
Abstract
Background
About 25–37% of resectable pancreatic ductal adenocarcinoma (PDAC) had a great chance of early recurrence after radical resection, which is mainly due to preoperative micrometastasis. We herein demonstrated the profiles of ctDNA in resectable PDAC and use of ctDNA to identify patients with potential micrometastasis.
Methods
A total of 113 and 44 resectable PDACs were enrolled in discovery and validation cohorts, separately. A panel containing 50 genes was used to screen ctDNA by an NGS-based assessment with high specificity.
Results
In the discovery cohort, the overall detection rate was 38.05% (43/113). Among positive ctDNA, KRAS mutation had the highest detection rate (23.01%, 26/113), while the others were <5%. Survival analysis showed that plasma KRAS mutations, especially KRAS G12D mutation, had significant association with OS and RFS of resectable PDAC. Plasma KRAS G12D mutation showed a strong correlation with early distant metastasis. In the validation cohort, survival analysis showed similar association between plasma KRAS G12D mutation and poor outcomes.
Conclusions
This study demonstrated that plasma KRAS mutations, especially KRAS G12D mutation, served as a useful predictive biomarker for prognosis of resectable PDAC. More importantly, due to high correlation with micrometastasis, preoperative detection of plasma KRAS G12D mutation helps in optimising surgical selection of resectable PDAC.
Similar content being viewed by others


Background
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive malignancies with a 5-year survival rate of <8%, and this is mainly due to late diagnosis and rapid progression.1 The majority, i.e., up to 80% of the patients have lost the chance of surgery at the time of diagnosis.2 Even for patients with resectable PDAC, a significant portion of them (25–37%) had great chances of early recurrence after undergoing radical resection.3,4 The presence of micrometastasis in patients that cannot be detected by current preoperative imaging techniques is the main reason for this quick relapse and as to why such patients cannot benefit from surgery.5,6 So, new-generation biomarkers are urgently needed to identify patients with micrometastasis, avoiding unnecessary surgical interventions.
Liquid biopsy biomarkers are gaining interest in cancer research due to their advantages of convenient and non-invasive nature.7 Exosomes play an important role in PDAC progression and can be used for early detection, especially Glypican-1-positive exosomes can identify patients with late-stage PDAC and distinguish from patients with precancerous pancreatic lesions.8 The value of circulating tumour cells (CTCs) in predicting prognosis has been broadly studied. The US Food and Drug Administration (FDA) has approved the use of CTC as a biomarker for prognostic evaluation in three types of cancers.9,10,11 Cell-free circulating tumour DNA (ctDNA) can be effectively distinguished from normal cell-free circulating DNA (cfDNA) by specific cancer-related mutations.12,13 Several studies have proved a high detection rate and potential prognostic value of ctDNA in PDAC.14,15,16,17 However, the data regarding the complexity of exosome isolation invalidated it in other centres, and heterogeneity as well as rarity of CTCs and ctDNA restrained the application mainly in advanced patients.18 Therefore, more stable methods and more validations are necessary before clinical application, especially for patients with early-stage PDAC who are commonly considered as resectable.
In recent years, different procedures for plasma ctDNA processing, library construction and analytical methods have led to incomparable results and confusion in clinical application.19 For NGS techniques, random errors usually happened during the exponential amplification process, leading to high false-positive rate, and this has been one of the most important defects.20 However, some high-fidelity linear amplification sequencing methods and optimised analytical methods may help to improve.21 Hence, in this study, a highly specific ctDNA detection method was used to explore the clinical value of hotspot mutations in resectable PDAC patients and hoped that a new ctDNA biomarker can be used to optimise the surgical selection of PDAC patients and eventually improve the survival time of PDAC.
Methods
Patients
Patients with resectable PDAC were collected from Changhai prospective database (Changhai Hospital, Shanghai, China). All patients underwent curative surgery for tumour resection and were histologically confirmed with PDAC. Patients who died of surgical complications within 1 month after surgery have been excluded. All patients provided written informed consent to use their clinical data. The study was conducted in accordance with the national guidelines, and acquired the approval of the Ethics Committee at Changhai Hospital.
Clinical samples
Approximately 0.5–1 cm3 tumour tissue was removed from the centre of the resected lesions, and then was rapidly maintained in liquid nitrogen until DNA extraction. In total, 10 mL of preoperative venous blood was collected from patients in ethylenediaminetetraacetic acid tubes (Vacutainer blood collection tubes, Becton, Dickinson and Company, NJ, USA). Within 2 h of collection, the blood was centrifuged at 1600g for 10 min at 4 °C to obtain plasma and a buffy coat layer (containing WBCs). The plasma was then further centrifuged at 16,000g for 10 min at 4 °C to pellet any remaining cells. Both plasma and the buffy coat layer were collected and stored at −80 °C. Patients were neither provided with the results of ctDNA sequencing nor any treatment decisions made based on ctDNA results.
Tumour tissue, white blood cell genomic DNA and ctDNA extraction
Before tumour tissue DNA extraction, at least one frozen resection of each sample was assessed by two specialist pathologists to confirm the tumour cellularity ≥20% (Supplementary Fig. 1). Tumour tissue genomic DNA was extracted from the freshly frozen mass using QIAamp DNA mini kit (Qiagen, Venlo, The Netherlands). WBC genomic DNA was extracted by using the QIAamp DNA blood maxi kit (Qiagen, Venlo, The Netherlands). ctDNA was extracted from 3 to 5 mL of plasma using the QIAamp circulating nucleic acid kit (Qiagen, Venlo, The Netherlands). DNA was extracted according to the manufacturer’s instructions, quantified by a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA), and maintained at −80 °C until subsequent use.
Next-generation sequencing (NGS) analysis in tumour tissue and white blood cell
The genomic NGS library of tumour tissue and WBC was constructed by using KAPA sequencing library construction kit (Kapa Biosystems, Boston, MA, USA). A panel consisting of all exons from 50 genes that are associated with pancreatic cancer (Supplementary Table 122) was used for capturing. The captured products were purified and sequenced on an Illumina Hi-Seq 2500 (Illumina, San Diego, CA, USA). The average coverage depth for all probes of NGS library was at least 500×. Sequencing results were aligned according to the human reference sequence hg19/GRCh37, and the background noise introduced by random NGS error was removed. The true variants were then identified, and the allele frequency was calculated by comparing the number of unique reads containing a variant to the total number of sequencing reads that mapped according to the position of the variant. Germline variants were identified from WBC genomic DNA sequencing, and were removed from tumour genomic DNA total variants to determine somatic mutations.
Firefly next-generation sequencing analysis in ctDNA
NGS-based assessment of ctDNA was performed using the Firefly platform (AccuraGen, Inc., Shanghai, China). ctDNA was circularised using CircLigase II single-strand DNA ligase (EpiCentre, Madison, WI, USA) with 10–30 ng of DNA per 20 μL of reaction. After ligation, the samples were treated with exonuclease (NEB, Ipswich, MA, USA) to remove the uncircularised DNA. The circularised DNA was then amplified in a rolling circle amplification reaction using Phi29 DNA polymerase (NEB, Ipswich, MA, USA), and the exonuclease-resistant random primers by using the manufacturer's instructions with slight modifications. The amplification product was then purified by AMPure beads (Beckman Coulter, Brea, CA, USA) and sonicated to short fragments that are suitable for NGS library 7/20 construction by using a Covaris sonicator. NGS sequencing libraries were generated from 100 ng of amplified ctDNA using the KAPA sequencing library construction kit (Kapa Biosystems, Boston, MA, USA) according to the manufacturer’s instructions. After library construction, a panel consisting of all exons from 50 genes associated with pancreatic cancer was used for capturing. The captured products were then purified, sequenced on an Illumina Hi-Seq 2500 (Illumina, San Diego, CA, USA) and the unique sequencing reads were determined by using an AccuraGen proprietary algorithm. The average coverage depth for all probes of Firefly NGS library was approximately 7000×. The sequencing results were aligned to the human reference sequence hg19/GRCh37, and the background noise introduced by random NGS error was removed by AccuraGen proprietary algorithms. The true variants were then identified, and the allele frequency was calculated by comparing the number of unique reads containing a variant to the total number of sequencing reads that mapped according to the position of the variant. Germline variants were identified from WBC genomic DNA sequencing and were removed from ctDNA total variants to determine somatic mutations.
Droplet digital polymerase chain reaction (ddPCR)
The QX200 system (Bio-Rad, Hercules, CA, USA) was utilised for ddPCR experiments. Multiplex KRAS products for ddPCR were utilised, which included six KRAS mutations (KRAS G12D/G12V/G12R/G12C/G12A/G12S). For conducting the experiments, a reaction volume of 20 µl consisting of cfDNA ranging 4–80 ng (8 µL of cfDNA input in volume) was used. Emulsified PCR reactions were run on a 96-well plate on a MJ thermal cycler (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions. Plates were read on a Bio-Rad QX200 droplet reader using QuantaSoft version 1.4.0 software (Bio-Rad, Hercules, CA, USA) to assess the number of droplets that was positive for wild-type KRAS and/or KRAS mutations. A sample without plasma ctDNA was used as a negative control. Three positive events were regularly used as a threshold for false positivity.
Clinicopathological characteristics
The preoperative clinical variables included age, sex, smoking history, drinking history, status of diabetes mellitus, the first clinical symptom (abdominal pain and jaundice), carcinoembryonic antigen (CEA), carbohydrate antigen 19-9 (CA19-9), surgical procedure and radiologic tumour size. Postoperative variables included the eighth edition of American Joint Committee on Cancer (AJCC) TNM classification, differentiation degree, perineural invasion, pathological margin status, nodal involvement and treatment with adjuvant chemotherapy. Overall survival (OS) was defined as the interval from the date of surgery till the date of patient death or the last follow-up visit after 1 month post surgery. Recurrence-free survival (RFS) was defined as the time interval from the date of surgery till the date of local or regional recurrence, distant metastases, death or the last follow-up visit after 1 month post surgery. The status of recurrence was recorded as no, local, distant and both.
Statistical analysis
Discrete variables were presented as number and/or percentage, whereas the continuous variables were presented as median and/or mean. Fisher’s exact test was used to report variable association of plasma KRAS mutations. Spearman’s rank correlation coefficient was used to determine the association between ddPCR and Firefly NGS in plasma KRAS G12D allele frequency (AF). Univariate and multivariate Cox regression analysis was used to assess the impact of prognostic factors on OS and RFS. P-values less than 0.05 were considered to be statistically significant. Statistical analysis was performed by using SPSS 22.0 for Windows (SPSS, Chicago, IL, USA). Curves for OS and RFS were generated using the R statistical package v.3.5.1 (http://www.r-project.org/), and figures were created using GraphPad Prism (version 7.0; GraphPad, San Diego, CA).
Results
Discovery cohort
Clinicopathological characteristics of patients
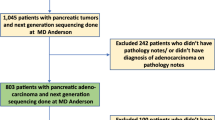
From August 2014 to August 2015, a total of 130 consecutive patients diagnosed with resectable PDAC who underwent curative pancreatectomy were enrolled from the Changhai prospective database (Fig. 1). Among these, 13 were found with metastasis during operation, 2 patients died due to surgical complications and 2 patients failed to follow up within 1 month after surgery, and so were excluded from the study. The remaining 113 patients with resectable PDAC were enrolled in this study. Clinicopathological characteristics of 113 patients are summarised in Table 1. The patient cohort enrolled had a median age of 61.5 years, and consisted of a little disproportionately male (69, 61.1%). Patients with stages IA, IB, IA, IIB and III (AJCC, 8th edition) accounted for 15 (13.3%), 34 (30.0%), 14 (12.4%), 43 (38.1%) and 7 (6.2%), respectively. All patients received adjuvant chemotherapy, except six patients due to physical conditions or refusal. Median follow-up of the discovery cohort was 23.6 months.

Workflow diagram of the study.
Detection of pancreatic research panel in ctDNA by Firefly NGS
The Firefly NGS technique allowed a detection of 0.02% mutated DNA.21 But according to the previous studies,18 we chose 0.1% AF as the threshold to avoid situations, like ageing, benign tumours and preneoplastic lesions, better reflecting the systematic tumour burden of the patients.
Thirty-three genes of the pancreatic research panel were found in the plasma of 43 patients, with an overall detection rate of 38.05% (43/113). Interestingly, except for KRAS, the other genes detected commonly had a pretty low positive rate of <5%. Plasma KRAS mutations were detected in 26 patients, while 87 patients did not have KRAS mutations in their plasma (Table 1). Among the 26 patients with KRAS mutations in ctDNA, KRAS G12D mutation was detected in 13 patients, KRAS G12V in 7 and KRAS G12R in 2. The remaining four patients were detected with KRAS G12C, KRAS G13D, KRAS Q61H and KRAS Q61R, separately.
Strong correlation between Firefly NGS and ddPCR in detection of ctDNA
Twenty-six KRAS-positive samples were screened again using multiplex ddPCR (KRAS G12D/G12V/G12R/G12C/G12A/G12S). We compared the AF of KRAS mutation between ddPCR and Firefly NGS. The results showed that the detection of plasma KRAS mutations by ddPCR and Firefly NGS technique was largely equivalent (Supplementary Table 2). For ddPCR-negative subgroup, no false positives were identified by Firefly NGS, suggesting 100% specificity for NGS-based KRAS detection. For ddPCR-positive group, the AF between ddPCR and Firefly NGS was highly concordant (R2 = 0.98, Supplementary Fig. 2). Moreover, Firefly NGS has successfully profiled three special mutations that were not included in the design of ddPCR (filled in blue, Supplementary Table 2), assisting the previously reported disadvantage of ddPCR for the requirement of detailed prior information on the mutational spectrum.23 These results demonstrated that NGS-based platform had similar detection accuracy with that of ddPCR.
Validation of plasma KRAS mutation by tissue NGS
Tumour tissues of 26 patients above underwent genomic DNA sequencing using pancreatic research panel. Surprisingly, the results showed that there was one patient who was detected with KRAS G12D mutation in the plasma turned out to be negative in the tissue (filled in green, Supplementary Table 3). This might be due to the spatial heterogeneity within a tumour, which is an advantage of ctDNA24,25 or the direct sequencing of surgical sample without microdissection. However, the most important point is that, except for this one, the remaining patients with detected plasma KRAS mutation by Firefly NGS were proven positive in tissue, and consistent in the mutation subtype (Supplementary Table 3). These results demonstrated high specificity of Firefly NGS platform.
Patient characteristics associated with plasma KRAS mutation status
A total of 113 patients were divided into two groups according to the presence of KRAS mutations in ctDNA. Clinicopathological characteristics of the two groups are shown in Supplementary Table 4. Most of the patients with KRAS mutation in ctDNA had a history of smoking and drinking, but showed no significant differences between these two groups. Besides, among the other clinicopathological variables included, no significant difference was observed.
Significant correlation between plasma KRAS mutations and patient outcomes
Next, the association between KRAS mutations in ctDNA and outcomes of patients was explored. The results showed significant differences in OS (17.1 vs 26.3 months, P = 0.002) and RFS (9.2 vs 18.9 months, P = 0.001) among patients with detectable KRAS mutations in ctDNA (n = 26) when compared with those without KRAS mutations (n = 87) (Fig. 2a, b). At 12 months post surgery, the OS was 73.1% (19/26) and 82.8% (72/87) in plasma KRAS mutation positive and negative patients, respectively. The corresponding RFS was 42.3% (11/26) and 65.5% (57/87), respectively.

a OS for resectable PDAC patients with (n = 26, red) and without (n = 87, blue) KRAS mutants in ctDNA. In total, 73.1% of patients with KRAS mutations survived at 12 months post surgery vs 82.8% for the patients without KRAS mutations. b RFS for resectable PDAC patients with (n = 26, red) and without (n = 87, blue) KRAS mutations in ctDNA. In all, 42.3% of patients with KRAS mutations were recurrence-free at 12 months post surgery vs 65.5% for the patients without KRAS mutations. c OS for resectable PDAC patients with (n = 13, red) and without (n = 100, blue) KRAS G12D mutant in ctDNA. d RFS for resectable PDAC patients with (n = 13, red) and without (n = 100, blue) KRAS G12D mutation in ctDNA. e Comparison for recurrence types of resectable PDAC patients with (n = 13) and without (n = 100) KRAS G12D mutation in ctDNA.
Among 26 patients with KRAS mutations in ctDNA, patients with plasma KRAS G12D mutation (n = 13) showed a significantly reduced OS (12.1 vs 24.9 months, P < 0.001) and RFS (6.3 vs 17.4 months, P < 0.001) relative to those without plasma KRAS G12D mutations (n = 100) (Fig. 2c, d). No such association was found among patients with other assayed KRAS mutations (n = 13) (Supplementary Fig. 3A, B).
Univariate and multivariate analysis of clinicopathological characteristics for patient outcomes
After that, whether the following patient characteristics were risk factors for OS and RFS were investigated: age, sex, smoking history, drinking history, the first clinical symptom (abdominal pain and jaundice), diabetes mellitus, surgery procedure, CEA, CA19-9, radiologic tumour size, pathological stage (AJCC, 8th edition), differentiation degree, perineural invasion, pathological margin status, nodal involvement and the use of adjuvant chemotherapy. Univariate analysis demonstrated radiologic tumour size, pathological stage (AJCC, 8th edition), nodal involvement, KRAS mutations and KRAS G12D mutation in ctDNA as considerable risk factors for RFS and OS (Table 2). Further multivariate analysis, in which nodal involvement was excluded due to representation in a more general variable (pathological stage), showed that radiologic tumour size, pathological stage (AJCC, 8th edition) and KRAS G12D mutation in ctDNA were significantly independent prognostic factors for RFS and OS (Table 3).
Plasma KRAS G12D mutation closely associated with preoperative potential micrometastasis
Survival analysis suggested that patients with KRAS G12D mutation in ctDNA showed significant trend of early recurrence, the median RFS of whom was only 6 months (Fig. 2d). Then a correlation analysis between plasma KRAS G12D mutation and recurrence types was performed, and found that patients with KRAS G12D mutation in ctDNA were more inclined to develop distant metastasis, while those without detectable plasma KRAS G12D mutation had a higher proportion of local recurrence (Table 4, Fig. 2e). The quick appearance of distant metastasis after radical surgery strongly indicated the presence of preoperative potential micrometastasis in patients with plasma KRAS G12D mutation. These results highly supported that plasma KRAS G12D mutation well reflected the systematic tumour burden in resectable PDAC, and ≥0.1% AF of KRAS G12D mutation in ctDNA was able to serve as a promising biomarker of preoperative potential micrometastasis.
Validation cohort
Clinicopathological characteristics of patients
According to the same standard with the discovery cohort, the clinical data of 47 consecutive patients from the Changhai prospective database during August 2016 and December 2016 were collected (Fig. 1). After excluding two patients with intraoperative metastasis and one patient died due to surgical complications within 1 month after surgery, forty-four patients formed the validation cohort. As shown in Table 1, the validation cohort had similar distribution to that of the discovery cohort in most of the clinicopathological variables, but there were no stage III patients in the validation cohort. Median follow-up of the validation cohort was 24.4 months.
Verification of the association between plasma KRAS G12D and patient outcomes
The association between plasma KRAS G12D and patient outcomes from the discovery cohort was tested in the validation cohort. We performed the same Firefly NGS for ctDNA detection to patients of the validation cohort. Among the 44 patients enrolled, 15 genes in the pancreatic research panel have been detected with an overall detection rate of 34.09% (15/44). These results were similar to the discovery cohort, where only plasma KRAS mutations had a detection rate of >5% (Table 1). Of the nine patients, five patients with KRAS mutations in ctDNA were detected as KRAS G12D subtype. Survival analysis showed that patients with plasma KRAS G12D mutation had significantly poorer prognosis than those without KRAS G12D mutation in ctDNA (Fig. 3a, b), which was consistent with the results of the discovery cohort. The median DFS of patients with plasma KRAS G12D mutation was merely 6 months, which highly suggested that an upfront surgery might not be suitable for such patients even though diagnosed as resectable PDAC.

a OS for resectable PDAC patients with (n = 5, red) and without (n = 39, blue) KRAS G12D mutant in ctDNA. b RFS for resectable PDAC patients with (n = 5, red) and without (n = 39, blue) KRAS G12D mutation in ctDNA.
Discussion
As a component of liquid biopsy, ctDNA has the most potential clinical application value due to its non-invasive, real-time and highly specific genomic result.26,27 Currently, the clinical utility of ctDNA is only approved in metastatic non-small-cell lung cancer (NSCLC) by FDA as a companion diagnostic,28,29 while studies on many other cancers have also shown good prospects.30,31,32,33,34,35,36 The fact that even little variability in process and analysis will result in completely different results restrains the further clinical utility of ctDNA.37 In order to solve these problems, some projects, such as the European CANCER-ID, European Liquid Biopsy Academy (ELBA) and European Liquid Biopsy Society (ELBS) networks or the US-based BloodPAC, have been initiated.38 In this study, we tried to use a detection method of high specificity to address whether ctDNA in patients with resectable PDAC could possibly serve as a decisive factor for neoadjuvant therapy.
The most popular methods such as ddPCR and NGS-based techniques have their own advantages and disadvantages, separately. ddPCR is associated with high analytical sensitivity down to single molecules and has lower turnaround time than NGS-based approaches when testing single mutations only,39,40 but the main disadvantages included sequential testing of mutations and detection of previously unknown mutations is impossible.18 In contrast, NGS-targeted sequencing method utilises enrichment of recurrently altered loci by PCR amplification or hybrid capture, allowing de novo identification of molecular alterations, but this inevitably introduces random errors in the amplification process.20 In our research, we used the ‘firefly’ method, circularised library and linear amplification to maximise the accuracy and specificity, as the false positive in clinical practice renders the patients to lose the chance to be cured by radical resection.
Different ctDNA concentrations reflect different stages of tumour development and progression (metastatic vs non-metastatic recurrence).41 cfDNA in plasma is highly fragmented DNA that is mainly derived from the apoptotic cells, perhaps predominantly from the apoptotic leukocytes (e.g. neutrophils).42 During the early stages of cancer, the total amount of ctDNA is usually <0.01% of the total cfDNA, while >10% concentration of ctDNA is believed to be in the clinical metastatic stages, which can be easily detected by imaging examination or tumour marker testing.18 The concentration of cfDNA between 0.1% and 10% is thought to be related to micrometastasis of primary tumours, which cannot be undetectable by conventional techniques and critical for surgical outcome and long-time survival.18 Therefore, based on Firefly NGS platform, we choose 0.1% as the threshold for ctDNA positive. The combination of a reasonable cut-off value and an accurate detection method with high specificity might be helpful for the stratification of resectable PDAC patients and further provides a proper individualised treatment.
Driver gene mutations represent both primary and metastatic genetic variation during the process of tumour progression. It has been proved that identical mutations in known driver genes, especially KRAS, were present in every metastatic lesion in all PDAC patients, and the genetic similarity among the cells of metastases was higher than that expected for any two random cells taken from a normal tissue.43 Moreover, several studies have reported that the correlation of ctDNA with tumour stage, metastatic burden and increased ctDNA mutant allele frequency was observed at the time of recurrence or progression.44,45,46 So, detection of credible tumour-driving gene mutations from ctDNA during the early-stage PDAC might reflect the formation of micrometastasis. In this study, we sequenced the whole exons of 50 genes, including KRAS, P53, SMAD4, CDKN2A and so on. However, only KRAS mutations were identified in 20–25% of patients with early-stage PDAC, while the mutation detection rates of the remaining genes, including driver genes,47,48 were significantly lower than that of KRAS, <5%. There might be two reasons: first, the alteration rates of these genes in PDAC tissue are correspondingly lower than KRAS;22,49 second, mutation type of suppressor genes like SMAD4 is usually deletion, which makes it difficult to detect in ctDNA.22,49 Survival analysis of the discovery cohort demonstrated that KRAS mutation, especially KRAS G12D mutation, in ctDNA was strongly correlated with poor OS and RFS, and this has been proven in tissue also.50 Further recurrence analysis showed that patients with plasma KRAS G12D mutation were more likely to develop distant metastasis, especially liver metastasis, in 6 months after surgery, and this highly suggested the existence of micrometastasis before surgery. Importantly, we found similar poor outcome in the validation cohort. Only a 6-month RFS strongly indicated that these patients should receive neoadjuvant therapy rather than upfront surgery, despite resectable PDAC in radiology.
Patients who did not harbour KRAS mutations in ctDNA but had a KRAS mutation in tissue accounted for 74.7% (74/99) (Supplementary Table 5), which indicated a little low sensitivity of 25.3% (25/99) compared with previous studies (30.0%–49.0%).16,17 However, this is due to the impairment caused by the threshold of 0.1% AF, which we chose to avoid situations, like ageing, benign tumours and preneoplastic lesions, and better reflect the systematic tumour burden of the patients.18 In fact, the Firefly NGS technique allowed a detection of 0.02% mutated DNA,21 which suggested that negative results in this study did not mean no detectable ctDNA in preoperative plasma. By maximising the specificity of our method, we could identify the resectable PDAC patients who are most suitable for neoadjuvant therapy instead of upfront surgery. It is more practical in clinical work due to the slim chance of making resectable PDAC patients lose the opportunity of radical surgery.
However, our study still has limitations. Firstly, we performed a retrospective study, in which the PDAC patients were enrolled from a single institution. The conclusions we made from this research still need to be verified in a prospective study. Secondly, the panel we used in this study was established based on a research about the mutational landscape of pancreatic cancer. Genes like GNAS, which was reported to be closely related to the development of pancreatic cyst51 and intraductal papillary mucinous neoplasm (IPMN)-associated invasive adenocarcinoma52 and might serve as an alternative oncogene in KRAS wild-type PDAC,49 were not included and needed to be paid more attention in further study.
In summary, a study screening ctDNA by Firefly NGS in resectable PDAC was performed, and the results showed that KRAS mutations, especially KRAS G12D mutation, in preoperative ctDNA were significantly associated with the outcomes of resectable PDAC. The presence of plasma KRAS G12D mutation showed a strong correlation with early distant metastasis, indicating that these patients had a high probability of preoperative micrometastasis and should possibly receive neoadjuvant therapy first instead of undergoing an upfront surgery. The preoperative detection of plasma KRAS G12D mutation will help in optimising surgical selection of resectable PDAC patients and improving survival time.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2018. CA: A Cancer J. Clin. 68, 7–30 (2018).
Ryan, D. P., Hong, T. S. & Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 371, 1039–1049 (2014).
Fischer, R., Breidert, M., Keck, T., Makowiec, F., Lohrmann, C. & Harder, J. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J. Gastroenterol. 18, 118–121 (2012).
Matsumoto, I., Murakami, Y., Shinzeki, M., Asari, S., Goto, T., Tani, M. et al. Proposed preoperative risk factors for early recurrence in patients with resectable pancreatic ductal adenocarcinoma after surgical resection: a multi-center retrospective study. Pancreatology 15, 674–680 (2015).
Komo, T., Murakami, Y., Kondo, N., Uemura, K., Hashimoto, Y., Nakagawa, N. et al. Prognostic impact of para-aortic lymph node micrometastasis in pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 23, 2019–2027 (2016).
Yamaguchi, K., Chijiiwa, K., Torato, N., Kinoshita, M. & Tanaka, M. Ki-ras codon 12 point and P53 mutations: a molecular examination of the main tumor, liver, portal vein, peripheral arterial blood and para-aortic lymph node in pancreatic cancer. Am. J. Gastroenterol. 95, 1939–1945 (2000).
Marrugo-Ramirez, J., Mir, M. & Samitier, J. Blood-based cancer biomarkers in liquid biopsy: a promising non-invasive alternative to tissue biopsy. Int. J. Mol. Sci. 19, 2877 (2018).
Melo, S. A., Luecke, L. B., Kahlert, C., Fernandez, A. F., Gammon, S. T., Kaye, J. et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523, 177–182 (2015).
Bidard, F. C., Peeters, D. J., Fehm, T., Nole, F., Gisbert-Criado, R., Mavroudis, D. et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 15, 406–414 (2014).
Sastre, J., Maestro, M. L., Gomez-Espana, A., Rivera, F., Valladares, M., Massuti, B. et al. Circulating tumor cell count is a prognostic factor in metastatic colorectal cancer patients receiving first-line chemotherapy plus bevacizumab: a Spanish Cooperative Group for the Treatment of Digestive Tumors study. Oncologist 17, 947–955 (2012).
de Bono, J. S., Scher, H. I., Montgomery, R. B., Parker, C., Miller, M. C., Tissing, H. et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 14, 6302–6309 (2008).
Zhou, B., Xu, J. W., Cheng, Y. G., Gao, J. Y., Hu, S. Y., Wang, L. et al. Early detection of pancreatic cancer: where are we now and where are we going? Int. J. Cancer 141, 231–241 (2017).
Riva, F., Dronov, O. I., Khomenko, D. I., Huguet, F., Louvet, C., Mariani, P. et al. Clinical applications of circulating tumor DNA and circulating tumor cells in pancreatic cancer. Mol. Oncol. 10, 481–493 (2016).
Sausen, M., Phallen, J., Adleff, V., Jones, S., Leary, R. J., Barrett, M. T. et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 6, 7686 (2015).
Kinugasa, H., Nouso, K., Miyahara, K., Morimoto, Y., Dohi, C., Tsutsumi, K. et al. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 121, 2271–2280 (2015).
Cohen, J. D., Javed, A. A., Thoburn, C., Wong, F., Tie, J., Gibbs, P. et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl Acad. Sci. USA 114, 10202–10207 (2017).
Groot, V. P., Mosier, S., Javed, A. A., Teinor, J. A., Gemenetzis, G., Ding, D. et al. Circulating tumor DNA as a clinical test in resected pancreatic cancer. Clin. Cancer Res. 25, 4973–4984 (2019).
Pantel, K. & Alix-Panabieres, C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424 (2019).
Volckmar, A. L., Sultmann, H., Riediger, A., Fioretos, T., Schirmacher, P., Endris, V. et al. A field guide for cancer diagnostics using cell-free DNA: from principles to practice and clinical applications. Genes, Chromosomes Cancer 57, 123–139 (2018).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Xu, T., Kang, X., You, X., Dai, L., Tian, D., Yan, W. et al. Cross-platform comparison of four leading technologies for detecting EGFR mutations in circulating tumor DNA from non-small cell lung carcinoma patient plasma. Theranostics 7, 1437–1446 (2017).
Waddell, N., Pajic, M., Patch, A. M., Chang, D. K., Kassahn, K. S., Bailey, P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 (2015).
Olmedillas-Lopez, S., Garcia-Arranz, M. & Garcia-Olmo, D. Current and emerging applications of droplet digital PCR in oncology. Mol. Diag. Ther. 21, 493–510 (2017).
Appierto, V., Di Cosimo, S., Reduzzi, C., Pala, V., Cappelletti, V. & Daidone, M. G. How to study and overcome tumor heterogeneity with circulating biomarkers: the breast cancer case. Semin. Cancer Biol. 44, 106–116 (2017).
Zhang, J., Fujimoto, J., Zhang, J., Wedge, D. C., Song, X., Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014).
von Bubnoff, N. Liquid biopsy: approaches to dynamic genotyping in cancer. Oncol. Res. Treat. 40, 409–416 (2017).
Ignatiadis, M., Lee, M. & Jeffrey, S. S. Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin. Cancer Res. 21, 4786–4800 (2015).
John, T., Akamatsu, H., Delmonte, A., Su, W. C., Lee, J. S., Chang, G. C. et al. EGFR mutation analysis for prospective patient selection in AURA3 phase III trial of osimertinib versus platinum-pemetrexed in patients with EGFR T790M-positive advanced non-small-cell lung cancer. Lung Cancer 126, 133–138 (2018).
Khozin, S., Blumenthal, G. M., Jiang, X., He, K., Boyd, K., Murgo, A. et al. U.S. food and drug administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist 19, 774–779 (2014).
O'Kane, G. M., Liu, G., Stockley, T. L., Shabir, M., Zhang, T., Law, J. H. et al. The presence and variant allele fraction of EGFR mutations in ctDNA and development of resistance. Lung Cancer 131, 86–89 (2019).
Coombes, R. C., Page, K., Salari, R., Hastings, R. K., Armstrong, A. C., Ahmed, S. et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin. Cancer Res. 25, 4255–4263 (2019).
Hahn, A. W., Stenehjem, D., Nussenzveig, R., Carroll, E., Bailey, E., Batten, J. et al. Evolution of the genomic landscape of circulating tumor DNA (ctDNA) in metastatic prostate cancer over treatment and time. Cancer Treat. Res. Commun. 19, 100120 (2019).
Siravegna, G. & Sartore-Bianchi, A. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin. Cancer Res. 25, 3046–3053 (2019).
Condoluci, A. & Rossi, D. The future of cell-free DNA testing to guide therapeutic decisions in B-cell lymphomas. Curr. Opin. Hematol. 26, 281–287 (2019).
Creemers, A., Krausz, S., Strijker, M., van der Wel, M. J., Soer, E. C., Reinten, R. J. et al. Clinical value of ctDNA in upper-GI cancers: a systematic review and meta-analysis. Biochimica et. Biophysica Acta Rev. Cancer 1868, 394–403 (2017).
Xu, R. H., Wei, W., Krawczyk, M., Wang, W., Luo, H., Flagg, K. et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 16, 1155–1161 (2017).
Cree, I. A., Uttley, L., Buckley Woods, H., Kikuchi, H., Reiman, A., Harnan, S. et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: a systematic mapping review. BMC Cancer 17, 697 (2017).
Grossman, R. L., Abel, B., Angiuoli, S., Barrett, J. C., Bassett, D., Bramlett, K. et al. Collaborating to compete: blood profiling atlas in cancer (BloodPAC) consortium. Clin. Pharmacol. Ther. 101, 589–592 (2017).
Zhang, R., Chen, B., Tong, X., Wang, Y., Wang, C., Jin, J. et al. Diagnostic accuracy of droplet digital PCR for detection of EGFR T790M mutation in circulating tumor DNA. Cancer Manag. Res. 10, 1209–1218 (2018).
van Ginkel, J. H., Huibers, M. M. H., van Es, R. J. J., de Bree, R. & Willems, S. M. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer 17, 428 (2017).
Bettegowda, C., Sausen, M., Leary, R. J., Kinde, I., Wang, Y., Agrawal, N. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra24 (2014).
Yang, M., Forbes, M. E., Bitting, R. L., O'Neill, S. S., Chou, P. C., Topaloglu, U. et al. Incorporating blood-based liquid biopsy information into cancer staging: time for a TNMB system? Ann. Oncol. 29, 311–323 (2018).
Makohon-Moore, A. P., Zhang, M. & Reiter, J. G. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 49, 358–366 (2017).
Pietrasz, D., Pecuchet, N., Garlan, F., Didelot, A., Dubreuil, O., Doat, S. et al. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin. Cancer Res. 23, 116–123 (2017).
Kruger, S., Heinemann, V., Ross, C., Diehl, F., Nagel, D., Ormanns, S. et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. 29, 2348–2355 (2018).
Cheng, H., Liu, C., Jiang, J., Luo, G., Lu, Y., Jin, K. et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int. J. Cancer 140, 2344–2350 (2017).
Qian, Z. R., Rubinson, D. A., Nowak, J. A., Morales-Oyarvide, V., Dunne, R. F., Kozak, M. M. et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 4, e173420 (2018).
Hayashi, H., Kohno, T., Ueno, H., Hiraoka, N., Kondo, S., Saito, M. et al. Utility of assessing the number of mutated KRAS, CDKN2A, TP53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas 46, 335–340 (2017).
Raphael, B. J., Hruban, R. H., Aguirre, A. J., Moffitt, R. A., Yeh, J. J., Stewart, C. et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203, e13 (2017).
Miglio, U., Oldani, A., Mezzapelle, R., Veggiani, C., Paganotti, A., Garavoglia, M. et al. KRAS mutational analysis in ductal adenocarcinoma of the pancreas and its clinical significance. Pathol., Res. Pract. 210, 307–311 (2014).
Wu, J., Matthaei, H., Maitra, A., Dal Molin, M., Wood, L. D., Eshleman, J. R. et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 3, 92ra66 (2011).
Tan, M. C., Basturk, O., Brannon, A. R., Bhanot, U., Scott, S. N., Bouvier, N. et al. GNAS and KRAS mutations define separate progression pathways in intraductal papillary mucinous neoplasm-associated carcinoma. J. Am. Coll. Surg. 220, 845–854 (2015).
Acknowledgements
All of the authors would like to thank all of the patients and their families for their support of this study. All of the authors thank AccuraGen, Inc. (Shanghai, China) for all its technical support.
Author contributions
S.W.G. and G.J. conceived and supervised the study; S.W.G., X.H.S. and J.S. designed the experiments; X.H.S., J.S., S.Z.G. and H.W. performed the experiments; S.S., Y.Q.P., B.L., X.F.X. and Z.S. provided all the clinical information; S.W.G., X.H.S. and J.S. analysed all the data; S.W.G., X.H.S. and J.S. wrote the paper; S.W.G. and G.J. revised the paper, and all authors reviewed the results and approved the final version of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki and approved by the Shanghai Changhai Hospital Ethics Committee. All patients provided written informed consent to use their clinical data.
Consent to publish
Written informed consent for publication was obtained.
Data availability
All data in this study are available upon request.
Competing interests
The authors declare no competing interests.
Funding information
This study was supported by National Natural Science Foundation of China (81672830), National Key R&D Program (2017YFC0908400), Science and Technology Commission of Shanghai Municipality (16140903800 and 19140902300), Top Projects of Military Medical Science and Technology Training Program (17QNP017) and the Shanghai ShenKang hospital development centre (SHDC12014108).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, S., Shi, X., Shen, J. et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br J Cancer 122, 857–867 (2020). https://doi.org/10.1038/s41416-019-0704-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0704-2
- Springer Nature Limited
This article is cited by
-
Detection and characterization of pancreatic and biliary tract cancers using cell-free DNA fragmentomics
Journal of Experimental & Clinical Cancer Research (2024)
-
Liquid biopsy techniques and pancreatic cancer: diagnosis, monitoring, and evaluation
Molecular Cancer (2023)
-
Serum lipidomic profiling by UHPLC-MS/MS may be able to detect early-stage endometrial cancer
Analytical and Bioanalytical Chemistry (2023)
-
Circulating tumour DNA: a challenging innovation to develop “precision onco-surgery” in pancreatic adenocarcinoma
British Journal of Cancer (2022)
-
Prognostic Value of KRAS Mutations in the Peripheral Blood of Patients with Pancreatic Cancer: a Systematic Review and Meta-analysis
Indian Journal of Surgery (2022)