
Abstract
Anti-apoptotic Bcl-2 proteins are upregulated in different cancers, including diffuse large B-cell lymphoma (DLBCL) and chronic lymphocytic leukemia (CLL), enabling survival by inhibiting pro-apoptotic Bcl-2-family members and inositol 1,4,5-trisphosphate (IP3) receptor (IP3R)-mediated Ca2+-signaling. A peptide tool (Bcl-2/IP3R Disruptor-2; BIRD-2) was developed to abrogate the interaction of Bcl-2 with IP3Rs by targeting Bcl-2′s BH4 domain. BIRD-2 triggers cell death in primary CLL cells and in DLBCL cell lines. Particularly, DLBCL cells with high levels of IP3R2 were sensitive to BIRD-2. Here, we report that BIRD-2-induced cell death in DLBCL cells does not only depend on high IP3R2-expression levels, but also on constitutive IP3 signaling, downstream of the tonically active B-cell receptor. The basal Ca2+ level in SU-DHL-4 DLBCL cells was significantly elevated due to the constitutive IP3 production. This constitutive IP3 signaling fulfilled a pro-survival role, since inhibition of phospholipase C (PLC) using U73122 (2.5 µM) caused cell death in SU-DHL-4 cells. Milder inhibition of IP3 signaling using a lower U73122 concentration (1 µM) or expression of an IP3 sponge suppressed both BIRD-2-induced Ca2+ elevation and apoptosis in SU-DHL-4 cells. Basal PLC/IP3 signaling also fulfilled a pro-survival role in other DLBCL cell lines, including Karpas 422, RI-1 and SU-DHL-6 cells, whereas PLC inhibition protected these cells against BIRD-2-evoked apoptosis. Finally, U73122 treatment also suppressed BIRD-2-induced cell death in primary CLL, both in unsupported systems and in co-cultures with CD40L-expressing fibroblasts. Thus, constitutive IP3 signaling in lymphoma and leukemia cells is not only important for cancer cell survival, but also represents a vulnerability, rendering cancer cells dependent on Bcl-2 to limit IP3R activity. BIRD-2 seems to switch constitutive IP3 signaling from pro-survival into pro-death, presenting a plausible therapeutic strategy.
Similar content being viewed by others


Introduction
Different malignancies, including B-cell cancers such as diffuse large B-cell lymphoma (DLBCL), are characterized by overexpression of the anti-apoptotic Bcl-2 protein [1]. This proto-oncogene is localized at the mitochondria and at the endoplasmic reticulum (ER). At the level of the mitochondria, Bcl-2 binds to and neutralizes pro-apoptotic BH3-only proteins via its hydrophobic cleft, thereby preventing Bak/Bax activation and mitochondrial outer membrane permeabilization [2]. BH3-mimetic compounds, like venetoclax, counteract Bcl-2′s anti-apoptotic function at the mitochondria [3]. These molecules trigger apoptosis in cancer cells that are primed to death due to high levels of Bax or Bim, and thus are addicted to Bcl-2 for their survival [4, 5].
However, some cancer cells with high Bcl-2 levels respond poorly to BH3 mimetics [6,7,8,9], suggesting that Bcl-2 promotes cell survival via a different mechanism. Indeed, the last decades, Bcl-2 proteins emerged as critical modulators of intracellular Ca2+ dynamics [10, 11]. As such, Bcl-2 also acts at the ER Ca2+ stores where it inhibits inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs), a major class of intracellular Ca2+-release channels [12, 13]. Bcl-2 impacts IP3Rs by binding with its N-terminal BH4 domain to the central, modulatory domain of the channel [14,15,16]. Furthermore, Bcl-2′s C-terminal transmembrane domain enables efficient IP3R inhibition within cells [17]. A cell-permeable peptide tool named Bcl-2/IP3R Disruptor-2 (BIRD-2) was developed, capable of stripping Bcl-2 from IP3Rs [18]. In contrast, the BH3-mimetic Bcl-2 inhibitor venetoclax is not able to disrupt Bcl-2/IP3R complexes [19]. In chronic lymphocytic leukemia (CLL) and DLBCL, BIRD-2 triggered pro-apoptotic Ca2+-release events, while sparing normal peripheral mononuclear blood cells [18, 20].
In a collection of DLBCL cell lines, we previously identified IP3R2-expression levels as an important determinant underlying BIRD-2 sensitivity [20]. Here, we investigated whether IP3R2 levels are the only determinant that dictates the BIRD-2 sensitivity of B-cell cancers. Of note, IP3R2 is the IP3R isoform that displays the highest sensitivity to its ligand IP3 [21, 22]. Interestingly, B-cell cancers, including DLBCL and CLL, display constitutive B-cell receptor (BCR) signaling [23,24,25]. A cascade of signaling proteins becomes activated downstream of the BCR, including phospholipase C gamma 2 (PLCγ2), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into IP3. We investigated whether constitutive PLCγ2/IP3 signaling occurs in B-cell cancer models and whether this contributes to survival and BIRD-2 sensitivity in DLBCL with elevated IP3R2-expression levels. Our results indicate that cancer cells are addicted to Bcl-2 acting at the ER Ca2+ stores to regulate IP3R-mediated Ca2+ release. We found that disrupting the Bcl-2/IP3R interaction with BIRD-2 switched Ca2+ signaling within cancer cells from pro-survival to pro-death, resulting in cancer cell death.
Results
IP3R2 expression is necessary but not sufficient for sensitivity to BIRD-2
Since the sensitivity of DLBCL cell lines to BIRD-2 correlated to IP3R2-expression levels [20], we questioned whether IP3R2 expression is sufficient to dictate BIRD-2 sensitivity. Via western-blot analysis, we measured the expression levels of IP3R2 and Bcl-2 in microsomes prepared from primary hepatocytes, which have a high IP3R2 density [26,27,28], in human liver carcinoma HepG2 cells and in the BIRD-2-sensitive (SU-DHL-4) and BIRD-2-resistant (OCI-LY-1) DLBCL cell lines (Fig. 1a). This analysis revealed that IP3R2 is expressed in SU-DHL-4 and HepG2 cells, as well as in primary hepatocytes, while IP3R2 is virtually absent in OCI-LY-1 (Fig. 1a). Furthermore, the DLBCL cell lines expressed high levels of the anti-apoptotic Bcl-2 protein, whereas Bcl-2 expression was very low in the HepG2 cells or even absent in the liver microsomes (Fig. 1a). We next asked whether HepG2 cancer cells and primary hepatocytes are sensitive to BIRD-2. Therefore, apoptosis was measured in the four different cell types after 2 and 24 h of BIRD-2 (10 µM) treatment (Fig. 1b-d). BIRD-2 induced cell death in about 50% of the SU-DHL-4 cells, whereas OCI-LY-1 cells were not sensitive to 10 µM BIRD-2. In HepG2 cells, BIRD-2 induced apoptosis in approximately 20% of the population, suggesting that tumorigenic cells expressing IP3R2 display BIRD-2 sensitivity. To further substantiate the importance of IP3R2 for BIRD-2 sensitivity, primary hepatocytes were treated with 10 µM BIRD-2. Despite expression of IP3R2 (Fig. 1a), the hepatocytes were resistant to BIRD-2-induced apoptosis (Fig. 1b-d). Consistent with our previous findings [20], these data indicate that IP3R2 expression is required for BIRD-2-evoked apoptosis since tumorigenic cells lacking IP3R2 (OCI-LY-1) were resistant to BIRD-2, whilst tumorigenic cells expressing IP3R2 (SU-DHL-4) were sensitive. However, IP3R2 expression per se is not sufficient for BIRD-2-evoked cell death, since hepatocyte cell viability was not significantly affected by BIRD-2.

High IP3R2-expression levels are not sufficient per se to render cells sensitive to BIRD-2. a The IP3R2- and Bcl-2-protein levels present in cell lysates from SU-DHL-4 (40 µg), OCI-LY-1 (40 µg), and HepG2 (40 µg) cells and from microsomes extracted from primary hepatocytes (20 µg) were determined by western-blot analysis. The expression level of calnexin was used as a control for equal loading. b Representative dot plots from flow cytometry analysis measuring apoptosis by staining SU-DHL-4, OCI-LY-1, HepG2 cells and primary hepatocytes with Annexin V-FITC and 7-AAD. Cells were treated with vehicle or 10 μM BIRD-2 for 2 h. The dot plots are representative of 3 independent experiments. c, d Quantitative analysis of 3 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained cells treated with vehicle or 10 µM BIRD-2. Apoptotic cell death was measured 2 h (c) and 24 h (d) after BIRD-2 treatment. Data are represented as average ± SEM (N = 3). Statistically significant differences were determined with a Student’s t-test (paired, two-tailed, *P < 0.05) (BIRD-2 versus vehicle)
SU-DHL-4 cells display enhanced basal IP3 signaling
Since the BCR is tonically active in DLBCL cells and since the BCR signalosome activates PLCγ2 [23,24,25], we investigated whether the BIRD-2 sensitive SU-DHL-4 cell line displayed elevated constitutive IP3 signaling. As a read-out for constitutive IP3 signaling, we monitored the cytosolic Ca2+ concentration ([Ca2+]cyt) with Fura-2 in SU-DHL-4 cells pre-treated with vehicle, the PLC inhibitor U73122 or its inactive enantiomer U73343 (Fig. 2a-b). The basal [Ca2+]cyt in vehicle-treated SU-DHL-4 cells was 59 ± 6.3 nM, which was lowered to 32 ± 5.6 and 38 ± 4.5 nM upon treatment with 1 and 2.5 μM U73122, respectively, while U73343 was without effect (Fig. 2c). Taken together, these results indicate that SU-DHL-4 cells are characterized by a constitutively active IP3 signaling, likely downstream to tonic BCR signaling.

SU-DHL-4 cells display constitutive IP3/Ca2+ signaling. a The basal Ca2+ level was used as a read-out for measuring the level of constitutive IP3 signaling downstream the BCR, which has been reported to be tonically active in germinal center DLBCL (BCR*) [23,24,25]. PLC activity was suppressed using U73122, whereas its inactive enantiomer U73343 did not affect PLC activity. b A typical fluorescent recording of the basal [Ca2+]cyt in SU-DHL-4 cells pre-treated with vehicle (black line), 1 μM U73122 (red line) or 1 µM U73343 (gray line) using the ratiometric Ca2+ indicator Fura-2 AM. The cells were present in Krebs medium supplemented with 1.5 mM CaCl2. The ratio of emitted fluorescence of Fura-2 (F340/F380) was monitored and Ca2+ values were calibrated by adding digitonin (50 µM) and a 20-fold excess of EGTA (33 mM) to determine Rmax and Rmin respectively (see Method section). Basal [Ca2+] (nM) are reported in c as the mean ± SEM (N = 5). The exact values of each independent experiment are represented in different colors. Statistically significant differences were determined using an analysis of variance (ANOVA, **P < 0.01, ***P < 0.001)
PLC inhibition suppresses BIRD-2-induced apoptosis in SU-DHL-4 cells
To assess the contribution of IP3 signaling to BIRD-2-induced cell death, we blocked PLC signaling with U73122 in SU-DHL-4. In these cells, U73122 suppressed IP3-induced Ca2+ release, since the anti-IgG/M-provoked cytosolic Ca2+ response was reduced in cells pre-treated for 30 min with 1 and 2.5 µM U73122, compared to vehicle- or U73343-treated cells (Fig. 3a). This Ca2+ response was quantified by measuring the area under the curve (AUC), which was significantly reduced upon treatment with U73122 (2.5 µM) (Fig. 3b). Next, it was determined whether PLC inhibition by itself impacted SU-DHL-4 survival by treating them for 30 min, 2 h or 24 h with different concentrations of U73122 (0.1, 0.5, 1, and 2.5 µM) or U73343. Interestingly, the highest concentrations of U73122 (1 and 2.5 µM), but not its inactive enantiomer, induced apoptotic cell death in SU-DHL-4 cells (Fig. 3c). These data indicate that PLC signaling has a pro-survival role in DLBCL cells. Subsequently, it was determined whether PLC inhibition protected against BIRD-2-induced cell death in SU-DHL-4 cells. Therefore, apoptosis induced by 10 µM BIRD-2 was measured in cells pre-treated for 30 min with vehicle, U73122 or U73343 (Fig. 3d). After 2 h (Fig. 3e) and 24 h (Fig. 3f) of peptide treatment, U73122 significantly protected SU-DHL-4 cells against BIRD-2-triggered apoptosis, while U73343 did not. Of note, since U73122 provoked cell death by itself, the ∆ apoptotic fraction was calculated for each condition (Fig. 3e,f). The ∆ apoptotic fraction was obtained by subtracting the % of cells undergoing cell death in U73122-treated conditions from the % of cells undergoing cell death upon BIRD-2 + U73122 treatment (Fig. 3d).

U73122 protects against BIRD-2-triggered apoptosis in SU-DHL-4. a Cell-population analysis of the cytosolic Ca2+ response, measured with Fura-2 AM, in SU-DHL-4 cells pre-treated for 30 min with U73122 (1 and 2.5 µM), U73343 (2.5 µM) or vehicle (DMSO). Addition of 3 mM EGTA and 12 µg/ml anti-IgG/M antibody is indicated by the first and second arrow, respectively. The curves are representative of 3 independent experiments. The cytosolic Ca2+ response after anti-IgG/M addition was quantified by measuring the area under the curve (AUC), which is shown in b. c Quantitative analysis of 3 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-4 cells. Cells were treated with varying concentrations of U73122 or 2.5 µM U73343. Apoptotic cell death was measured 30 min, 2 h and 24 h after treatment. On the y-axis the percentage of living cells is plotted. Data are shown as the average ± SEM (N = 3). d Representative dot plots from flow cytometry analysis detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-4 cells treated for 2 h with vehicle or 10 µM BIRD-2. Cells were pre-treated for 30 min with U73122 or U73343. e, f Quantitative analysis of 4 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-4 cells. Apoptotic cell death was measured as the percentage of Annexin V-FITC-positive cells. Cells were pre-treated with U73122 or U73343 for 30 min. Cell death was measured 2 h (e) and 24 h (f) after BIRD-2 treatment. On the y-axis, the ∆ apoptotic fraction is plotted, which is the difference in apoptosis between the BIRD-2-treated and the vehicle-treated fraction, and between the BIRD-2 + U73122-treated and the U73122-treated fraction, and finally between the BIRD-2 + U73343-treated and the U73343-treated fraction. Data are shown as the average ± SEM (N = 5). g Quantitative analysis of 4 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-4 cells treated with 1 or 2.5 µM U73122, 2.5 µM U73343, 5 µM BIRD-2 (blue), 3 µM venetoclax (green) or a combination of U73122/U73343 with BIRD-2/venetoclax. For the conditions without Bcl-2 inhibitor (indicated with a ‘-’), the green bars indicate the use of the vehicle control for venetoclax, while the blue bars indicate the use of vehicle control for BIRD-2 treatment. A ‘+’ indicates that the Bcl-2 inhibitor (BIRD-2/venetoclax) was added in this condition. Cell death was measured 24 h after treatment. On the y-axis the percentage of living cells, which corresponds to the Annexin V-FITC- and 7-AAD-negative fraction, is shown. Data are expressed as the average ± SEM (N = 4). h CI derived from SU-DHL-4 cells treated with U73122/U73343 in combination with BIRD-2/venetoclax. The CI was calculated (see Method section) from the data shown in g. Statistically significant differences were determined using an analysis of variance (ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001)
To exclude that the protective effect of U73122 against BIRD-2-induced apoptosis was due to the artifact that less living cells were available for BIRD-2 upon U73122 treatment, we performed cell death assays in which SU-DHL-4 cells were treated with U73122 in combination with venetoclax, a Bcl-2-selective BH3-mimetic drug that provokes cell death independently of Ca2+ overload [19]. If U73122 and venetoclax work independently, the effect should be additive, providing a clear distinction with the data obtained with BIRD-2. After 24 h of venetoclax treatment, approximately 60% of the cells were alive (Fig. 3g). Combined treatment of venetoclax with U73122 (1 and 2.5 µM) further decreased the percentage of living cells compared to single treatment with venetoclax due to the cell death induced by U73122, while U73343 did not display this effect. In contrast, BIRD-2-induced cell death was decreased in combination with the PLC inhibitor, indicating that U73122 protected against BIRD-2-triggered apoptosis (Fig. 3g). Thus, the reduction in BIRD-2-induced cell death provoked by PLC inhibition is not due to a decreased availability of living cells upon U73122 treatment. To substantiate this further, we calculated the combination index (CI), which quantifies whether a drug combination is synergistic (CI < 0.8), additive (0.8 ≤ CI ≤ 1.2), or antagonistic (CI > 1.2) (Fig. 3h). The CI for the combined treatment of venetoclax with U73122 was approximately 1, indicating this drug combination is additive. In contrast, combined treatment of U73122 with BIRD-2 resulted in a CI of around 1.7, indicating an antagonistic drug combination. Hence, the ∆ apoptotic fraction provides a bona fide analysis for the protective effects of U73122 against BIRD-2-induced cell death (Fig. 3e,f).
Thus, these data indicate that PLC activity contributes to BIRD-2-induced DLBCL cancer cell death. This suggests that disrupting Bcl-2/IP3R complexes results in excessive, pro-apoptotic Ca2+ signals that are driven by endogenous IP3 signaling, whereby Bcl-2 suppresses such pro-death Ca2+ fluxes by tuning-down IP3R activity. Moreover, the increased basal PLC activity in DLBCL cells is a pro-survival signal, which can be changed to pro-death signaling by BIRD-2.
PLC inhibition blunts the BIRD-2-induced cytosolic [Ca2+] rise in SU-DHL-4 cells
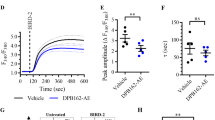
Next, we investigated in more depth how PLC inhibition prevented the BIRD-2-evoked death of SU-DHL-4 cells. As reported previously [20], BIRD-2 caused an IP3R-dependent increase in cytosolic Ca2+ levels in SU-DHL-4 cells. Here, we assessed BIRD-2-induced Ca2+ elevations in Fura-2-loaded SU-DHL-4 cells in the presence of U73122 using single cell (Fig. 4a,b) and cell population (Fig. 4c,d) Ca2+ measurements. BIRD-2, but not a TAT-control peptide, caused a rise in the cytosolic Ca2+ levels in SU-DHL-4 single cells as measured by fluorescence microscopy. This Ca2+ rise was less prominent in cells pre-treated with 1 μM U73122, but not with U73343 (Fig. 4a,b). Similar findings were obtained in SU-DHL-4 cell populations analyzed using a FlexStation 3 microplate reader (Fig. 4c). The peak amplitude of the BIRD-2-evoked Ca2+ rise was significantly lower in SU-DHL-4 cells pre-treated with 1 µM U73122 compared to cells treated with vehicle or U73343 (Fig. 4d).

U73122 reduces the BIRD-2-induced cytosolic Ca2+ rise in SU-DHL-4 cells. a Single-cell analysis of the BIRD-2-induced Ca2+ response in SU-DHL-4 cells using the ratiometric Ca2+ indicator Fura-2 AM. Representative pseudo-color images before (2 s) and after (500 s) BIRD-2 treatment are shown. Vehicle and TAT-Ctrl were used as negative control conditions. The pseudo-color scale bar indicates increasing ratio fluorescence. b Single-cell cytosolic Ca2+ signals (gray lines) and their respective mean (black line) upon addition of vehicle, TAT-ctrl peptide or 10 µM BIRD-2 to SU-DHL-4 cells without or with pre-treatment of 1 μM U73122/U73343. c Cell-population analysis of the cytosolic Ca2+ response induced by 10 µM BIRD-2 in SU-DHL-4 cells pre-treated without (black line) or with 1 µM U73122 (green line) or 1 µM U73343 (gray line). The curves are representative of 4 independent experiments. Data were quantified by calculating the peak amplitude (d). In d, data are represented as mean ± SEM (N = 4). Statistically significant differences were determined using an analysis of variance (ANOVA, *P < 0.05)
Buffering intracellular IP3 suppresses BIRD-2-induced apoptosis in SU-DHL-4 cells
Next, we aimed to confirm these findings by transfecting SU-DHL-4 cells with a high-affinity IP3 sponge that efficiently buffers intracellular IP3 [29]. Of note, approximately 40% of the SU-DHL-4 cells could be successfully transfected with our transfection method (Fig. 5a). BIRD-2-induced apoptosis was reduced in SU-DHL-4 cells expressing the IP3 sponge compared to mock-transfected or empty vector-transfected cells (Fig. 5b,c). The ∆ apoptotic fraction was approximately 22 and 16% in mock-transfected cells and SU-DHL-4 cells expressing a control vector, respectively (Fig. 5c). In contrast, the ∆ apoptotic fraction was only around 10% in SU-DHL-4 cells expressing the IP3 sponge. We also performed single-cell Ca2+ measurements, in which cells expressing the IP3 sponge displayed reduced BIRD-2-induced Ca2+ signals compared to empty vector-expressing cells (Fig. 5d). Hence, our pharmacological (U73122) and genetic (IP3 sponge) approaches indicate that constitutive IP3 signaling is an important determinant underlying BIRD-2 sensitivity in DLBCL. Moreover, the effect of the IP3 sponge demonstrates that IP3, rather than another messenger arising from upstream PLC activity, is critical for BIRD-2-evoked cell death.

SU-DHL-4 cells are protected from BIRD-2-triggered apoptosis by genetically manipulating the IP3 signaling pathway. a Examples of flow cytometry analysis showing the percentage of GFP-positive transfected SU-DHL-4 cells, visible as a shift in the BL-1 (515–545 nm) channel, while the values in the BL-3 (665–715 nm) channel remained unaffected. b Representative flow cytometry analysis of BIRD-2-induced apoptosis in SU-DHL-4 cells transfected with the IP3 sponge or a control vector compared to mock-transfected cells. Apoptosis was detected via Annexin V-APC-positive staining (RL1+ = red laser, see Method section) of the cells. c Quantification of the apoptotic fraction (%) after treatment with 10 µM BIRD-2 (red histogram in panel b) or vehicle (black histogram panel b) in mock-transfected SU-DHL-4 cells or cells transfected with the IP3 sponge or a control vector. Apoptotic cells were identified as the Annexin V-APC-positive fraction (RL1+). Data are represented as mean ± SEM of 3 independent experiments. d Single-cell cytosolic Ca2+ measurements performed in SU-DHL-4 cells utilizing Fura-2 AM. Cells were transfected with an IP3 sponge vector or with an empty vector as negative control condition (pcDNA3.1). The addition of 10 µM BIRD-2 is indicated by the arrow. Data are represented as the average ± SEM of 3 independent experiments (n > 100 cells/condition)
Pharmacological PLC inhibition also suppresses BIRD-2-induced apoptosis in other DLBCL cell lines
It was examined whether constitutive IP3 signaling also contributes to BIRD-2-triggered apoptosis in other DLBCL cell lines besides SU-DHL-4, including Karpas 422 and SU-DHL-6 as germinal center DLBCL and RI-1, characterized as activated B-cell DLBCL. First, the U73122 sensitivity of these cells was determined by measuring apoptosis 24 h after treatment with U73122 (1 or 2.5 µM) or U73343 (2.5 µM) (Fig. 6a-c). U73122, but not the inactive enantiomer, induced cell death in the three cell lines. U73122-triggered apoptosis was the lowest in SU-DHL-6 (Fig. 6a), whereas RI-1 cells (Fig. 6c) were the most sensitive to PLC inhibition. These results indicate that all three DLBCL cell lines, like the SU-DHL-4 cells, depend on constitutive PLC/IP3 signaling for their survival. Next, it was investigated whether BIRD-2-induced apoptosis depends on this constitutive IP3 signaling (Fig. 6). In each cell line, the IC50 value of BIRD-2, previously determined in a subset of DLBCL [9], was used. This corresponds to 15 µM BIRD-2 for SU-DHL-6 (Fig. 6a-e) and Karpas 422 cells (Fig. 6b-f), and 26 µM for RI-1 (Fig. 6c-g). To determine U73122-mediated protection against BIRD-2 in these cell lines, the ∆ apoptotic fraction was used. This analysis was again validated using venetoclax (Fig. 6a-d) as before (Fig. 3g-h), showing that U73122 treatment displayed additive cell-death effects with venetoclax, whereas U73122 + BIRD-2 is an antagonistic drug combination (Fig. 6d). In all three cell lines tested, BIRD-2-induced cell death was significantly suppressed by U73122 (Fig. 6e-g). In conclusion, these data indicate that not only SU-DHL-4 but also other DLBCL cell lines depend on constitutive IP3 signaling for their survival, and that this pro-survival signaling can be turned into pro-death signaling by BIRD-2.

PLC inhibition also suppresses BIRD-2-triggered apoptosis in other DLBCL cell lines. a, b, c Quantitative analysis of at least 3 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-6 (a), Karpas 422 (b), and RI-1 (c) cells treated with U73122, U73343, BIRD-2 (blue), venetoclax (green) or a combination of U73122/U73343 with BIRD-2/venetoclax. For the conditions without Bcl-2 inhibitor (indicated with a ‘-’), the green bars indicate the use of the vehicle control for venetoclax, while the blue bars indicate the use of vehicle control for BIRD-2 treatment. A ‘+’ indicates that the Bcl-2 inhibitor (BIRD-2/venetoclax) was added in this condition. SU-DHL-6 and Karpas 422 cells were treated with 15 µM BIRD-2, whereas 26 µM BIRD-2 was used to treat the RI-1 cells. SU-DHL-6 cells were treated with 500 nM venetoclax, Karpas 422 cells with 1 µM venetoclax and RI-1 cells were treated with 10 nM venetoclax. Cell death was measured 24 h after treatment. On the y-axis the percentage of living cells, which corresponds to the Annexin V-FITC- and 7-AAD-negative fraction, is shown. Data are expressed as the average ± SEM (N ≥ 3). d CI derived from cells treated with U73122/U73343 in combination with BIRD-2/venetoclax. The CI was calculated from the data shown in a, b, c. e, f, g Quantitative analysis of 4 independent experiments detecting apoptosis in Annexin V-FITC/7-AAD-stained SU-DHL-6 (e), Karpas 422 (f), and RI-1 (g) cells. Apoptotic cell death was measured as the percentage of Annexin V-FITC-positive cells. Cells were pre-treated with U73122 (1 or 2.5 µM) or U73343 (2.5 µM) for 30 min. Cell death was measured 24 h after BIRD-2 treatment. Data are shown as the ∆ apoptotic fraction, which is the difference in apoptosis between the BIRD-2-treated and the vehicle-treated fraction, and between the BIRD-2 + U73122-treated and the U73122-treated fraction, and finally between the BIRD-2 + U73343-treated and the U73343-treated fraction. Statistically significant differences were determined using an analysis of variance (ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001)
Pharmacological PLC inhibition suppresses BIRD-2-induced apoptosis in primary CLL patient cells
Finally, we aimed to translate our findings to primary peripheral blood cells obtained from patients diagnosed with CLL, another B-cell malignancy characterized by constitutively active BCR signaling. BIRD-2 (30 µM) treatment for 2 h triggered apoptosis in all 14 CLL samples analyzed, though with potencies ranging from ~20% to ~70% of the cells being apoptotic (Fig. 7). To assess whether IP3 signaling contributes to the BIRD-2 response in the CLL cells, BIRD-2-triggered apoptosis was measured in samples pre-treated for 30 min with U73122. The lowest U73122 concentration for which an effect could be observed was used in each sample (0.1/0.5/2.5 µM, Supplemental Table 1). The CLL patient samples were stratified in groups according to their sensitivity towards U73122 and to the CI calculated for the combined treatment of U73122 with BIRD-2. In this way, four different groups are recognized: CI > 1.2 and cell death U73122 < 10% (Fig. 7a), CI > 1.2 and cell death U73122 > 10% (Fig. 7b), 0.8 ≤ CI ≤ 1.2 (Fig. 7c), and CI < 0.8 (Fig. 7d). In 9 out of 14 CLL samples, we found that the drug combination was antagonistic (CI > 1.2), suggesting that PLC inhibition protected against BIRD-2-induced apoptosis. However, because the CLL cells displayed varying sensitivity to U73122, the samples were further subdivided according to U73122-induced cell death. U73122 did not induce apoptosis in 5 of these samples (Fig. 7a), whereas cell viability of the other 4 samples was reduced by PLC inhibition (Fig. 7b). To determine whether U73122 significantly protected against BIRD-2-induced apoptosis in these groups, the ∆ apoptotic fraction for BIRD-2-treated and U73122 + BIRD-2-treated cells was calculated (Supplemental Table 1). This analysis indicates that U73122 significantly protected against BIRD-2-induced apoptosis in both groups (Fig. 7a-b). Next, in 2 out of 14 CLL samples, U73122 did not protect against BIRD-2 (0.8 ≤ CI ≤ 1.2) (Fig. 7c), whereas in 3 out of 14 samples BIRD-2-induced apoptosis was even increased by U73122 (CI < 0.8) (Fig. 7d). The ∆ apoptotic fraction analysis was also validated for these CLL samples using venetoclax (Supplemental Fig. 1).

PLC inhibition suppresses BIRD-2-induced cell death in a subset of primary CLL patient cells. (a-d) Results from flow cytometry analysis of Annexin V-FITC/7-AAD-stained CLL patient samples treated for 2 h with 30 µM BIRD-2 with or without U73122 pre-treatment. For each CLL sample, the individual bar graph, plotting the apoptotic fraction (%) measured in untreated cells (black bar), cells treated with BIRD-2 (dark gray bar), U73122 (gray bar) or a combination of U73122 and BIRD-2 (light gray bar), is shown. The lowest U73122 concentration for which an effect could be observed was used (0.1/0.5/2.5 µM; see Supplemental Table 1). The CLL samples were stratified in 4 categories, according to the CI calculated for the combined treatment of U73122 and BIRD-2 and according to their sensitivity towards U73122: (a) CI > 1.2 & cell death U73122 < 10%; (b) CI > 1.2 and cell death U73122 > 10%; (c) 0.8 ≤ CI ≤ 1.2; (d) CI < 0.8. At the bottom of each panel, the ∆ apoptotic fraction (%), which corresponds to the difference in apoptotic fraction between the BIRD-2-treated and the control condition, and between the BIRD-2 + U73122-treated and the U73122-treated conditions, is shown for each CLL sample belonging to that category. In the dot plots, each CLL sample is represented with a different color, which is shown in the titles of the individual bar graphs. Statistically significant differences were determined using a one-tailed paired t-test (**P < 0.01)
Finally, we measured the effect of U73122 on the BIRD-2 response in CLL cells co-cultured with CD40L-expressing fibroblasts to allow for longer BIRD-2 treatments. Co-cultured CLL cells were treated for 20 h with BIRD-2 and/or U73122, after which the cells were collected and cell viability was assessed. Co-cultured CLL cells appeared better protected from spontaneous apoptosis than CLL cells in non-supported cultures (Fig. 8a). In these co-cultured CLL cells, BIRD-2 remained capable to induce cell death and PLC inhibitor U73122 significantly reduced BIRD-2-induced apoptosis (Fig. 8b). Thus, CLL cells in both unsupported and supported cultures share a common sensitivity to BIRD-2, as disruption of the Bcl-2/IP3R interaction leads to death of the primary cells. Furthermore, in most CLL patient samples suppressing basal PLC activity with U73122 protected against BIRD-2-induced apoptosis. However, for some CLL cells, enhanced basal PLC signaling is very critical for their survival, and solely inhibiting PLC is sufficient to cause cell death.

Pharmacological PLC inhibition protects against BIRD-2-induced apoptosis in co-cultured CLL cells. a Results from flow cytometry analysis of Annexin V-FITC/PI-stained CLL samples that were either unsupported or co-cultured with CD40L-expressing fibroblasts. The co-cultured CLL cells were treated for 20 h with vehicle or 30 µM BIRD-2 with or without 0.1 µM U73122 pre-treatment. Data are shown as the percentage of apoptotic cells (%). For each CLL sample, the CI calculated for U73122 + BIRD-2 treatment is indicated. b Plot of the ∆ apoptotic fraction (%) for BIRD-2 and U73122 + BIRD-2 treatment of each CLL sample in co-cultured conditions. The ∆ apoptotic fraction corresponds to the difference in apoptotic fraction between the BIRD-2-treated and the control condition, and between the BIRD-2 + U73122-treated and the U73122-treated conditions. Statistically significant differences were determined using a one-tailed paired t-test (**P < 0.01)
Discussion
The main finding of this study is that constitutive IP3 signaling, besides high IP3R2-expression levels, is an important determinant that underlies cancer cells’ addiction to Bcl-2 at the ER Ca2+ stores. Constitutive IP3 signaling is therefore an additional determinant of the sensitivity of B-cell cancers, like DLBCL and CLL, to BIRD-2, a Bcl-2 inhibitor that targets its BH4 domain and alleviates Bcl-2′s inhibitory role on IP3R channels. As such, BIRD-2 can be applied as a tool to exploit pro-survival constitutive IP3 signaling occurring in B-cell cancers and switch it into pro-apoptotic signaling.
BIRD-2 disrupts endogenous Bcl-2/IP3R complexes, thereby triggering Ca2+-driven apoptosis in different malignancies, including CLL [18, 20, 30], DLBCL [20], multiple myeloma [31], follicular lymphoma [31], and small cell lung cell carcinoma [32]. DLBCL cells displayed a varying sensitivity to BIRD-2, which correlated to the expression levels of IP3R2 [20]. Furthermore, a reciprocal sensitivity between BIRD-2 and venetoclax has been reported for DLBCL cells, indicating that cancer cells less sensitive to BH3 mimetics are more sensitive to BIRD-2 and vice versa [9]. Additionally, low BIRD-2 concentrations sensitized DLBCL cells towards venetoclax by upregulating the pro-apoptotic BH3-only protein Bim [9]. BIRD-2 also sensitized multiple myeloma cells to BH3 mimetics through a mechanism that involved the Ca2+-dependent upregulation of Bim [31]. In this study, we also measured Bim expression in SU-DHL-4 cells treated with higher concentrations of BIRD-2 (Supplemental Fig. 2). After 24 h of treatment with the IC50 value of BIRD-2 (10 µM), we observed a significant increase in Bim expression, suggesting that this BH3-only protein may contribute to BIRD-2-triggered cytotoxicity. However, further work is needed to elucidate the role of Bim in BIRD-2-induced apoptosis.
It is well established that DLBCL and CLL cells display chronic BCR signaling, leading to constitutive activation of different signaling pathways, including the PLCγ2 pathway, which leads to the production of IP3 in basal conditions [23,24,25]. We here show that IP3 levels are likely elevated in SU-DHL-4 cells, as a PLC inhibitor was able to lower basal [Ca2+]. We also attempted to directly measure IP3 levels using an IP3 FRET sensor [33], but the dynamic range of this sensor appeared insufficient to reliably assess a decrease in basal IP3 levels using our microscopy systems. The role of constitutive IP3 signaling in DLBCL cell survival requires further study, since pharmacological PLC inhibition using U73122 affected the survival of several DLBCL cell lines. We anticipate that this is an on-target effect of U73122 on PLC, since its inactive enantiomer U73433 did not display this effect. These findings indicate a pro-survival role of basal IP3/Ca2+ signaling, but further work is needed to document this in other B-cell cancers and lymphoproliferative malignancies. Nevertheless, these results converge with findings obtained in solid tumors, showing that tumorigenic, but not non-tumorigenic, cells depend on basal IP3R function for their survival [34,35,36]. In these cancer models, IP3Rs provide a constitutive ER-mitochondrial Ca2+ flux to drive mitochondrial metabolism and the production of mitochondrial substrates needed for nucleotide synthesis critical for cancer cell proliferation. Normal cells are less dependent on IP3Rs for their survival, as they can tune down proliferation to accommodate the compromised mitochondrial bio-energetics [34,35,36]. However, further research is needed to determine whether constitutive IP3 signaling and basal IP3R function are both essential for B-cell cancer cell survival by mediating ER-mitochondrial Ca2+ fluxes that sustain mitochondrial metabolism, thereby accounting for U73122-induced cell death [34, 35].
An important implication of this study is that although IP3R2 expression is important for BIRD-2-induced apoptosis, it is not sufficient per se. It is clear that a constitutively increased level of IP3, the ligand that activates IP3R channels, is needed as well. Of interest, IP3R2 channels display the highest IP3 sensitivity [22]. Thus, the combination of high IP3R2-expression levels and constitutive IP3 signaling makes DLBCL cells particularly addicted to Bcl-2 inhibition of IP3Rs at the ER, and thus sensitive to BIRD-2. This is supported by data obtained in primary hepatocytes, cells expressing relatively high levels of IP3R2, but which are resistant to BIRD-2, suggesting that IP3R2 alone is not sufficient for BIRD-2 sensitivity. These data are of high importance, as IP3R2 channels are expressed in different organs and tissues in the human body, where they exert important physiological functions [22]. Our data therefore suggest that BIRD-2-derived or BIRD-2-mimetic molecules may be well tolerated in the human body and may not cause a general toxicity in normal cells or tissues that express high IP3R2 levels.
The concept of constitutive IP3 signaling contributing to BIRD-2 sensitivity was also observed in primary CLL samples, where low concentrations of U73122 protected against BIRD-2-induced apoptosis. In the first place, we focused on the cell-autonomous response of the CLL cells towards BIRD-2. However, interactions with bystander cells in micro-environmental niches support CLL cells by providing survival and proliferative signals [37,38,39]. Hence, these unsupported experiments were restricted towards short-term BIRD-2 application to limit spontaneous cell death, correlating with loss of Bcl-2-family members such as anti-apoptotic Mcl-1 due to rapid loss of supportive signals [37]. Therefore, we also performed experiments in CLL cells supported by CD40L-expressing fibroblasts. These co-culture conditions protected against spontaneous apoptosis, but did not antagonize BIRD-2-induced cell death. Furthermore, PLC inhibition with U73122 remained capable of suppressing BIRD-2-induced apoptosis in CLL cells irrespective of whether they were exposed to BIRD-2/U73122 in unsupported or co-cultured conditions.
We observed variability in the sensitivity of individual CLL samples to BIRD-2, though the underlying mechanisms remain elusive. The BIRD-2 sensitivity of CLL cells did not correlate to their BCR mutational status, suggesting that basal IP3 signaling might be increased in B-cell cancers irrespective of their BCR mutational status (Supplemental Table 1). The varying BIRD-2 sensitivity could be due to differences in IP3R2 expression, due to varying deficiencies in regulators of IP3R function [40], or due to different degrees of coupling between the ER and the mitochondria [41]. For instance, phosphatase and tensin homolog (PTEN) and protein kinase B (Akt/PKB) control Ca2+-dependent apoptosis via IP3R3 [42,43,44]. Since reduced PTEN levels have been reported in CLL, as well as in DLBCL [45, 46], we measured PTEN expression in the primary CLL samples and DLBCL cell lines used in this study. All CLL samples expressed PTEN at similar levels (Supplemental Fig. 3a), indicating that differences in PTEN expression do not account for the varying BIRD-2 sensitivity of CLL cells. Furthermore, PTEN was detected in SU-DHL-4, SU-DHL-6 and RI-1 cells, but not in Karpas 422 (Supplemental Fig. 3b). Thus, BIRD-2 sensitivity of DLBCL cells appears unrelated to PTEN expression, since both PTEN-proficient SU-DHL-4 and PTEN-deficient Karpas 422 cells respond well to BIRD-2 (IC50 values around 10 µM) [9].
Overall, our study indicates that constitutive IP3 signaling, likely a pro-survival mechanism in B-cell malignancies, is an important contributor for BIRD-2-induced apoptosis in cancer cells that express high IP3R2 levels. Although IP3R2 is important for BIRD-2-induced cell death, its high expression alone is not sufficient per se for BIRD-2 sensitivity. This is important given the pivotal physiological functions of IP3R2 channels in normal tissues and cells. Hence, Bcl-2 antagonism via the BH4 domain might be a promising strategy to target B-cell cancers, in particular those displaying high IP3R2-expression levels and constitutive IP3 signaling.
Materials and methods
Reagents, antibodies, and constructs
Reagents were as follows: ethylene glycol tetraacetic acid (EGTA) (Acros Organics, Geel, Belgium, 409910250), Fura-2 AM (Biotium, Kampenhout, Belgium, 50033), Annexin V-Fluorescein isothiocyanate (FITC) (Becton Dickinson, Franklin Lakes, NJ, USA, 556419), 7-aminoactinomycin D (7-AAD) (Becton Dickinson, 555815), U73122 (Enzo Life Sciences, Farmingdale, NY, USA, BML-ST391-0005), U73343 (Enzo Life Sciences, BML-ST392-0005), venetoclax (ChemieTek, Indianapolis, IN, USA, CT-A199), anti-human IgG/M (Jackson ImmunoResearch, West Grove, PA, USA, 109-006-127). The following antibodies were used: anti-IP3R2 (Abiocode, Agoura Hills, CA, USA, R2872-3); anti-calnexin (Enzo Life Sciences, Farmingdale, NY, USA, ADI-SPA-865-D); anti-Bcl-2 (Santa Cruz Biotechnology, Dallas, TX, USA, sc7382HRP); anti-Bim (Bioké, Leiden, The Netherlands, 2819 S); anti-GAPDH (Sigma-Aldrich, St. Louis, MO, USA, G8795); anti-vinculin (Sigma-Aldrich, Munich, Germany, V9131). The sequences of the peptides used in this study were: BIRD-2 (RKKRRQRRRGGNVYTEIKCNSLLPLAAIVRV) and TAT-Ctrl (RKKRRQRRRGGSIELDDPRPR). These peptides were synthesized by LifeTein (South Plainfield, New Jersey, USA) with a purity of at least 85%. The IP3 sponge (pEF-GSTm49-IRES-GFP) is a protein constructed from the IP3-binding core of the type 1 IP3R with a single amino acid substitution (R441Q) that has a very high affinity for IP3 [29].
CLL patient samples
CLL was defined by clinical examination of the patients and immunophenotypic analysis of the blood samples. Only samples where > 80% of the cells were CD19 + were considered. The tumor immunoglobulin heavy chain variable (IGHV) sequence was determined to designate the BCR status (unmutated or mutated). The collection of blood samples from CLL patients has been approved by the ethical committee of the UZ Leuven (Belgian Number: B3222001536) and by the ethical committee of the Università Cattolica del Sacro Cuore, Fondazione Policlinico A. Gemelli, Rome, Italy (protocol number 14563/15). Blood samples were collected according to the principles established by the International Conference on Harmonization Guidelines on Good Clinical Practice. An informed consent was obtained from all patients. Primary lymphocytes were separated using a Ficoll Hypaque density gradient from the peripheral blood of adult patients with B-CLL, and re-suspended in RPMI-1640 medium. For co-culture experiments, human CLL cells (1 × 107/ml) were cultured for 24 h in the presence of CD40L-expressing fibroblasts (2 × 104/condition), which were previously treated for 2 h with 10 µg/ml mitomycin C. The CLL co-cultures were then pre-treated for 90 min with U73122 prior to the addition of BIRD-2. After 20 h of BIRD-2 treatment, CLL cells were collected and analyzed.
Cell culture
The SU-DHL-4, OCI-LY-1, Karpas 422, and SU-DHL-6 DLBCL cell lines were kindly obtained from Dr. Anthony Letai (Dana-Farber Cancer Institute, Boston, Massachusetts, USA). The RI-1 DLBCL cell line was obtained from DSMZ (Braunschweig, Germany). All these cell lines were authenticated by the University of Arizona Genetics Core (Tucson, AZ, USA) using autosomal short tandem repeat (STR) profiling utilizing the Science Exchange platform (www.scienceexchange.com). All cell lines fully matched the DNA fingerprint present in reference databases, except for SU-DHL-6 cells, which matched 7 out of 8 tested alleles. OCI-LY-1 cells were cultured in suspension in Iscove’s modified Dulbecco’s medium (Invitrogen, Merelbeke, Belgium), while the other DLBCL cell lines were cultured in suspension in RPMI-1640 medium (Invitrogen, Merelbeke, Belgium). The human hepatocellular carcinoma cell line HepG2 was cultured in Dulbecco’s Modified Eagle medium (DMEM). Media were supplemented with 10% heat-inactivated fetal bovine serum, L-glutamine (100 × GlutaMAX, Gibco/Invitrogen, 35050) and penicillin and streptomycin (100 × Pen/Strep, Gibco/Invitrogen, 15070-063). Cells were cultured at 37 °C in the presence of 5% CO2. Primary hepatocytes were isolated from mice using a two-step collagenase perfusion as previously described [47]. Subsequently, the primary cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, L-glutamine, penicillin and streptomycin.
Cell transfection
Twenty-four hours after seeding, the indicated vectors were introduced into the SU-DHL-4 cells utilizing the Amaxa® Cell Line Nucleofector® Kit L (Lonza, Basel, Switzerland), program C-05. Briefly, 3 × 106 cells were transfected with 3 μg of pEF-GSTm49-IRES-GFP (IP3 sponge vector), 3 μg pcDNA3.1 (control vector), or 1.5 μg pmaxGFP® vector (to assess transfection efficiency). GFP expression was checked by flow cytometry 24 h after transfection. A pcDNA 3.1(-) mCherry expressing vector was co-transfected at a 1:3 ratio as a selection marker for single-cell cytosolic Ca2+ imaging.
Apoptosis assay
DLBCL cells (5 × 105 cells/ml) were treated as indicated, pelleted by centrifugation, and incubated with Annexin V-FITC/7-AAD or Annexin V-APC. Cell suspensions were analyzed with an Attune® Acoustic Focusing Flow Cytometer (Applied Biosystems). Cell death by apoptosis was scored by quantifying the population of Annexin V-FITC-positive cells (blue laser; BL-1) or Annexin V-APC-positive cells (red laser; RL-1). The latter was used in combination with pEF-GSTm49-IRES-GFP. To assess the effect of U73122 on BIRD-2-induced cell death, the ∆ apoptotic fraction was obtained by subtracting the % of cells undergoing cell death in U73122-treated conditions from the % of cells undergoing cell death upon BIRD-2 + U73122 treatment. Flow-cytometric data were plotted and analyzed using Attune version 2.1.0 (Applied Biosystems) or FlowJo version 10 software. The CI was calculated in order to determine mathematically whether a drug combination is synergistic (CI < 0.8), additive (0.8 ≤ CI ≤ 1.2), or antagonistic (CI > 1.2). The CI was determined by making the ratio of the sum of the individual effects (EffectCompound A + EffectCompound B) with the effect of the combined treatment (EffectCompound A + Compound B).
Western-blot analysis
Cells were washed with phosphate-buffered saline and incubated at 4 °C with lysis buffer (25 mM HEPES, pH 7.5, 1% Triton X-100, 300 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol, and protease inhibitor tablets (Roche, Basel, Switzerland)) for 30 min on a head-over-head rotor. Cell lysates were centrifuged for 5 min at 10,000 r.p.m. and analyzed by western blotting as previously described [16]. Microsomes were prepared from primary hepatocyte as previously described [48].
Basal [Ca2+]cyt measurements
Basal Ca2+ levels were monitored with the cytosolic Ca2+ indicator Fura-2 AM. Cells (10 × 106/sample) were loaded for 30 min with 1.25 µM Fura-2 AM at room temperature in modified Krebs solution (containing 150 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 11.6 mM HEPES (pH 7.3), 11.5 mM glucose and 1.5 mM CaCl2), followed by a de-esterification step of 30 min in the absence of Fura-2 AM. During the de-esterification step, cells were treated with vehicle, U73343 (1 and 2.5 µM) or U73122 (1 and 2.5 µM). Fluorescence was monitored on a luminescence spectrometer (AMINCO-Bowman Series 2, Spectronic Unicam) by alternately exciting the Ca2+ indicator at 340 and 380 nm and collecting emitted fluorescence at 510 nm. Basal [Ca2+]cyt was derived after in situ calibration according to the Grynkiewicz equation: [49]
Kd is the dissociation constant of Fura-2 for Ca2+ at room temperature (241 nM), q is the fluorescence ratio of the emission intensity in the absence of Ca2+ (F380 max), to that in the presence of saturating Ca2+ (F380 min), R is the fluorescence ratio, and Rmin and Rmax are the minimal and maximal fluorescence ratios, respectively. Rmax was obtained by administrating 50 µM digitonin, subsequently Rmin was measured by adding 33 mM EGTA in Ca2+-free modified Krebs solution.
Ca2+ measurements in cell populations
To perform Ca2+ measurements in intact cells, DLBCL cells were seeded in poly-L-lysine-coated 96-well plates (Greiner) at a density of 5 × 105 cells/ml. The cells were loaded for 30 min with 1.25 µM Fura-2 AM at 25 °C in modified Krebs solution, followed by a 30 min de-esterification step in the absence of Fura-2 AM. Fluorescence was monitored on a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) by alternately exciting the Ca2+ indicator at 340 and 380 nm and collecting emitted fluorescence at 510 nm, as described previously [50]. All data were obtained in triplicate and are plotted as F340/F380. At least three independent experiments were performed.
Single-cell Ca2+ imaging
The IP3 sponge and mCherry constructs were introduced into SU-DHL-4 cells as described above. A Zeiss Axio Observer Z1 Inverted Microscope equipped with a 20x air objective and a high-speed digital camera (Axiocam Hsm, Zeiss, Jena, Germany) were used for these measurements. Fura-2 AM measurements were performed as previously described [15].
Statistical analysis
Results are expressed as average ± SD or SEM as indicated. The number of independent experiments is always indicated. Significance was determined using a one-tailed or two-tailed paired Student’s t-test or an analysis of variance (ANOVA) as appropriate. Differences were considered significant at P < 0.05.
References
Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40.
Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–41.
Besbes S, Mirshahi M, Pocard M, Billard C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862–71.
Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000.
Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32.
Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol J Am Soc Clin Oncol. 2017;35:826–33.
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A, et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Vervloessem T, Akl H, Tousseyn T, De Smedt H, Parys JB, Bultynck G, et al. Reciprocal sensitivity of diffuse large B-cell lymphoma cells to Bcl-2 inhibitors BIRD-2 versus venetoclax. Oncotarget. 2017;8:111656–71.
Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016;35:5079–92.
Vervliet T, Clerix E, Seitaj B, Ivanova H, Monaco G, Bultynck G. Modulation of Ca2+ signaling by anti-apoptotic B-cell lymphoma 2 proteins at the endoplasmic reticulum-mitochondrial interface. Front Oncol. 2017;7:75.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203.
Joseph SK, Hajnóczky G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis. 2007;12:951–68.
Rong Y-P, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31:255–65.
Rong Y-P, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA. 2009;106:14397–402.
Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19:295–309.
Ivanova H, Ritaine A, Wagner L, Luyten T, Shapovalov G, Welkenhuyzen K, et al. The trans-membrane domain of Bcl-2α, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget. 2016;7:55704–20.
Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011;117:2924–34.
Vervloessem T, Ivanova H, Luyten T, Parys JB, Bultynck G. The selective Bcl-2 inhibitor venetoclax, a BH3 mimetic, does not dysregulate intracellular Ca2+ signaling. Biochim Biophys Acta. 2017;1864:968–76.
Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013;4:e632.
Foskett JK, White C, Cheung K-H, Mak D-OD. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658.
Vervloessem T, Yule DI, Bultynck G, Parys JB. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim Biophys Acta. 2015;1853:1992–2005.
Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12:229–43.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92.
Dühren-von Minden M, Übelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489:309–12.
De Smedt H, Missiaen L, Parys JB, Bootman MD, Mertens L, Van Den Bosch L, et al. Determination of relative amounts of inositol trisphosphate receptor mRNA isoforms by ratio polymerase chain reaction. J Biol Chem. 1994;269:21691–8.
Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G, et al. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014;1843:2164–83.
Wojcikiewicz RJ. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem. 1995;270:11678–83.
Uchiyama T, Yoshikawa F, Hishida A, Furuichi T, Mikoshiba K. A novel recombinant hyperaffinity inositol 1,4,5-trisphosphate (IP3) absorbent traps IP3, resulting in specific inhibition of IP3-mediated calcium signaling. J Biol Chem. 2002;277:8106–13.
Akl H, La Rovere RML, Janssens A, Vandenberghe P, Parys JB, Bultynck G, et al. HA14-1 potentiates apoptosis in B-cell cancer cells sensitive to a peptide disrupting IP3 receptor / Bcl-2 complexes. Int J Dev Biol. 2015;59:391–8.
Lavik AR, Zhong F, Chang M-J, Greenberg E, Choudhary Y, Smith MR, et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget. 2015;6:27388–402.
Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW, et al. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015;6:e2034.
Matsu-ura T, Michikawa T, Inoue T, Miyawaki A, Yoshida M, Mikoshiba K, et al. Cytosolic inositol 1,4,5-trisphosphate dynamics during intracellular calcium oscillations in living cells. J Cell Biol. 2006;173:755–65.
Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2016;14:2313–24.
Bultynck G. Onco-IP3Rs feed cancerous cravings for mitochondrial Ca2+. Trends Biochem Sci. 2016;41:390–3.
Morciano G, Marchi S, Morganti C, Sbano L, Bittremieux M, Kerkhofs M, et al. Role of mitochondria-associated ER membranes in calcium regulation in cancer-specific settings. Neoplasia N Y N. 2018;20:510–23.
van Attekum MHA, Terpstra S, Slinger E, von Lindern M, Moerland PD, Jongejan A, et al. Macrophages confer survival signals via CCR1-dependent translational MCL-1 induction in chronic lymphocytic leukemia. Oncogene. 2017;36:3651–60.
van Attekum MH, Eldering E, Kater AP. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica. 2017;102:1469–76.
Davids MS, Deng J, Wiestner A, Lannutti BJ, Wang L, Wu CJ, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–9.
Bittremieux M, Parys JB, Pinton P, Bultynck G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca2+ signaling. Biochim Biophys Acta. 2016;1863:1364–78.
Decuypere J-P, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB, et al. The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta. 2011;1813:1003–13.
Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun. 2008;375:501–5.
Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012;3:e304.
Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013;20:1631–43.
Leupin N, Cenni B, Novak U, Hügli B, Graber HU, Tobler A, et al. Disparate expression of the PTEN gene: a novel finding in B-cell chronic lymphocytic leukaemia (B-CLL). Br J Haematol. 2003;121:97–100.
Pfeifer M, Grau M, Lenze D, Wenzel S-S, Wolf A, Wollert-Wulf B, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110:12420–5.
Dirkx R, Vanhorebeek I, Martens K, Schad A, Grabenbauer M, Fahimi D, et al. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatol Baltim Md. 2005;41:868–78.
Vanlingen S, Sipma H, De Smet P, Callewaert G, Missiaen L, De Smedt H, et al. Modulation of inositol 1,4,5-trisphosphate binding to the various inositol 1,4,5-trisphosphate receptor isoforms by thimerosal and cyclic ADP-ribose. Biochem Pharmacol. 2001;61:803–9.
Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50.
Decuypere J-P, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgó J, et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy. 2011;7:1472–89.
Acknowledgements
We thank Marina Crabbé, Anja Florizoone, Benny Das and Tomas Luyten for their excellent technical assistance. We thank Dr. A. Letai (Dana-Farber Cancer Institute, United States) for providing the DLBCL cell lines. We are grateful to the Laboratory of Ion Channel Research (KU Leuven, Belgium) for providing HepG2 cells. This work was supported by grants from the Fund for Scientific Research-Flanders (FWO) (grants G.0571.12 N, G.0634.13 N, G.0C91.14 N, and G.0A34.16 N) and the Research Council-KU Leuven (OT14/101). Ma.Bi. is holder of a Ph.D. Fellowship from the FWO. GB, JBP, and PP are part of the FWO–Scientific Research Community CaSign (W0.019.17 N). DGE is supported by grants from the Italian Association for Cancer Research (AIRC IG2016 Id.19236). PP is grateful to Camilla degli Scrovegni for continuous support and is supported by the Italian Ministry of Education: University and Research, the Italian Ministry of Health, Telethon (GGP15219/B), the Italian Association for Cancer Research (AIRC IG-18624) and by local funds from the University of Ferrara.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by A. Villunger
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bittremieux, M., La Rovere, R.M., Akl, H. et al. Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ 26, 531–547 (2019). https://doi.org/10.1038/s41418-018-0142-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0142-3
- Springer Nature Limited
This article is cited by
-
IP3R2-mediated Ca2+ release promotes LPS-induced cardiomyocyte pyroptosis via the activation of NLRP3/Caspase-1/GSDMD pathway
Cell Death Discovery (2024)
-
Bcl-xL acts as an inhibitor of IP3R channels, thereby antagonizing Ca2+-driven apoptosis
Cell Death & Differentiation (2022)
-
Dynamic control of mitochondria-associated membranes by kinases and phosphatases in health and disease
Cellular and Molecular Life Sciences (2021)
-
BDA-366, a putative Bcl-2 BH4 domain antagonist, induces apoptosis independently of Bcl-2 in a variety of cancer cell models
Cell Death & Disease (2020)
-
Recent advances in uncovering the mechanisms contributing to BIRD-2-induced cell death in B-cell cancer cells
Cell Death & Disease (2019)