon-driven transcriptional responses to influenza A virus in murine fibroblasts. PLoS ONE. 2016;11:e0158774.
Downey J, Pernet E, Coulombe F, Allard B, Meunier I, Jaworska J, et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017;13:e1006326.
Moriwaki K, Bertin J, Gough PJ, Chan FK. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194:1938–44.
Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, et al. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity. 2016;45:46–59.
Saleh D, Najjar M, Zelic M, Shah S, Nogusa S, Polykratis A, et al. Kinase activities of RIPK1 and RIPK3 can direct IFN-beta synthesis induced by lipopolysaccharide. J Immunol. 2017;198:4435–47.
Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell. 2017;169:301–13.e11.
Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567–78.
Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J Immunol. 2018;200:1489–95.
Hartmann BM, Albrecht RA, Zaslavsky E, Nudelman G, Pincas H, Marjanovic N, et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat Commun. 2017;8:1931.
Moriwaki K, Chan FK. The inflammatory signal adaptor RIPK3: functions beyond necroptosis. Int Rev Cell Mol Biol. 2017;328:253–75.
Rathinam VAK, Chan FK. Inflammasome, inflammation, and tissue homeostasis. Trends Mol Med. 2018;24:304–18.
Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1–RIP3–DRP1 signaling pathway. Nat Immunol. 2014;15:1126–33.
Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, et al. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. 2016;196:407–15.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79.
Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515.
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40.
Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–46.
Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2017;2:16258.
Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against Influenza A virus. Cell Host Microbe. 2016;20:13–24.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90.
Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. 2013;8:e76841.
Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54:273–80.
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–7.
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65.
Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21.
Acknowledgements
This work is funded by NIH grant AI128197 and AI119030 (FKMC). The authors declare no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nailwal, H., Chan, F.KM. Necroptosis in anti-viral inflammation. Cell Death Differ 26, 4–13 (2019). https://doi.org/10.1038/s41418-018-0172-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0172-x
- Springer Nature Limited
This article is cited by
-
Implications of inflammatory cell death-PANoptosis in health and disease
Archives of Pharmacal Research (2024)
-
Role of necroptosis in kidney health and disease
Nature Reviews Nephrology (2023)
-
OASL phase condensation induces amyloid-like fibrillation of RIPK3 to promote virus-induced necroptosis
Nature Cell Biology (2023)
-
Differential effects of CMV infection on the viability of cardiac cells
Cell Death Discovery (2023)
-
Regulated cell death pathways and their roles in homeostasis, infection, inflammation, and tumorigenesis
Experimental & Molecular Medicine (2023)