
Abstract
The tumour microenvironment (TME) determines vital aspects of tumour development, such as tumour growth, metastases and response to therapy. Cancer-associated fibroblasts (CAFs) are abundant and extremely influential in this process and interact with cellular and matrix TME constituents such as endothelial and immune cells and collagens, fibronectin and elastin, respectively. However, CAFs are also the recipients of signals—both chemical and physical—that are generated by the TME, and their phenotype effectively evolves alongside the tumour mass during tumour progression. Amid a rising clinical interest in CAFs as a crucial force for disease progression, this review aims to contextualise the CAF phenotype using the chronological framework of the CAF life cycle within the evolving tumour stroma, ranging from quiescent fibroblasts to highly proliferative and secretory CAFs. The emergence, properties and clinical implications of CAF activation are discussed, as well as research strategies used to characterise CAFs and current clinical efforts to alter CAF function as a therapeutic strategy.
Similar content being viewed by others

Background
Conceptualising cancer as an exclusively tumour cell-centric disease is no longer an effective approach for identifying novel therapeutic targets.1 Invasive cancer in particular, has been described as a ‘derangement in the proper sorting of cell populations, causing a violation of normal tissue boundaries’,2 and requires a two-way, tumour-permissive relationship between host stroma and tumour epithelium. The stromal components of a tumour have thus graduated in our appreciation from bystanders, to enablers, to indispensable and determinant players with respect to disease evolution and response to therapy.3 Thanks mainly to more sophisticated in vivo models and analysis techniques,4,5 a focused effort is currently underway to consider the tumour-cell phenotype as an emergent property of both the tumour microenvironment (TME) and the activity of stromal cell populations that surround and infiltrate the tumour.
The stromal content of solid tumours can vary significantly, both in amount and composition; in certain tumour types, such as pancreatic, stomach, colon and breast cancer, the stroma contributes 60–90% of the total tumour mass.6,7,8 Tumour stromal tissue comprises fibroblasts, endothelial and immune cells, and extracellular matrix (ECM) components, which all differ markedly from their quiescent counterparts in homoeostatic tissues. Fibroblasts are of particular interest owing to their extensive matrix-synthesising and matrix-remodelling capacities, both of which are pivotal for establishing an invasion-permissive TME.9 In fact, levels of fibroblast markers within tumours are known to be predictive of disease outcome, with the number and properties of fibroblasts in the tumour stroma influencing overall survival, time to recurrence and resistance to therapy.10,11 Targeting fibroblast function for therapeutic benefit is therefore an area of intensive research.11
The term ‘fibroblast’ describes a heterogeneous group of spindle-shaped cells of mesenchymal origin with a spectrum of phenotypes characterised by varying degrees of contractility, ranging from the resting fibroblast to the activated myofibroblast observed in wound healing.12 Fibroblasts that have become activated during wound healing are referred to as ‘myofibroblasts’, while those activated in the context of tumours are called carcinoma-associated fibroblasts (CAFs) or peritumour fibroblasts.13 Though different in nomenclature, activated fibroblasts display analogous marker expression and very similar phenotypes, with the important distinction that in contrast to acute inflammation, fibroblasts in chronic inflammation and tumour tissue fail to inactivate.10 A great number of studies have identified the signalling mechanisms by which CAFs influence tumour cell properties (we refer the reader to several comprehensive reviews),11,14 and it has become clear that CAFs play a pivotal role in the TME. However, our understanding of the diverse properties of CAFs throughout the different stages of tumour progression remains incomplete. The function of CAFs has remained especially elusive in light of their heterogeneity and different effects on tumour development; while their pro-tumorigenic role has been well documented across tumour types,15 CAFs have also been reported to restraining tumour growth in several studies.16,17,18
In this review, we describe the dynamics of fibroblast activation in acute and chronic wounds, focusing on solid cancers. Most studies investigating the CAF–tumour cell relationship concentrate on carcinomas at the invasive stage, but we attempt to look before and after this stage to provide insights into the wider life cycle of fibroblasts located in the tumour stroma. Specifically, we describe the properties of fibroblasts as they progress from their resting state in tissue homoeostasis to the different magnitudes of activation occurring during early, invasive and metastatic tumour growth. Finally, we consider the clinical relevance of the activated fibroblast phenotype and opportunities for targeting fibroblast activation therapeutically.
Fibroblasts in homoeostasis
During tissue homoeostasis, epithelial cells are strictly segregated from fibroblasts in the adjacent connective tissue19 by the basement membrane (BM), a specialised ECM structure that presents the biophysical cues necessary for cell attachment, polarity and cell movement within and across tissue compartments.20,21 Resting or quiescent fibroblasts are dispersed as single cells within the stromal tissue compartment and are found proximal to the BM, where they orient themselves in the direction of matrix fibres, but have no direct association with the BM.22 During homoeostasis, quiescent fibroblasts show minimal metabolic and transcriptional activity but play a pivotal role in defining the differentiation status of adjacent epithelia via the secretion of specific signalling factors, including among others Wnt and bone morphogenic proteins,23,24 and the production and organisation of matrix proteins, including collagens, fibronectin and elastin, at a minimal rate, to maintain a compliant, yet tension-resistant BM.4
Although the structural characteristics of quiescent stroma are relatively well defined, the molecular characteristics of quiescent fibroblasts have remained elusive. From a collection of several markers that are not exclusive nor obligate to fibroblasts across tissues, fibroblast-specific protein 1 (FSP-1), a cytoplasmic calcium-binding protein, has been proposed as the most accurate marker for resting fibroblasts to date (even though FSP-1 is also found on cancer cells that have undergone EMT,25 as well as being expressed on macrophages26); other markers include vimentin, filamin A and Thy-1/CD90.4,27 Consequently, the quiescent fibroblast is generally considered to be a fibroblast that does not express the markers observed in activated fibroblasts, as defined primarily from studies of fibrosis and wound repair. As such, perhaps the most apt characterisation of a quiescent fibroblast is that of a mesenchymal cell with the potential to become activated by growth factors, and drastically increase its contractility, proliferation and secretory phenotype in response to an appropriate stimulus.4 Note that the lack of data specific to resting fibroblasts might be attributable to two factors. First, the focus is typically placed on their tissue-specific, activated counterparts (myofibroblasts or CAFs).4 Second, the inability to establish fibroblast cultures with a truly ‘resting’ phenotype has hampered the investigation of these cells in vitro; although fibroblasts can readily be cultured in vitro,4 they become at least partially activated owing to the properties of culture plastic, which is reminiscent of stiffened, fibrotic ECM.28,29
Fibroblasts in non-homoeostatic conditions
Wound healing
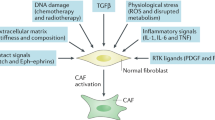
The acute inflammation that occurs during wound healing is a classic example of the loss of the homoeostatic ‘separation’/sorting behaviour of epithelial and stromal-cell populations and is characterised by the appearance of activated fibroblasts (myofibroblasts); indeed, much of what we know about fibroblast activation was first observed in the context of acute inflammation. During wound healing, the goal of fibroblast activation is to re-establish the pre-injury tissue architectural and homoeostatic states,4,12 and accordingly the fibroblast content in wounds increases via proliferation and recruitment.30 Fibroblasts become activated (as myofibroblasts) to perform a number of functions. They regulate the de novo deposition of matrix proteins and remodelling of the existing matrix via the secretion of matrix-degrading enzymes (e.g. matrix metalloproteinases) and through enhanced contractility.31 Their contractile apparatus also enables them to regulate interstitial fluid pressure.24 Through a plethora of secreted factors, fibroblasts can also interact with other cell types,32 in order to re-vascularise the healing tissue, and furthermore they secrete and absorb metabolites to rebalance the tissue niche.12 A key functional difference between fibroblasts and myofibroblasts therefore involves the cell secretome, with myofibroblasts demonstrating an increased production of proteins, including signalling molecules. Furthermore, on a molecular basis, while resting fibroblasts are characterised by the absence of activation markers and markers typical for other cell types,12 myofibroblasts acquire, in addition to FSP-1, and among others, the expression of α-smooth muscle actin (α-SMA), desmin, fibroblast activation protein (FAP), discoidin domain-containing receptor 2 (DDR2), collagens (e.g. collagen 1a1) and fibronectin, as well as numerous other growth factors and receptors, cytokines and ECM components,10 many of which are listed in Fig. 1.

Fibroblasts respond to physical cues and chemical signalling factors in the tumour microenvironment (TME) (‘input signals’) by producing a number of molecules that signal in an autocrine and paracrine fashion, as well as by altering the physical properties of the TME (‘output functions’). In contrast to acute wounds, fibroblasts in desmoplasia establish a self-activating and perpetual feedback loop (bottom arrow), which forms the basis of their pro-tumorigenic capacities. Moving from the quiescent phenotype (green, top), to the CAFs in desmoplasia (orange, bottom), the number of known inputs and outputs increases cumulatively. ROS reactive oxygen species, ET endothelin, TGF-β transforming growth factor β, bFGF basic fibroblast growth factor, PDGF platelet-derived growth factor, MMPs matrix metalloproteinases, TIMPs tissue inhibitors of metalloproteinases, VEGF vascular endothelial growth factor, HGF hepatocyte growth factor, IGF insulin-like growth factor, EGF epidermal growth factor, CXCL CXC motif chemokine ligand, SDF-1 stromal-cell-derived factor-1, TNF tumour necrosis factor, IFN-γ interferon-γ, IL interleukin, Shh sonic hedgehog, SPARC secreted protein acidic and rich in cysteine, EDA-FN EDA-containing cellular fibronectin, PGE2 prostaglandin E2, CTGF connective tissue growth factor; NF-κB nuclear factor κB.
Tumours
CAFs in tumours acquire the same features as do myofibroblasts in inflammation, and no systematic stratification or differences have been identified between these fibroblast populations. Figure 1 summarises the similarities and differences in functional properties of fibroblasts as they transition from quiescence to acute inflammation (myofibroblasts) and chronic inflammation/tumours (CAFs), and Table 1 describes the common functional assays used to assess the properties of activated fibroblasts; for a more exhaustive list of common assays to assess invasion, refer to ref. 33 A crucial difference between chronic wounds/tumours and acute inflammation however, is the failure to abate the fibrotic response in the former. Whereas the properties of myofibroblasts lead to the re-establishment of tissue architecture in acute inflammation, the acquisition of these same properties over prolonged periods in chronic wounds/tumours leads to further disorganisation of tissue architecture and tissue desmoplasia, a term that specifically denotes an increase in stromal cells and matrix components within an injured tissue.3
During both inflammation and tumour growth, the number of activated fibroblasts at the injured tissue site increases via recruitment of progenitor cells from different sources and proliferation of these cells, as well as tissue-resident fibroblasts,34,35 which represent the most immediate pool of cells to be activated into highly proliferative CAFs and recruited to the site of tumour growth. It is unclear whether these different CAF sources give rise to different CAF subpopulations that maintain distinct properties within the tumour over time.
Tissue desmoplasia is common in cases of chronic inflammation and tumours, highlighting the parallels between the two states, and positions the activated fibroblasts as an important mechanistic link between cancer and fibrosis.36 Indeed, conditions of chronic inflammation, such as inflammatory bowel disease, hepatic fibrosis or prostatitis, can be pre-emptive of tumour development at these anatomical sites.37 Furthermore, because of the similarities between chronic wounds and tumours in establishing fibrotic tissue niches, certain signalling pathways have been identified in both conditions as potential therapeutic targets for decreasing fibrosis and CAF-driven tumour aggressiveness, including the platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) pathways.38,39
The life cycle of CAFs during disease progression
The above discussion has compared the two best-known states of fibroblasts—quiescence versus activation in chronic wounds/tumours. In this section, we focus on the transition that occurs between these states during tumour growth, which is concurrent with the breakdown of local tissue structures and tumour cell invasion. In addition to highlighting the properties of CAFs in invasive disease, we emphasise studies that have provided insight into what precedes and what follows the establishment of a mature CAF and this timeline is illustrated in Fig. 2.

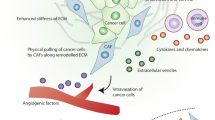
Schematic overview of the changes in tumour cell and CAF phenotypes in the context of an evolving tumour microenvironment (TME). The bottom panel lists open questions in CAF biology with potential implications for therapeutic interventions. The black arrow in the ‘metastatic spread’ panel indicates metastatic spread of tumour cells.
Fibroblast recruitment during carcinoma in situ
To date, it is unclear whether changes in stromal properties precede or are a result of the hyperproliferation of transformed epithelial cells,2,40 and the mechanisms and timing of fibroblast recruitment and activation at the preinvasive stage are poorly understood. The local dysregulation of epithelial signalling and proliferation in an otherwise homoeostatic tissue potentially result in altered chemical signalling to the surrounding tissue stroma and mechanical changes to the BM. Other than paracrine signalling from epithelial cells,41 potential sources of fibroblast-activating signals include cues from immune cells that are recruited to the site of epithelial hyperproliferation,42 microRNAs (miRNAs) produced by the growing mass of transformed epithelial cells43 and mechanical cues arising from a local increase in tissue tension.44 These activating signals potentially permeate through the ECM and BM structures of certain porosities (BM pore diameter ranges from 5 nm to 8 µm21); alternatively, tumour-derived exosomes carrying, for example, miRNAs or TGF-β family members, have been implicated in establishing tumour-promoting signalling in tumour niches and fibroblast activation.45,46 Immune cells, especially macrophages, B cells and regulatory T cells, can directly activate fibroblasts, and increased densities of these cells are found in tumour stroma,47 where they accumulate in response to the release of inflammatory mediators by hyperproliferative, transformed epithelial cells.48,49 Fibroblast activation might also result from an external insult responsible for initiation of the neoplastic growth itself: one study showed that dermal fibroblasts could be primed by UV radiation, which resulted in an epigenetic loss of Notch signalling, increasing their synthesis of growth factors and capacity to remodel the matrix.50
A few specific cancers, such as breast ductal carcinoma in situ (DCIS), offer the opportunity to explore fibroblast status prior to BM breaching. For example, one study examined the gene expression signatures of stromal cells in DCIS versus invasive cancer51 and identified a 66-gene signature common to fibroblasts from invasive breast cancer and a subset (40%) of DCIS tissues, suggesting that fibroblast activation occurs at the preinvasive tumour stage. Analogously, a comparison of the expression of angiogenic genes between the stroma of non-malignant, preinvasive, invasive and metastatic breast tissues revealed a clustering of these genes in all but the healthy tissues.52 Furthermore, a study specifically investigating the appearance of α-sma+ myofibroblasts in normal, benign and malignant breast tissue observed a shift from the presence of exclusively CD34+ fibrocytes in normal tissue to the emergence of α-sma+-reactive myofibroblasts in benign lesions and invasive tumours concurrent with the loss of the CD34+ cell populations.53 These results further suggest that stromal-cell activation can occur at the preinvasive stages of the disease, and that the tumour stroma as a whole is then ‘primed’ for the subsequent stages of tumour development.
CAF maturation during basement membrane breaching and local invasion
The transition from in situ to locally invasive disease is a crucial step for tumour growth and requires breaching of the BM. This process is carried out actively by the tumour epithelium, immune cells and CAFs54 and exposes these cell populations to each other, allowing for direct contact and increased short-range signalling between them. CAFs are capable of digesting BM components and might therefore be responsible for mediating significant breaching of this structure.2 Previous studies, for example, have revealed the capacity of CAFs to break down BM via matrix-metalloproteinase (MMP)-mediated digestion to generate ‘tracks’ that were subsequently occupied by invading carcinoma cells.55 Moreover, CAFs that were presented with murine mesentery (a BM substitute) induced cell permeability in the membrane via an MMP-independent mechanism; they remodelled the matrix by applying contractile forces, creating gaps between BM fibres that allowed cancer cells to pass through.56 Importantly, a caveat of such studies is the use of fully mature CAFs sourced from tumours that were already invasive and our appreciation of the contribution of stage-matched CAFs to BM breaching during the transition to locally invasive tumour growth is limited.
As a consequence of the changes in tissue structure (ECM composition and organisation), tissue architecture/composition (intermingling of CAFs with other cellular fractions) and activated fibroblast secretome during local tissue invasion (see Fig. 1), the parameters that dictate the gradients of CAF-produced signals are likely to change to more readily influence other TME cellular components, leading to an increasingly activated TME. It is useful at this point to draw a parallel with fibroblast activation during acute wound healing, a process in which the temporal dynamics of activation have been well defined, and a two-stage model of myofibroblast differentiation has been proposed.57 In inflamed tissue, fibroblasts increase their production of stress fibres (composed of cytoplasmic actin) and organised fibronectin on their cell surface, due to the mechanical tension present. These features promote the transition of a normal fibroblasts to an intermediate stage known as a protomyofibroblast. This cell type is characterised by an enhanced contractile capacity compared with quiescent fibroblasts, and has been observed in 2–4 day-old wounds within the granulation tissue (the new connective tissue created to seal the wound).58 Protomyofibroblasts have not yet incorporated α-SMA into their stress fibre network in order to generate the potent traction forces on the surrounding matrix that are characteristic of a mature myofibroblast.31 However, continued mechanical tension induces the production of specific splice variants of fibronectin, mainly ED-A and ED-B, which have been shown to guide transdifferentiation of protomyofibroblasts into myofibroblasts through poorly understood mechanisms.58 Concurrently, autocrine or paracrine signalling through the TGF-β pathway results in the synthesis of α-SMA and collagen type 1, ultimately giving rise to fully mature myofibroblasts.57
In contrast to the process described above, the temporal dynamics of fibroblast activation during tumour progression have yet to be firmly pinpointed. However, it is reasonable to surmise that a fibroblast entity equivalent to the protomyofibroblasts found in acute wounds might exist, which can be thought of as an intermediately activated fibroblast with lower contractility that arises at some point during the early stages of tumorigenesis. The further activation of such intermediates could then be achieved by the several CAF-mediated signalling pathways known to perpetuate further CAF activation.10 For example, the production of TGF-β and stromal-cell-derived factor-1 (SDF-1, also known as CXC motif chemokine 12 [CXCL12]), by CAFs results in further fibroblast activation.59 Similarly, CAF-mediated expression of leukaemia inhibitory factor (LIF) is responsible for initiating, within CAFs themselves, the constitutive activation of Janus kinase 1 (JAK1)/signal transducer and activator of transcription 3 (STAT3) signalling via p300-mediated histone acetylation, resulting in enhanced CAF contractility and consequent ECM remodelling. These properties lead to further CAF activation, consequent tumour-cell invasion and further breakdown of tissue architecture.60,61
As exemplified above, epigenetic regulation is considered, for a number of reasons, to be a major contributor to CAF activation within the TME.62 First, fibroblast transition from the quiescent to the activated state is not based on the acquisition of genetic errors (CAFs are mostly considered genetically stable, although debate on this topic continues35,63). Second, the epigenetic status is heritable and microenvironmentally induced, and is therefore likely to fit the dynamics of fibroblast activation.64 For example, TGF-β and other growth factor signals result in global changes in the methylation status of fibroblast genes.62,65 In lung cancer stroma, SMAD3 was one of the genes found to be silenced by hypermethylation, which elicited a hyper-responsiveness of CAFs to TGF-β signalling, leading to an increased expression of wound-response genes such as COL1A1, thereby linking TME methylation to increased ECM deposition by CAFs.66
During local invasion, the physical interaction and bidirectional movement between epithelial and stromal tissue components that follows BM breakdown creates a ‘zone of mixing’, from which metastatic cells eventually emerge.2 These mixing zones attract a continuous supply of fibroblast progenitors from different cell pools (see above), which differentiate/become activated, proliferate and infiltrate the tumour mass.10,67 Such interface zones of tumour cells and tumour stromal-cell populations are arguably sites in which intense TME remodelling occurs, and which are characterised by unique functionalities and marker expression, including markers of invasion or EMT in epithelial cells.68,69 Utilising a clonal tracing strategy, a recent study found that tumour cells with cancer stem cell properties were located along the tumour margin and regulated by CAF-secreted osteopontin.70 With respect to CAFs, a study investigating the differences between fibroblasts at increasing distances from the surgical tumour margin identified increased pro-proliferative and EMT-inducing capacities between all CAFs compared with fibroblasts taken at least 10 mm from the tumour margin.71 More importantly, however, CAFs sourced from the interface zone were more potent inducers of tumour progression than CAFs present within the tumour core, as assessed in vitro, indicating that interface CAFs possessed a pronounced tumour modulatory capacity.71
Mature CAFs during local and metastatic spread
The complete CAF response to the presence of tumour epithelia and remodelled matrix takes place in tumour–stroma mixing zones. The local tissue architecture becomes highly distorted by CAF infiltration, with aberrant de novo fibrinogenesis and matrix cross-linking (via CAF-secreted lysyl oxidase).72,73 The consequent rise in tissue tension continues to catalyse the activation of CAFs, which enter the feed-forward loop of contractility, ECM stiffening and TGF-β production.31 Analyses of CAFs present in invasive carcinomas have revealed the presence of diverse activation markers and associated functions, which might reflect differences in CAFs across tissues, differences in the proportions and origins of CAF progenitor cells recruited, and a possible spectrum of CAF ‘maturity’ within the TME.74
A number of classifications of CAFs have been recently proposed. At a minimum, and based on the most polarising effects of the CAF phenotype, a stratification between tumour-promoting and tumour-restraining CAFs has been identified.18,75 The tumour-promoting activities classically identified in CAFs across tumour tissues include promoting tumour cell survival, growth and stemness, as well as metastatic capacity.10 Furthermore, CAFs have been implicated in affecting chemoresistance via specific CAF paracrine signalling (e.g. interleukin [IL]-6 76 or plasminogen activator inhibitor [PAI-1] 77), as well as CAF-mediated drug scavenging and the establishment of a biophysical ECM barrier (the latter two were more commonly identified in highly desmoplastic solid tumours such as PDAC). These CAF activities hamper drug delivery and reduce drug efficacy.78,79 CAFs with tumour-restraining activity, on the contrary, have been named the ‘F1’ subtype, and a landmark study has shown their tumour-restraining qualities in the pancreatic tumours in a transgenic mouse model, where depletion of α-SMA+ CAFs resulted in tumours with enhanced aggressiveness via an upregulation of CD4+ regulatory T cells.16 Meflin, a marker of perivascular mesenchymal stromal cells, was recently proposed as a marker of tumour-restraining CAFs in pancreatic cancer.18 By contrast, the ‘F2’ CAF phenotype denotes pro-tumorigenic fibroblasts, which have been the focus of most studies and described above.75 It remains unclear whether a choice exists in different organs between an F1 and F2 fate, or how the F1 phenotype persists in certain cases, and whether a switch from an F2 phenotype to an F1 phenotype is possible. Further stratification between CAF subtypes has been provided, again in pancreatic cancer, by Tuveson et al., who revealed the presence of CAF populations termed ‘iCAFs’, ‘myCAFs’ and antigen-presenting CAFs, which have distinct roles in direct interactions with tumour cells, paracrine signalling and modulation of the immune niche, respectively.80,81 Other studies have further assessed CAF heterogeneity at the single-cell level.82,83 Based on the expression patterns of specific sets of genes in breast cancer, distinct but overlapping clusters of ‘protomyofibroblasts’, ‘ECM-regulating myofibroblasts’ and ‘secretory myofibroblasts’ were identified and a fibroblast differentiation model could be constructed, which suggested a hierarchical organisation of fibroblast populations.82 RNA-Seq revealed further CAF heterogeneity with CAF subgroups expressing independent prognostic biomarkers and arising through EMT from distinct cellular sources, including tumour cells, resident fibroblasts and perivascular cells.83 Overall, this heterogeneous impact of CAFs on tumour progression might depend on the properties of the tumour cells themselves, or be reflective of tumour stage.35
Fibroblast senescence
Another stage in the life cycle of CAFs that has been identified is senescence—that is, the decline of the proliferative and differentiation capacity of CAFs and CAF progenitors, with a concurrent increase in secretory capacity.84,85 In terms of the positioning of senescence along the CAF life cycle timeline, no information is available; however, in vitro, senescent CAFs are encountered after several culture passages—particularly rapidly when the cells were obtained from older tissues.4 Senescence is a process associated with cellular ageing that results from telomere dysfunction and senescence stimuli, such as the accumulation of reactive oxygen species and DNA damage,86 eventually leading to growth arrest in G1, an enlarged morphology in vitro, non-responsiveness to mitogens and resistance to apoptosis. Importantly, apoptosis and senescence are viewed as alternative cell fates, and the balance between them appears to be cell specific.87 In CAFs, senescence has a reported dual effect: it arrests the proliferation of CAFs in the TME, which could therefore prove beneficial; however, senescent CAFs secrete inflammatory mediators and growth factors, such as IL-888 and MMP-2,89 which have pro-tumorigenic effects.90 SDF-1/CXCL12, a chemokine present in the secretome of senescent CAFs, was found to mediate tumour cell survival and proliferation both directly and through the recruitment of other TME components.91
Because senescent fibroblasts accumulate in ageing tissues, it stands to reason that they could be responsible for conditioning stromal tissue over time, transforming it into a premetastatic niche.86 However, the question of whether senescence is the obligate fate of a CAF in desmoplasia remains open. Notably, senescence was observed in fibroblasts cultured in the presence of doxorubicin and paclitaxel, and therapy-induced senescence is a well-known phenomenon,90 which highlights an important issue: although it is desirable to therapeutically induce senescence in cancer cells, the senescence of stromal-cell components might induce further inflammation, and ultimately, tumour relapse.90
CAF inactivation and depletion through apoptosis
During wound resolution, the reconstituted ECM eventually takes over the mechanical load of the healing tissue, allowing a large portion of the now stress-released myofibroblast population to undergo apoptosis or inactivation.92 Spontaneous myofibroblast inactivation was specifically described in a model of regressing liver fibrosis, which demonstrated that half of the myofibroblast population is inactivated while the remainder undergoes apoptosis.93 However, very little is known about the role of inactivation or apoptosis in regulating CAF abundance and function within the TME. CAF loss of function, or at least partial inactivation, has been observed in hypoxic tumours, in which CAFs lose their capacity for contraction and matrix deposition owing to the inhibition of propyl hydroxylase domain protein 2 (PHD2) and stabilisation of hypoxia-inducible factor (HIF)-1α,94 which leads to a reduced metastatic potential. The apoptotic sensitivity of CAFs in vivo has also been described and found to be analogous to myofibroblast apoptosis in wound resolution or activation-induced T-cell death, although the underlying basis and timing of the process is unclear.95 It remains to be investigated under which circumstances apoptotic sensitivity, rather than senescence, is engaged, and questions surrounding CAF apoptosis, its role in regulating CAF abundance in the TME and its implications in tumour progression are yet to be answered. The addition of all-trans retinoic acid (ATRA)96 or the vitamin D analogue calcipotriol97 has been shown to inactivate CAFs in genetically engineered mouse models of pancreatic cancer. Beyond these studies however, CAF inactivation and significant levels of apoptosis are not reported in tumours in vivo, most likely as a result of the perpetual activating signals delivered to CAFs by the TME and its dysfunctional matrix,92 and possibly also due the induction of a senescent CAF phenotype after a certain, unknown, point in the CAF life cycle. These observations raise the question of whether treatments that aim to achieve CAF inactivation will succeed while the TME ‘wound’ persists.
Targeting activated CAFs as a therapeutic strategy
While therapies targeting cancer cells directly encounter issues with drug resistance, due to the cells’ genetic instability, the concept of targeting CAFs therapeutically has received growing attention, since CAFs are characterised by relative genetic stability.35,98,99 In this section, we summarise the attempts made at targeting or modulating the activated CAF phenotype within the context of the CAF life cycle. Included are compounds in preclinical and clinical stages, which can be roughly categorised, based on the point of activity within the life cycle that they target, as ‘blocking CAF activation’, ‘blocking the CAF hyperproliferative phenotype’, ‘blocking mature CAF function’, ‘inactivating the CAF phenotype’, and ‘directly depleting the CAF population’. Compounds that target CAF-mediated chemoresistance are classified under sections relevant to the mature CAF life cycle and its function. Of note, all five methods ultimately result in a reduced number of CAFs with pro-tumorigenic activity within the TME.
Blocking initial and further CAF activation has been attempted by intercepting signalling pathways between CAFs and/or between cancer cells and CAFs,100,101 such as the Rho/Rho kinase (ROCK) pathway and STAT3-mediated activation of insulin-like growth factor type 1 receptor (IGF-1R). As the hyperproliferative phenotype is also observed in fibrosis, anti-fibrotic treatments, such as pirfenidone and tranilast, which target CAF hyperproliferation in maturing and mature CAF populations, have actively made their way towards applications in anticancer therapies.102,103,104 Blocking mature CAF function is currently the focus of a large research area for anti-cancer, CAF-centric therapies that target the cells’ secretome and inhibit growth factor signalling, such as IL-6 and PDGFR signalling.105,106 Inactivation of the CAF phenotype attempts to transdifferentiate CAFs towards fates reminiscent of their quiescent counterparts, by leveraging mechanisms involved in apoptosis and/or inactivation, which have been observed to spontaneously occur in fibrotic models, as well as in acute wound healing, and include, for example, calcipotriol (a vitamin D receptor ligand) and minnelide (which de-regulates the TGF-β pathway).97,107 Finally, directly depleting the intratumoural CAF population has been accomplished by targeting cell surface markers, such as FAP108 and by inducing cell death through inhibition of the Bcl-2 pathway.95 Selected drugs and compounds within each category are summarised in Table 2. It should be noted that many of these therapies are currently administered in combination with standard-of-care chemotherapeutic treatments in an attempt to enhance their effectiveness and/or to reduce CAF-mediated chemoresistance.103,109 However, besides compounds in preclinical and clinical stages, there is currently no specific CAF-based adjuvant therapy on the market, which is undoubtedly a result of the inability to specifically target CAFs, due to a lack of CAF-exclusive surface markers and the functional heterogeneity within the CAF population.11
Outlook
The growth and phenotypic changes that occur in tumour cells during disease progression are concurrent with, and enabled by, a co-evolving tumour stroma. In this review, we have outlined the properties of fibroblasts at different stages in their life cycle within this stroma, from fibroblast quiescence in homoeostasis to CAF senescence, and highlighted the parallels between activated states in acute injury and in chronic wounds/tumours. Based on this life cycle, we have also highlighted the therapeutic opportunities available to leverage CAF inactivation to achieve CAF depletion from tumours. Similar to the situation in chronically fibrotic tissue, in which a critical mass of myofibroblasts is responsible for extreme levels of matrix deposition (which ultimately leads to the loss of organ function), understanding how to interfere with the recruitment and proliferative mechanisms that lead to increased fibroblast densities in the TME has the potential to be hugely beneficial, as proliferation of CAFs leads to an exponential amplification of the amount of pro-tumorigenic interactions present within the TME.
The overview presented here highlights that the life cycle of a CAF is strewn with knowledge gaps, particularly in its temporal trajectory (Fig. 2). Studies examining fibroblast activation in preinvasive tumours suggest that initial activation must occur very early on in tumour progression, although the mechanisms of this fibroblast priming are unclear. Furthermore, provided that the stages of myofibroblast activation in wound healing are recapitulated in tumour stroma, it remains uncertain how the transient entity of protomyofibroblasts fits into this picture, both chronologically during recruitment, and spatially at increasing distances from the stiffened, desmoplastic tumour lesion. In addition, biomarkers for identifying and targeting fibroblasts during their maturation period are currently not available, although they could have interesting therapeutic implications for inactivating fibrotic stroma. Similarly, in invasive disease, although a consensus exists on the causal relationship between the persistence of activating signals in the cancer wound and the inability of CAFs to undergo inactivation, the mechanism responsible for this within CAF cells is unknown—albeit of great interest for therapeutic purposes.
Our review also emphasises that CAF heterogeneity represents another challenging aspect of tumour biology. The concurrent existence of CAFs with different properties within the TME might depend on the origin of the precursor cell or the evolutionary stage or clonal composition of the TME. At the same time, myofibroblasts/CAFs have been described as cellular entities of a particular functional state, rather than as a singular cell type.110 These views imply the convergence of multiple progenitor populations towards a specific set of functionalities within the TME, such as enhanced contractility and protein synthesis. They also raise the question of whether current research efforts are merely capturing snapshot data from tumours, which in reality harbour a much wider hierarchical network of CAF maturation stages. Future studies aiming at revealing the ‘differentiation hierarchy’ of CAFs during tumour evolution must focus on stage-specific and stage-matched in vitro models of CAF–cancer cell co-cultures and untangle intratumoural heterogeneity on a single-cell or CAF subpopulation basis, as well as answering the question of whether in vitro culture approaches are supporting the full spectrum of CAFs present in vivo, and if so, which of these types of CAF might be clinically relevant.
The unknowns scattered throughout the CAF life cycle represent challenges for therapeutic intervention. Understanding these aspects could have implications for therapies that aim to reduce the presence of activated, pro-tumorigenic CAFs in tumour stroma. Such therapies could act on CAFs during their maturation phase, halting their differentiation into fully and irreversibly activated fibroblasts, and thereby cutting off further supply of CAFs for the growing tumour. Finally, a paramount consideration for any CAF-centred therapy will be the question of which part of the CAF spectrum remains unaffected by, or is specifically induced by, a given treatment. Given both the pro- and anti-tumorigenic potential of CAFs, understanding how to appropriately modulate these phenotypes at a given tissue/cancer stage will probably be critical in determining the failure or success of a fibroblast-targeting therapy.
References
Rodenhizer, D., Dean, T., D’Arcangelo, E. & McGuigan, A. P. The current landscape of 3D in vitro tumor models: what cancer hallmarks are accessible for drug discovery? Adv. Healthc. Mater 7, 1–36 (2018).
Liotta, L. A. The miucroenvironment of the tumour-host interface. Nature. 411, 375–379 (2001).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat Rev. Cancer 6, 392–401 (2006).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat Rev. Cancer 16, 582–598 (2016).
Hanahan, D. & Coussens, L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 21, 309–322 (2012).
Ronnov-Jessen, L., Petersen, O. W. & Bissell, M. J. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 76, 69–125 (1996).
Dvorak, H. F. Tumors: wounds that do not heal. N. Engl. J. Med. 315, 1650–1659 (1986).
Powell, D. W., Adegboyega, P. A., Di Mari, J. F. & Mifflin, R. C. Epithelial Cells and Their Neighbors I. Role of intestinal myofibroblasts in development, repair, and cancer. AJP Gastrointest. Liver Physiol. 289, G2–G7 (2005).
Tao, L., Huang, G., Song, H., Chen, Y. & Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 14, 2611–2620 (2017).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model Mech. 11, dmm029447 (2018).
Chen, X. & Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov 18, 99–115 (2019).
McAnulty, R. J. & Laurent, G. J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 39, 666–671 (2002).
Liu T., Han C., Wang S., Fang P., Ma Z., Xu L., et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 12, 86 (2019)
Kobayashi, H., Enomoto, A., Woods, S. L., Burt, A. D., Takahashi, M. & Worthley, D. L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol 16, 282–295 (2019).
Fiori, M. E., Franco, S. Di, Villanova, L., Bianca, P., Stassi, G. & Maria, R. De. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. 18, 1–16 (2019).
Özdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C.-C., Simpson, T. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with diminished survival. Cancer Cell. 25, 719–734 (2014).
Rhim, A. D., Oberstein, P. E., Thomas, D. H., Mirek, E. T., Palermo, C. F., Sastra, S. A. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 25, 735–747 (2014).
Mizutani Y., Kobayashi H., Iida T., Asai N., Masamune A., Hara A., et al. Meflin-Positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 79, 5367–5382 (2019)
Armstrong, P. B. & Armstrong, M. T. Intercellular invasion and the organizational stability of tissues: a role for fibronectin. Biochim. Biophys. Acta 1470, O9–O20 (2000).
Sawant, S., Dongre, H., Singh, A. K., Joshi, S., Costea, D. E., Mahadik, S. et al. Establishment of 3D co-culture models from different stages of human tongue tumorigenesis: Utility in understanding neoplastic progression. PLoS ONE. 11, 1–20 (2016).
Sekiguchi, R. & Yamada, K. M. Basement membranes in development and disease. Curr. Top Dev. Biol. 130, 143–191 (2018).
Tschumperlin, D. J. Fibroblasts and the ground they walk on. Physiology. 28, 380–390 (2013).
Ootani, A., Li, X., Sangiorgi, E., Ho, Q. T., Ueno, H., Toda, S. et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 15, 701–706 (2009).
Roberts, K. J., Kershner, A. M. & Beachy, P. A. The stromal niche for epithelial stem cells: a template for regeneration and a brake on malignancy. Cancer Cell. 32, 404–410 (2017).
Kahounov, Z., Kurf, D., Bouchal, J., Kharaishvili, G., Navratil, J., Remsik, J. et al. The fibroblast surface markers FAP, anti-fibroblast, and FSP are expressed by cells of epithelial origin and may be altered during epithelial-to-mesenchymal transition. Cytom. Part A. 93A, 941–951 (2018).
Österreicher C. H., Penz-österreicher M., Grivennikov S. I., Guma M., Koltsova E. K., Datz C., et al. Fibroblast-specific protein 1 identifies aninflammatory subpopulation of macrophages in the liver. Proc. Natl Acad. Sci. USA 108, 308–313 (2011)
Sun, L., Sun, C., Liang, Z., Li, H., Chen, L., Luo, H. et al. FSP1+fibroblast subpopulation is essential for the maintenance and regeneration of medullary thymic epithelial cells. Sci. Rep. 5, 1–16 (2015).
Liu, F., Lagares, D., Choi, K. M., Stopfer, L., Marinković, A., Vrbanac, V. et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L344–L357 (2014).
Balestrini, J. L., Chaudhry, S., Sarrazy, V., Koehler, A. & Hinz, B. The mechanical memory of lung myofibroblasts. Integr. Biol. 4, 410–421 (2012).
Park, J. E. & Barbul, A. Understanding the role of immune regulation in wound healing. Am J Surg. 187, S11–S16 (2004).
Otranto, M., Sarrazy, V., Bonté, F., Hinz, B., Gabbiani, G. & Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 6, 203–219 (2012).
Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 22, 407–412 (2013).
Kramer, N., Walzl, A., Unger, C., Rosner, M., Krupitza, G., Hengstschläger, M. et al. In vitro cell migration and invasion assays. Mutat. Res. 752, 10–24 (2012).
Lebleu, V. S., Taduri, G., Teng, Y., Vesselina, G., Woda, C., Sugimoto, H. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 (2014).
Ishii, G., Ochiai, A. & Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 99, 186–196 (2016).
Yazdani, S., Bansal, R. & Prakash, J. Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv. Drug Deliv. Rev. 121, 101–116 (2017).
Colotta, F., Allavena, P., Sica, A., Garlanda, C. & Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 30, 1073–1081 (2009).
Takebe, N., Ivy, P., Timmer, W., Khan, N., Schulz, T. & Harris, P. J. Review of cancer – associated fibroblasts and therapies that interfere with their activity. Tumor Microenviron. Ther. 1, 19–36 (2013).
Li X., Zhu L., Wang B., Yuan M., Zhu R. Drugs and targets in fibrosis. Front. Pharmacol. 8, 855 (2017)
Aboseif, S., El-Sakka, A., Young, P. & Cunha, G. Mesenchymal reprogramming of adult human epithelial differentiation. Differentiation. 65, 113–118 (1999).
Avgustinova A., Iravani M., Robertson D., Fearns A., Gao Q., Klingbeil P., et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 7, 10305 (2016)
Erez, N., Truitt, M., Olson, P. & Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell. 17, 135–147 (2010).
Pang, W., Su, J., Wang, Y., Feng, H., Dai, X., Yuan, Y. et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 106, 1362–1369 (2015).
Räsänen, K. & Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 316, 2713–2722 (2010).
Maia, J., Caja, S., Carolina, M., Moraes, S., Couto, N. & Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front Cell Dev Biol 6, 1–19 (2018).
Ning, X., Zhang, H., Wang, C. & Song, X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer- associated fibroblasts. Med. Sci. Mon. 24, 2350–2359 (2018).
Komohara, Y. & Takeya, M. CAFs and TAMs: maestros of the tumour microenvironment. J. Pathol. 241, 313–315 (2017).
DeNardo, D. G. & Coussens, L. M. Inflammation and breast cancer. Balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 9, 1–10 (2007).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature. 454, 436–444 (2008).
Vanharanata, S. & Massagué, J. Field cancerization: something new under the sun. Cell 149, 1179–1181 (2012).
Sharma, M., Beck, A. H., Webster, J. A., Espnosa, I., Montgomery, K., Varma, S. et al. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res. 123, 397–404 (2010).
Brown, L. F., Guidi, A. J., Schnitt, S. J., Van De Water, L., Iruela-Arispe, M. L., Tognazzi, K. et al. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin. Cancer Res. 5, 1041–1056 (1999).
Barth, P. J., Ebrahimsade, S., Ramaswamy, A. & Moll, R. CD34+ fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows Arch. 440, 298–303 (2002).
Kelley, L. C., Lohmer, L. L., Hagedorn, E. J. & Sherwood, D. R. Traversing the basement membrane in vivo: a diversity of strategies. J. Cell Biol. 204, 291–302 (2014).
Gaggioli, C., Hooper, S., Hidalgo-Carcedo, C., Grosse, R., Marshall, J. F., Harrington, K. et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 9, 1392–1400 (2007).
Glentis, A., Oertle, P., Mariani, P., Chikina, A., El Marjou, F., Attieh, Y. et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 9, 1–13 (2018).
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C. & Brown, R. A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 (2002).
Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 (2003).
Kojima, Y., Acar, A., Ng Eaton, E., Mellody, K. T., Scheel, C., Ben-Porath, I. et al. Autocrine TGF- and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor promoting mammary stromal myofibroblasts. Proc. Natl Acad. Sci. USA 107, 20009–20014 (2010).
Albrengues, J., Bertero, T., Grasset, E., Bonan, S., Maiel, M., Bourget, I. et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 6, 1–15 (2015).
Bhowmick, Na, Neilson, E. G. & Moses, H. L. Stromal fibroblasts in cancer initiation and progression. Nature. 432, 332–337 (2004).
Marks, D. L., Olson, R. Lo & Fernandez-Zapico, M. E. Epigenetic control of the tumor microenvironment. Epigenomics. 8, 1671–1687 (2016).
Corver, W. E., Haar, N. T. Ter, Fleuren, G. J. & Oosting, J. Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations. Cell Oncol. 34, 553–563 (2011).
Kang, N., Shah, V. H. & Urrutia, R. Membrane-to-nucleus signals and epigenetic mechanisms for myofibroblastic activation and desmoplastic stroma: potential therapeutic targets for liver metastasis? Mol. Cancer Res. 13, 604–612 (2015).
Zeisberg, E. M. & Zeisberg, M. The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J. Pathol. 229, 264–273 (2013).
Vizoso, M., Puig, M., Carmona, F. J., Maqueda, M., Velásquez, A., Gómez, A. et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis. 36, 1453–1463 (2015).
De Wever, O., Van Bockstal, M., Mareel, M., Hendrix, A. & Bracke, M. Carcinoma-associated fibroblasts provide operational flexibility in metastasis. Semin. Cancer Biol. 25, 33–46 (2014).
Kang, S., Shim, H. S., Lee, J. S., Kim, D. S., Kim, H. Y., Hong, S. H. et al. Molecular proteomics imaging of tumor interfaces by mass spectrometry. J. Proteome Res. 9, 1157–1164 (2010).
De Wever, O., Pauwels, P., De Craene, B., Sabbah, M., Emami, S., Redeuilh, G. et al. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 130, 481–494 (2008).
Lenos, K. J., Miedema, D. M., Lodestijn, S. C., Nijman, L. E., van denvBosch, T., Ros, X. R. et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 20, 1193–1202 (2018).
Gao, M.-Q., Kim, B. G., Kang, S., Choi, Y. P., Park, H., Kang, K. S. et al. Stromal fibroblasts from the interface zone of human breast carcinomas induce an epithelial-mesenchymal transition-like state in breast cancer cells in vitro. J. Cell Sci. 123, 3507–3514 (2010).
Levental, K. R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 139, 891–906 (2009).
Decitre, M., Gleyzal, C., Raccurt, M., Peyrol, S., Aubbert-Foucher, E., Csiszar, K. et al. Lysyl oxidase-like protein localizes to sites of de novo fibrinogenesis in fibrosis and in the early stromal reaction of ductal breast carcinomas. Lab Investig. 78, 143–151 (1998).
Öhlund, D., Elyada, E. & Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 211, 1503–1523 (2014).
Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 4, 62 (2014).
Shintani, Y., Fujiwara, A., Kimura, T., Kawamura, T., Funaki, S., Minami, M. et al. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J. Thorac. Oncol. 11, 1482–1492 (2016).
Che Y., Wang J., Li Y., Lu Z., Huang J., Sun S., et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fi broblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 9, 759 (2018)
Hessmann, E., Patzak, M. S., Klein, L., Chen, N., Kari, V., Ramu, I. et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 67, 497–507 (2018).
Adiseshaiah P. P., Crist R. M., Hook S. S., McNeil S. E. Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat. Rev. Clin. Oncol. 13, 750–765 (2016)
Biffi, G., Oni, T. E., Spielman, B., Hao, Y., Elyada, E., Park, Y. et al. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Elyada, E., Bolisetty, M., Laise, P., Flynn, W. F., Courtois, E. T., Burkhart, R. A. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Busch, S., Andersson, D., Bom, E., Walsh, C., Ståhlberg, A. & Landberg, G. Cellular organization and molecular differentiation model of breast cancer-associated fibroblasts. Mol. Cancer. 16, 1–12 (2017).
Bartoschek M., Oskolkov N., Bocci M., Lövrot J., Larsson C., Sommarin M., et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150 (2018)
Mellone, M., Hanley, C. J., Thirdborough, S., Mellows, T., Garcia, E., Woo, J. et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 9, 114–132 (2017).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Alspach, E., Fu, Y. & Stewart, S. A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 18, 549–558 (2013).
Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J. & Van Deursen, J. M. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153 (2014).
Wang, T., Notta, F., Navab, R., Joseph, J., Ibrahimov, E., Xu, J. et al. Senescent carcinoma-associated fibroblasts upregulate IL8 to enhance prometastatic phenotypes. Mol. Cancer Res. 15, 3–14 (2017).
Hassona, Y., Cirillo, N., Heesom, K., Parkinson, E. K. & Prime, S. S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 111, 1230–1237 (2014).
Schosserer M., Grillari J., Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front. Oncol. 7, 278 (2017)
Begley, L., Monteleon, C., Shah, R. B., MacDonald, J. W. & Macoska, J. A. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell. 4, 291–298 (2005).
Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 (2007).
Kisseleva, T., Cong, M., Paik, Y., Scholten, D., Jiang, C., Benner, C. et al. Myo fi broblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl Acad. Sci. USA 109, 9448–9453 (2012).
Madsen, C. D., Pedersen, J. T., Venning, F. A., Singh, L. B., Moeendarbary, E., Charras, G. et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 16, 1394–1408 (2015).
Mertens, J. C., Fingas, C. D., Christensen, J. D., Smoot, R. L., Bronk, S. F., Werneburg, N. W. et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 73, 897–907 (2013).
Froeling, F. E. M., Feig, C., Chelala, C., Dobson, R., Mein, C. E., Tuveson, D. A. et al. Retinoic Acid–induced pancreatic stellate cell quiescence reduces paracrine Wnt–b-catenin signaling to slow tumor progression. Gastroenterology. 141, 1486–1497 (2011).
Sherman, M. H., Yu, R. T., Engle, D. D., Ding, N., Atkins, A. R., Tiriac, H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 159, 80–93 (2014).
Boesch M., Baty F., Rumpold H., Sopper S., Wolf D. & Brutsche M. H. Fibroblasts in cancer: defining target structures for therapeutic intervention. BBA Rev. Cancer. 1872, 111–121 (2019)
Kwa, Q. M., Herum, K. M. & Brakenbusch, C. Cancer-associated fibroblasts: how do they contribute to metastasis? Clin. Exp. Metastasis 36, 71–86 (2019).
Masamune, A., Kikuta, K., Satoh, M., Satoh, K. & Shimosegawa, T. Rho kinase inhibitors block activation of pancreatic stellate cells. Br. J. Pharmacol. 140, 1292–1302 (2003).
Sanchez-Lopez, E., Flashner-Abramson, E., Shalapour, S., Zhong, Z., Taniguchi, K., Levitzki, A. et al. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene. 35, 2634–2644 (2016).
Ohshio, Y., Hanaoka, J., Kotani, K. & Teramoto, K. Tranilast inhibits the function of cancer-associated fibroblasts responsible for the induction of immune suppressor cell types. Scand J. Immunol. 80, 408–416 (2014).
Mediavilla-Varela M., Boateng K., Noyes D., Antonia S. J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer. 16, (2016). https://doi.org/10.1186/s12885-016-2162-z.
Kozono, S., Ohuchida, K., Eguchi, D., Ikenaga, N., Fujiwara, K., Cui, L. et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 73, 2345–2356 (2013).
Xu, S., Yang, Z., Jin, P., Yang, X., Li, X., Wei, X. et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol. Cancer Ther. 17, 1291–1302 (2018).
Jain, R. K., Lahdenranta, J. & Fukumura, D. Targeting PDGF signaling in carcinoma-associated fibroblasts controls cervical cancer in mouse model. PLoS Med. 5, e24 (2008).
Dauer, P., Zhao, X., Gupta, V. K., Sharma, N., Kesh, K., Gnamlin, P. et al. Inactivation of cancer-associated-fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res. 78, 1321–1333 (2018).
Fang, J., Xiao, L., Joo, K. Il, Liu, Y., Zhang, C., Liu, S. et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int. J. Cancer 138, 1013–1023 (2016).
Cazet, A. S., Hui, M. N., Elsworth, B. L., Wu, S. Z., Roden, D., Chan, C. L. et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 9, 2897 (2018).
Madar, S., Goldstein, I. & Rotter, V. “Cancer associated fibroblasts” - more than meets the eye. Trends Mol. Med. 19, 447–453 (2013).
Yu, Y., Xiao, C.-H., Tan, L.-D., Wang, Q.-S., Li, X.-Q. & Feng, Y.-M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 110(October 2013), 724–732 (2014).
Labernadie, A., Kato, T., Brugués, A., Serra-Picamal, X., Derzsi, S., Albertazzi, L. et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 19, 224–237 (2017).
Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A. S., Ponz-Sarvise M., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596 (2017)
Camci-Unal, G., Newsome, D., Eustace, B. K. & Whitesides, G. M. Fibroblasts enhance migration of human lung cancer cells in a paper-based coculture system. Adv. Healthc. Mater. 5, 641–647 (2016).
Sung, K. E., Yang, N., Pahlke, C., Keely, P. J., Eliceiri, K. W., Friedl, A. et al. Transition to invasion in breast cancer; a microfluidic in vitro model enables examination of spatial and temporal effects. Integr. Biol. 4, 439–450 (2011).
Newman, A. C., Nakatsu, M. N., Chou, W., Gershon, P. D. & Hughes, C. C. W. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol. Biol. Cell 22, 3791–3800 (2011).
Calvo, F., Ege, N., Grande-Garcia, A., Hooper, S., Jenkins, R. P., Chaudhry, S. I. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Hinz, B., Celetta, G., Tomasek, J. J., Gabbiani, G. & Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 12, 2730–2741 (2001).
Narra, K., Mullins, S. R., Lee, H. O., Strzemkowski-Brun, B., Magalong, K., Christiansen, V. J. et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 6, 1691–1699 (2007).
Duluc, C., Moatassim-Billah, S., Chalabi-Dchar, M., Perraud, A., Samain, R., Breibach, F. et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 7, 735–753 (2015).
Diop-Frimpong, B., Chauhan, V. P., Krane, S., Boucher, Y. & Jain, R. K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl Acad. Sci. USA 108, 2909–2914 (2011).
Izumi, D., Ishimoto, T., Miyake, K., Sugihara, H., Eto, K., Sawayama, H. et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int. J. Cancer 138, 1207–1219 (2016).
Ren, Y., Zhou, X., Liu, X., Jia, H. H., Zhao, X. H., Wang, Q. X. et al. Reprogramming carcinoma associated fibroblasts by AC1MMYR2 impedes tumor metastasis and improves chemotherapy efficacy. Cancer Lett. 374, 96–106 (2016).
Haubeiss S., Schmid J. O., Mürdter T. E., Sonnenberg M., Friedel G., van der Kuip H., et al. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary Lung Carcinomas to a Phenotype comparable to that of normal fibroblasts. Mol. Cancer. 9, 168 (2010)
Chronopoulos, A., Robinson, B., Sarper, M., Cortes, E., Auernheimer, V., Lachowski, D. et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 7, 12630 (2016).
Author information
Authors and Affiliations
Contributions
E.D.A. designed manuscript content and structure and generated all content, except Table 1 (content by J.L.C.), Table 2 (content by N.C.W) and the section on ‘CAF targeting as therapeutic strategy’ (content by N.C.W.); A.P.M. designed and edited manuscript content/structure.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Data availability
Not applicable.
Competing interests
The authors declare no competing interests.
Funding information
This work was funded by a Natural Science and Engineering Research Council (NSERC) and Canadian Institute of Health (CIHR) Collaborative Health Research Program (CHRP) grant (FRN 146469) to APM, an Ontario Trillium Scholarship and a NSERC CREATE M3 fellowship to ED, a NSERC CREATE M3 fellowship to J.L.C. and a NSERC CREATE M3 fellowship to N.C.W.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Arcangelo, E., Wu, N.C., Cadavid, J.L. et al. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br J Cancer 122, 931–942 (2020). https://doi.org/10.1038/s41416-019-0705-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0705-1
- Springer Nature Limited
This article is cited by
-
Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment
Molecular Cancer (2023)
-
Crosstalk and plasticity driving between cancer-associated fibroblasts and tumor microenvironment: significance of breast cancer metastasis
Journal of Translational Medicine (2023)
-
RETRACTED ARTICLE: Systematic pan-cancer analysis identifies transmembrane protein 158 as a potential therapeutic, prognostic and immunological biomarker
Functional & Integrative Genomics (2023)
-
Helicobacter pylori–activated fibroblasts as a silent partner in gastric cancer development
Cancer and Metastasis Reviews (2023)
-
Comprehensive bioinformatic analysis reveals a cancer-associated fibroblast gene signature as a poor prognostic factor and potential therapeutic target in gastric cancer
BMC Cancer (2022)