
Abstract
Revised diagnostic criteria for myeloid neoplasms (MN) issued by the International Consensus Classification (ICC) and the World Health Organization (WHO) recommended major change pertaining to TP53-mutated (TP53mut) MN. However, these assertions have not been specifically examined in therapy-related myeloid neoplasm (t-MN), a subset enriched with TP53mut. We analyzed 488 t-MN patients for TP53mut. At least one TP53mut with variant allele frequency (VAF) ≥ 2% with or without loss of TP53 locus was noted in 182 (37.3%) patients and 88.2% of TP53mut t-MN had a VAF ≥10%. TP53mut t-MN with VAF ≥ 10% had a distinct clinical and biological profile compared to both TP53mut VAF < 10% and wild-type TP53 (TP53wt) cases. Notably, TP53mut VAF ≥ 10% had a significantly shorter survival compared to TP53wt (8.3 vs. 21.6 months; P < 0.001), while the survival of TP53mut VAF < 10% was comparable to TP53wt. Within TP53mut VAF ≥ 10% cohort, the inferior outcomes persisted irrespective of the single- or multi-hit status, co-mutation pattern, or treatments received. Finally, survival of TP53mut patients was poor across all the blast categories and MDS patients with >10% blasts had inferior survival compared to <5%. In summary, TP53mut VAF ≥10% signified a clinically and molecularly homogenous cohort regardless of the allelic status.
Similar content being viewed by others


Introduction
Tumor protein p53 (TP53) located on chromosome 17p13 is frequently mutated in cancer, including myeloid neoplasms (MN). Approximately 7–11% of de novo myelodysplastic syndromes (MDS) and 10–13% of acute myeloid leukemia (AML) patients harbor TP53 mutations (TP53mut) [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. TP53mut MN are often associated with the features of genomic instability such as complex and monosomal karyotype (CK and MK, respectively) and are universally associated with adverse outcomes [1, 2, 4,5,6,7,8,9,10,11, 13, 15]. Therapy-related myeloid neoplasms (t-MN) are rare, but often fatal MN that develop following exposure to cytotoxic therapies [7, 12, 16] and are highly enriched in TP53mut, CK, and MK [17].
The recently published 5th edition of the World Health Organization classification of MN (WHO-5) [18], and the International Consensus Classification (ICC) [19] recommended major reorganization of the MN. Among the congruent changes, both WHO-5 and ICC recognize the poor prognostic impact of biallelic TP53mut defined by the presence of ≥2 mutations or 1 mutation with the loss of residual wild-type TP53 (TP53wt). Both classifications consider variant allele frequency (VAF) ≥ 50% as presumptive evidence of biallelic/multi-hit TP53mut, and biallelic TP53mut MDS to be AML equivalent for therapeutic purposes. However, there are critical differences between the two classifications about allelic-status, VAF cut-off and blast categories [18, 19].
Another major change highlights the importance of genetic driver(s) and reduces the importance of the antecedent history and/or therapy. For example, ICC [19] removed t-MN as a distinct category and replaced it with a diagnostic qualifier, whereas WHO-5 [18] grouped t-MN with secondary MN as MN-post cytotoxic therapy (AML-pCT and MDS-pCT).
The underlying assumption of these changes is that TP53mut MN are characterized by similar characteristics and outcomes. However, the studies driving these changes were highly enriched in de novo MN [2], excluded patients with <10% blasts [3] or >20% blasts [4], or only included MN with CK [14]. For example, in a cohort of predominantly of de novo MDS, single-hit TP53mut had outcomes similar to TP53wt, whereas the association with CK, high risk of AML transformation, and poor survival were limited to multi-hit TP53mut patients [2]. Meanwhile, TP53mut AML and MDS with excess blasts (MDS-EB) had equally poor survival irrespective of single or multi-hit TP53mut status [3]. Furthermore, in MDS and AML with CK, the single- or multi-hit TP53mut was the only disease-related factor predicting survival [14]. We recently demonstrated that TP53mut t-MN is associated with poor survival irrespective of single or multi-hit status [20], suggesting that the prognostic impact of allelic loss of TP53mut MN is context dependent.
Hence, we performed a comprehensive analysis of a TP53mut t-MN cohort to: (i) define the genomic landscape of TP53mut t-MN; (ii) study the interaction of TP53mut with BM blast % and structural chromosomal changes; (iii) study the impact of 17p loss in the absence of a concurrent TP53mut, and (iv) identify the optimal TP53mut VAF threshold in t-MN.
Methods
This retrospective multi-center study was conducted by Mayo Clinic, Rochester (USA) and the South Australia MDS Registry (SA-MDS, Australia) and includes all t-MN patients who had conventional G-banding chromosome analysis (CBA) and mutation testing using targeted sequencing of the most recurrently mutated genes in MN (please refer to supplementary section for details) [12, 21]. The respective databases captured patient-level information that included diagnostic characteristics, treatments including the use of allogeneic stem cell transplant, the response to therapies, and long-term follow-up.
Integrated genomic analysis that includes acquired copy-number abnormalities (CNA) analysis based on NGS data, SNP-array and FISH was performed in a subset of patients.
Statistical methods
Comparisons were performed using Mann-Whitney U-test for non-normally distributed variables. Fisher’s exact test was used to determine associations between categorical variables. Overall survival (OS) was calculated from date of t-MN diagnosis to the last follow-up or the date of death. Post-transplant survival for patients who underwent allogeneic stem cell transplantation (SCT) was assessed from day of transplantation. Kaplan-Meier estimations were used with comparisons using log-rank tests. Cox regression multivariable analysis with backward selection was undertaken. P values < 0.05 were considered statistically significant. Further details of statistical analysis are provided in the Supplementary Methods section.
Data Sharing Statement
Additional methods and data can be found in the Supplementary Methods section. For original data, please contact devendra.hiwase@sa.gov.au or Shah.Mithun@mayo.edu.
Results
Patient cohort
This international cohort of 488 t-MN patients included 318 with t-MDS (65.2%) and 170 with t-AML (34.8%). The median age at t-MN diagnosis was 68 (IQR 60, 74) years. The most common primary cancers were lymphoproliferative disorders (n = 142, 29.1%), plasma cell neoplasms (n = 64, 13.1%), breast cancer (n = 61, 12.5%), and prostate cancer (n = 39, 8%). Most common DNA-damaging therapies used for treating the primary disease were chemotherapy (n = 230, 47.1%), chemotherapy plus radiotherapy (n = 160, 32.8%), autologous SCT (n = 95, 19.5%), or radiation therapy alone (n = 79, 16.2%, Table S1).
The median latency from the time of primary disease to t-MN diagnosis, was 81 (IQR 40, 149) months. Following t-MN diagnosis, most patients were treated with disease-modifying therapies (DMT) including hypomethylating agents (HMA) (n = 160, 33.8%), intensive chemotherapy (n = 100, 20.5%), venetoclax-based therapies (n = 70, 14.3%), and 91 (18.6%) patients underwent allogeneic SCT (Table S1).
We first analyzed genome-wide allelic imbalances to include arm-level alterations detected by CBA. In agreement with prior studies [22, 23], 365 (76%) had at least one chromosomal aberration including CK (n = 190, 39.3%), MK (n = 183, 37.90%), deletion 7q or monosomy 7 (n = 148, 30.6%), deletion 5q or monosomy 5 (n = 108, 22.4%), and deletion 17p across TP53 locus (referred to as 17p loss hereafter, n = 58, 12%) (Figure S1A). We next analyzed the somatic mutation landscape of TP53mut in t-MN.
Genomic landscape of TP53 mut t-MN
The next generation sequencing (NGS) identified 253 putative oncogenic mutations in TP53 at VAF ≥ 2% in 182 (37.29%) patients (Fig. 1A, B). A diverse spectrum of TP53mut including highly frequent missense mutations followed by frameshift insertion-deletions, splice-site, and nonsense mutations was observed (Fig. 1C, D and Figure S1B, C). Nearly all missense mutations occurred in the TP53 DNA-binding domain (Fig. 1C and Figure S1B).

A Oncoplot showing cytogenetic and mutational landscape of TP53mut and wild-type TP53 (TP53wt) t-MN. Patient-related factors (t-MN phenotype, abnormal karyotype, primary disease, treatment status, and gender) are shown in the upper panel, and the distribution of somatic gene mutations (including TP53 mutation status) in the lower panel. Each column represents an individual patient, and the presence of the aberration or mutation is colored as indicated above; B Consort diagram of the mutant TP53 status of 488 t-MN patients analyzed by integrated analysis employing next gene sequencing (NGS), conventional cytogenetics, FISH, SNP-array and CNA analysis. *Of the 48 patients with single TP53mut VAF 10–50% LOH information was available in 33 patients. Importantly, 80% of the remaining TP53mut patients (n = 15) without LOH information has complex karyotype and are considered equivalent to multi-hit by ICC; C Distribution of TP53mut along the gene. Mutations from single-hit patients are shown at the bottom and those from multi-hit patients are shown at the top. Missense mutations are shown as blue circles, truncated mutations corresponding to nonsense mutations as orange circles, frameshift deletions or insertions as red circles, and splice site variants are shown as purple circles. Other types of mutations are shown as green circles. Functional protein domains are indicated in yellow (transactivation motif), brown (transactivation domain 2), blue (DNA binding domain), and green (tetramerization motif); D Summary of TP53mut separated by mutation type and frequency of the mutations.
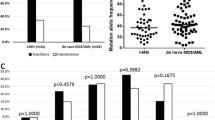
In the majority of the TP53mut t-MN with available information (n = 178; 97.8%), TP53mut was a dominant driver clone with VAF ≥ 40% (n = 124; 69.7%), while 20 (11.2%), 14 (7.8%) and 20 (11.2%) patients had VAF ≥ 22–40%, 10–22% and <10%, respectively (Fig. 2A).

A Distribution of cases according to TP53mut VAF; B Volcano plot comparing cytogenetic aberration and somatic mutations in TP53mut and TP53wt t-MN. Chromosomal aberrancies highly prevalent in TP53mut (red) and somatic mutations enriched in TP53wt cohort (green). Genomic changes that are not differentially expressed between the two groups are shown in gray color; C Frequency of cytogenetic aberrations or driver oncogenic gene mutations in TP53wt and TP53mut t-MN; D Number of co-mutations in TP53wt and TP53mut t-MN; E Overall survival (OS) of TP53mut with VAF ≥10% or loss of TP53 locus was significantly poor compared to wild-type TP53 (TP53wt) and TP53mut with VAF < 10% t-MN; F Multivariate Cox-regression analysis of factors predicting overall survival in t-MN; G OS of TP53mut t-MN according to VAF cut-offs; H Frequency of loss of heterozygosity (LOH) and copy neutral LOH (cnLOH) according to number of TP53mut; I Density estimation of VAF of single-hit and multi-hit TP53mut; J OS is equally poor in single- and multi-hit in t-MN.
Next, we determined the TP53mut VAF threshold associated with poor prognosis in t-MN. The receiver operating characteristics analysis identified 8% as the optimal VAF threshold (Figure S2A–C). As only 5 (2.3%) patients had VAF 8–10%, we adapted the ICC VAF threshold of 10% for further analyses (Figure S2C, D).
TP53 mut VAF ≥ 10% is associated with distinct presentation, features of genomic instability, and outcomes in t-MN
The majority of TP53mut t-MN had VAF ≥10% (n = 158; 88.8%) (Fig. 1B). The loss of 17p across TP53 locus (LOH) or copy neutral LOH (cnLOH) was detected in 73 (38.4%) patients including TP53mut VAF ≥10% (n = 60), VAF < 10% (n = 5), and in the absence of TP53mut (n = 8). Collectively, 171 (35%) had TP53mut VAF ≥ 10% or LOH/cnLOH across the TP53 locus.
We next compared cytogenetics abnormalities and somatic mutations in 30 genes analyzed in both (South Australian and Mayo) cohorts. Genomic instability was highly evident in TP53mut with VAF ≥ 10% and/or loss of TP53 locus compared to TP53wt t-MN. CK, MK, chromosome 5 aberrancies, and marker chromosomes were enriched in TP53mut t-MN (Fig. 2B, C, Table S2). In contrast, recurrent oncogenic mutations such as ASXL1, DNMT3A, FLT3-ITD, IDH1, IDH2, NPM1, PTPN11, RAS, RUNX1 and TET2 were less frequent in TP53mut t-MN (Figs. 1A, 2B, C, Table S2). Moreover, total number of co-mutations were significantly less in TP53mut cases compared to TP53wt (P < 0.001) (Table S2 and Fig. 2D). Enrichment of chromosomal aberrancies and lower frequency of somatic mutation was observed in TP53mut complex karyotype MDS [4] and multi-hit TP53mut compared to single-hit TP53mut and TP53wt de novo MDS [2]. TP53mut not only influenced the genomic instability but also dictated the clinical presentation. TP53mut t-MN had more severe anemia (P < 0.001), leukopenia (P < 0.001), and thrombocytopenia (P < 0.001) (Table S2).
Importantly, patients with TP53mut VAF ≥ 10% with or without loss of TP53 locus had significantly shorter survival compared to TP53wt (8.3 vs. 21.6 months; P < 0.001) (Fig. 2E). The three-year overall survival was 7% in patients with TP53mut compared to 34% in TP53wt patients (P < 0.0001). Poor survival of TP53mut was observed in t-MDS (9.9 vs. 24.1 months; P < 0.001) and t-AML (3.6 vs. 13.2; P < 0.001) (Figure S3A, B). The inferior outcomes of TP53mut persisted across all the t-MN treatment types, including supportive care (3.9 vs. 19.1; P < 0.0001), intensive chemotherapy (7.3 vs. 23.1; P < 0.0001), hypomethylating agents (10.9 vs. 20.5; P = 0.001), venetoclax-based combination therapies (8.1 vs. 23.3; P = 0.01) and allogenic SCT (20.6 vs. not reached; P = 0.01) (Figure S4A–F). Univariate Cox-regression analysis suggested that TP53mut, specific chromosomal abnormalities, complex karyotype, bone marrow blasts, age and allogeneic SCT predicted OS of t-MN (Table S3). Furthermore, multivariable multivariate Cox regression analyses validated inferior survival of TP53mut (HR 2.18, 95% CI 1.47–3.25; P < 0.001), independent of BM blast percentage (P = 0.003), chromosome 19 (P < 0.001) and allogeneic SCT (P < 0.001) (Fig. 2F). Poor prognosis with associated with TP53mut and higher blast counts is known in AML and MDS, however prognostic impact of abnormalities in chromosome 19 are not well known. In contrast to de novo AML [24] and MDS [25], chromosome 19 was associated with poor survival in t-MN.
In contrast to TP53mut with VAF ≥ 10%, majority of the clinical, cytogenetic, and mutation profile were similar between TP53mut VAF < 10% and TP53wt t-MN, except lower BM blasts and higher frequency of del 5q, CK, and ring chromosome. While somatic mutations were more prevalent in TP53wt t-MN (Table S4). Importantly, OS was not significantly different between the two groups (26.8 vs. 21.6 months, P = 0.69; Fig. 2E). In contrast, chromosomal aberrancies and OS were significantly different in TP53mut patients when categorized according to VAF < 10% vs. ≥10% (Fig. 2E and Table S5). Surprisingly, OS was equally poor in TP53mut when VAF 10–22%, 22–40%, and ≥40% (6.15 vs. 7.96 vs. 8.78 months, Fig. 2G) cut-offs were used.
Majority of the TP53 mut t-MN harbor biallelic loss of TP53
Among the 158 patients with TP53mut VAF ≥ 10%, 70% had TP53mut plus LOH/cnLOH of TP53 locus (n = 52; 32.9%) or ≥2 TP53mut (n = 59; 37.3%), while 29.1% (n = 47) had single TP53mut (Fig. 2H). Of the 47 patients with single TP53mut, 26 (55.3%) and 21 (44.7%) patients had VAF > 50% and 10–50%, respectively (Fig. 1B). Additional 13 patients had loss of the TP53 locus without evidence of TP53mut (n = 8) or with TP53mut VAF < 10% (n = 5) (Fig. 1B). Frequency of LOH/cnLOH was significantly higher in cases with single TP53mut compared to cases with ≥2 TP53mut (33% vs. 5%, P = 0.0001; Fig. 2H).
Next, we compared the clinical features, profiles of genome stability and patterns of co-mutation for each TP53 allelic state. Integrated cytogenetic, copy number and somatic mutation analysis classified TP53mut as multi-hit if there is: (1) presence of ≥2 distinct TP53mut, each with VAF ≥ 10%, or (2) a single TP53mut associated with either: (i) cytogenetic deletion of 17p13 involving the TP53 locus; (ii) a VAF of >50%; or (iii) copy-neutral loss of heterozygosity (cnLOH) at the TP53 locus. Single TP53mut with VAF 10%-50% or loss of 17p13 involving TP53 locus without TP53mut were defined as single-hit. In total, 34 (19.9%) of the 171 patients with TP53mut and/or loss of TP53 locus were considered single-hit and 137 (80.1%) were multi-hit (Fig. 1B). In single-hit TP53mut cases, the median VAF was significantly lower compared to multi-hit TP53mut (34% vs. 38.2%, P = 0.006) (Table S6 and Fig. 2I). Overall, the spectrum of TP53mut was shared among single- and multi-hit states (Fig. 1C).
Unlike de novo MDS [2], there was no significant difference in CK, MK, CK plus MK, chromosome 5 aberrancy, or co-mutations between single- and multi-hit TP53mut (Table S6). We did not observe significant differences in clinical features, age, latency, blood counts, BM blast percentage and cytogenetics when stratified by the allelic status except that the multi-hit TP53mut were enriched for marker chromosome. In contrast to previous publications [2, 14], the distribution of single- and multi-hit TP53mut was not different across the t-MN phenotype and the BM blast categories (Figure S5A, B).
Consistent with our previous observation [20], the OS was not significantly different between the single- and multi-hit TP53mut t-MN (Fig. 2J). Similarly, there was no survival difference between single- and multi-hit TP53mut when stratified by t-MDS vs. t-AML, according to the blast cut-off proposed by ICC (Figure S6A–D), or the type of treatment received (Figure S7A–C). Finally, there was no difference in the incidence of progression to AML in single versus multi-hit TP53mut t-MDS (Figure S7D).
TP53 mut burden increases with number of chromosomal aberrancies
The proportion of TP53mut increased from 4.5% in normal karyotype cases to 17.3% in cases with two chromosomal aberrancies (P = 0.019; Fig. 3A) and 76.8% in cases with CK (P < 0.0001). Even within the CK group, enrichment of TP53mut was observed with the increasing number of cytogenetic abnormalities: from 26.3% in cases with three chromosomal abnormalities to 75%, 96.6%, and 94% in cases with 4–6, 7–9, and >9 chromosomal aberrancies (Fig. 3A and Fig. S8). More than 80% of TP53mut were segregated in t-MN with >4 cytogenetic abnormalities (Figure S8). Furthermore, TP53mut were significantly high in typical- compared to atypical-CK (Fig. 3B). Typical CK is defined as CK with ≥3 abnormalities that include 5q, 7q, and/or 17p loss and atypical-CK as CK with ≥3 abnormalities without these specific abnormalities [26]. Conversely, TP53mut were enriched for CK compared to TP53wt (84.8% vs. 12.0%, P < 0.0001; Fig. 3C).

A Frequency of TP53mut according to number of structural cytogenetic abnormalities; B High frequency of TP53mut in typical-CK compared to atypical-CK t-MN; C TP53mut cases were enriched for CK; D In the absence of CK, TP53mut were prevalent in cases with 5q loss compared to 7q loss (+, present; -; absent; +/-, present or absent). Loss of 5q was defined as monosomy 5 or del 5q, while loss of 7q was defined as monosomy 7 or del 7q; E CK is associated with significantly poor survival in t-MN; F TP53mut status further stratify CK with very poor outcome.
The enrichment of TP53mut was also noted in cases with del 5q without CK. However, such enrichment of TP53mut was not observed in del 7q without CK (Fig. 3D). Together these findings indicate that TP53mut burden increases not only with number but with type of chromosomal aberrancies.
Complex karyotype is associated with poor survival (10.1 vs. 19.5 months; P < 0.001) (Fig. 3E). We assessed if TP53mut can further stratify by CK t-MN. Even within CK, TP53mut is associated with a higher structural genomic instability. In particular, MK, marker chromosomes, ring chromosome, 5q del/monosomy 5, chromosome 12, and 18 abnormalities were highly prevalent in TP53mut-CK compared to TP53wt-CK (Table S7). While number of somatic mutations, including RAS, ASXL1, and RUNX1 were enriched in TP53wt-CK (Table S7). Importantly, TP53mut further stratified outcomes for CK, with inferior survival of TP53mut-CK compared to the TP53wt-CK (8.3 vs. 19.7 months; P < 0.001, Fig. 3F). Conversely, CK-status did not influence the poor outcome of TP53mut t-MN (8.3 vs. 7.7 months, P = 0.29; Fig. 3F). Furthermore, OS of TP53wt CK was not significantly different than non-CK TP53wt. Together these findings suggest that poor prognosis of CK is driven by its association with prognostically adverse TP53mut (Fig. 3F). Similar association was previously reported in CK-MDS [4].
TP53 mut t-MDS can be stratified according to ICC BM blast categories
Next, we evaluated the frequency of TP53mut according to the disease phenotype. TP53mut burden was significantly higher in t-MDS compared to t-AML (40.3% vs. 29.3%, P = 0.021; Fig. 4A). However, the frequency of TP53mut in t-MDS was similar across the BM blast categories: 37.3 vs. 38.2 vs. 38.1% in <5%, 5–9% and 10–19% blasts, respectively (Fig. 4B and Table S8). There was no significant difference in age at t-MN diagnosis, latency, the type of the primary disease, or the degree of cytopenia at t-MN diagnosis across the blast % categories (Table S8). Moreover, genomic instability as evidenced by CK, MK, and number of cytogenetic abnormalities were also similar across all four categories (Table S8, Fig. 4C). Finally, the proportion of patients with single- vs. multi-hit, TP53 VAF, and co-mutations were comparable across the four blast categories (Table S8).

A Distribution of t-AML and t-MDS according to TP53mut status; B TP53mut frequency in t-MDS was similar across blast categories at diagnosis; C Chromosomal aberrancies and TP53mut allelic status across BM blast %; D Multivariate Cox-regression analysis showing BM blast >10%, chromosomal 18/19 abnormalities and disease modifying therapies were independent predictors of TP53mut OS; E In TP53mut t-MN, BM blast 10–19% and ≥20% were associated with poor OS compared to BM blast <5%; (F) OS of TP53mut t-MN according to ICC blast categories (0–9% vs. 10–19% vs. ≥20%).
Due to the molecular homogeneity of TP53mut across the blast categories, we investigated prognostic impact of BM blast %, variants in cancer-related genes, cytogenetics, and the severity of cytopenia in TP53mut t-MN. BM blasts 10–19%, ≥20%, hemoglobin, chromosome 19 abnormalities, and the first line therapies including allogeneic SCT were independent predictors of outcome in TP53mut t-MN (Fig. 4D; Table S9). Importantly, OS of TP53mut patients was poor across the blast categories with statistically significant difference between the BM blasts <5% vs. 10–19% and <5% vs. ≥20% categories (Fig. 4E). The OS of TP53wt t-MN significantly worsened with increasing blast % (P = 0.001) (Figure S9).
Recently, ICC proposed three categories of TP53mut MN according to BM blast % [19]. OS was significantly different in t-MDS with blast 0–9% vs. 10–19% blasts (10.9 vs. 8.2 months; P = 0.01). Similarly, OS was significantly different in TP53mut t-MN with BM blast 0–9% vs. ≥20% (10.9 vs. 4.6 months, P = 0.0008) (Fig. 4F). Together these findings suggest that TP53mut t-MN with >10% is uniformly associated with poor survival.
Discussion
The results above collectively demonstrate that the classification of t-MN based on TP53 status is clinically and biological relevant. This conclusion is supported by our findings that (1) TP53mut t-MN with ≥10% is uniformly associated with poor survival; (2) the presence of TP53mut was an independent risk factor for poor survival and was associated with inferior outcome, even within the traditionally known high-risk subsets such as CK; (3) the inferior outcomes persisted irrespective of the single- or multi-hit status, the co-mutation pattern, or the treatments received; (4) poor survival of single and muti-hit TP53mut was observed across the blast categories of t-MN.
TP53mut influenced the clinical presentation and survival. Although severe cytopenia is reported across multiple studies [2, 4, 14], the association with BM blast percentage is debated [2, 4, 10, 27]. TP53mut de novo MDS present with high BM blast [4, 27], especially those with the multi-hit alterations [2]. However, MDS with TP53mut/CK present with low BM blast burden [14]. We observed that TP53mut patients were more likely present as t-MDS. Furthermore, within the t-MDS cohort, BM blast burden was lower in TP53mut cases. In contrast to de novo MDS [2], frequency of multi-hit TP53mut remained similar across the BM blast categories. Secondly, poor prognosis of multi-hit TP53mut MDS/AML was reported irrespective of the BM blast and therapy-relatedness [14]. In our cohort, OS of TP53mut t-MN was poor across all the blast categories irrespective single- or multi-hit status and type of disease modifying therapies. However, OS of TP53mut MDS BM blasts >10% and AML was significantly inferior compared to TP53mut BM blasts <10%. Thus, the interaction between BM blast and TP53mut appears to be disease ontogeny specific. Overall, our findings support the ICC stratification of TP53mut MN using three blast cut-offs.
Though poor survival of TP53mut is well known, the prognostic implication TP53mut VAF remains an active area of research. Multiple studies reported VAF > 40% is associated with poor survival in high-risk MDS [4, 10, 28] while one study suggested that poor survival is a direct function of increasing VAF as a continuous variable [7]. In contrast to these findings, other studies showed an inferior OS irrespective of TP53mut VAF [3, 14, 29, 30]. Furthermore, prognostic implication of TP53mut VAF also depend upon TP53mut allelic status. Single-hit TP53mut MDS with VAF > 22% had poor survival and the favorable survival comparable to the TP53wt was restricted to the single-hit cases with VAF ≤ 22%. Conversely multi-hit patients had poor outcome across the range of TP53 VAF [2]. In t-MN, OS was significantly poor in TP53mut with VAF ≥ 10% compared to VAF < 10%. There was no survival difference in cases with VAF 10–22% vs. >22–40% vs. >40%. Together these findings suggest prognostic implication of TP53mut VAF is context dependent and varies significantly between de novo and therapy-related MN.
As expected, TP53mut and CK/MK were highly enriched in t-MN compared to de novo MDS [2] and AML [3, 5, 6, 31]. Within t-MN, CK was more frequent with TP53mut than TP53wt. Conversely, increasing genomic instability was associated with enrichment of TP53mut: 75% and ~90% of patients with ≥4–6 and ≥7 chromosomal abnormalities harbored TP53mut, respectively, compared to only 26% of patients with 3 chromosomal abnormalities. In addition to number, type of chromosomal abnormalities also influences enrichment of TP53mut. Critical understanding of the relationship between TP53mut and chromosomal aberrancies can be harnessed for prioritization of TP53mut testing in limited resources, and screening/counselling appropriate patients for clinical trials of novel therapies as waiting time for mutation results can be up to 3–4 weeks. Importantly, within CK t-MN, patients with co-existent TP53mut had evidence of profound genomic complexities and structural aberrancies [14, 32], and had poor outcomes compared to CK-TP53wt in our t-MN cohort and other MN [10], emphasizing the importance of assessing TP53mut alongside complex karyotype for an accurate risk estimation. Conversely, poor prognosis of biallelic TP53 loss compared to single allelic loss was evident only in non-CK, while presence of CK was invariably associated with poor survival irrespective of TP53mut allelic status [33].
In contrast to the findings in predominantly de novo MDS, we did not observe a difference in the frequency of structural chromosomal aberrancies including CK, MK, or co-mutation pattern between single- and multi-hit TP53mut in t-MDS and as such in the whole t-MN cohort. Moreover, there was no difference in the transformation rate to AML and the OS between the multi- and the single-hit TP53mut t-MDS. These findings have significant impact on the classification and management of t-MN patients especially considering the other recent changes in the WHO classification. The WHO has grouped t-MN with secondary MN and renamed it as “myeloid neoplasm post cytotoxic therapy”, with the assertion that a majority of MDS and AML occurring post-cytotoxic therapy have TP53mut and that only multi-hit TP53mut had a poorer outcome compared to single-hit [2, 18], thus undermining the poor prognosis of single-hit TP53mut t-MN. Exclusion of single-hit TP53mut t-MDS from the TP53 mutated MDS have huge impact on management such as consideration for allogeneic stem cell transplantation [34], and exclusion from clinical trials targeted toward TP53mut MDS. For example, allogeneic SCT may not be offered to fit single-hit TP53mut t-MDS with BM blast 5–9% (according to ICC) and <20% (according to WHO) as they are considered to have OS similar to TP53wt MDS. Similarly, these patients would be excluded from enrolment in clinical trials.
The apparent discrepancies in the conclusions among prior studies can be on the account of the characteristics of the study cohort, technical aspects, as well as true biological differences. For example, t-MN constituted a small subset of the patients in some large studies [2, 4]. Others excluded patients with low blasts [3], or only included patients with CK [4, 14]. Secondly, variable VAF thresholds have been used: 1% [3], 2% [2, 14], or 10% in the ICC guidelines. Thirdly, and likely the most significant difference is the criteria used to designate single vs. multi hit status. For example, in the absence of a detailed analysis of the TP53 locus (using CBA, FISH, or chromosomal microarray), both WHO and ICC consider >50% VAF as presumptive evidence multi-hit, though the evidence suggests that VAF estimation is a poor surrogate for the allelic status [2]. In the absence of LOH information, the presence of a single TP53mut in the context of CK is considered equivalent to a multi-hit TP53mut by ICC, but not in WHO-5. Key studies driving recent classifications variably used CBA, FISH, NGS, and SNP array to determine the hit status [2, 3]. In contrast, a recent study used WGS to confer hit status [35]. Therefore, what LOH assessment is considered minimal or optimal is unclear at this time. In summary, there is an urgent need to define a uniform diagnostic genetic tools and criteria for determining allelic status of TP53mut and VAF in all types of MN including t-MN, secondary and de novo MN.
Finally, the universally poor outcome of TP53mut t-MN highlights the great unmet need for these patients and the necessity for novel therapies. Unfortunately, none of the routinely offered leukemia-directed therapies were able to overcome the impact of TP53mut. TP53mut are noted to be present long before the eventual development of t-MN—sometimes even before the original exposure to DNA-damaging therapies. Recent evidence suggests a deterministic order of genetic and genomic changes following TP53 mutation/loss [36, 37]. A comprehensive characterization of the genomic changes, and its correlation with the resultant morphological changes, may help identify patients at the risk of imminent leukemic transformation and devise effective preventive strategies. Hence, the identification of individuals at high-risk of developing t-MN and employment of preventative approaches may improvement outcomes for this difficult-to-treat group of patients [16, 38,39,40].
References
Bally C, Ades L, Renneville A, Sebert M, Eclache V, Preudhomme C, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res. 2014;38:751–5.
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26:1549–56.
Grob T, Al Hinai ASA, Sanders MA, Kavelaars FG, Rijken M, Gradowska PL, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2022;139:2347–54.
Haase D, Stevenson KE, Neuberg D, Maciejewski JP, Nazha A, Sekeres MA, et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia. 2019;33:1747–58.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl J Med. 2013;368:2059–74.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686–98.
Montalban-Bravo G, Kanagal-Shamanna R, Benton CB, Class CA, Chien KS, Sasaki K, et al. Genomic context and TP53 allele frequency define clinical outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv. 2020;4:482–95.
Montalban-Bravo G, Takahashi K, Patel K, Wang F, Xingzhi S, Nogueras GM. et al. Impact of the number of mutations in survival and response outcomes to hypomethylating agents in patients with myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Oncotarget. 2018;9:9714–27.
Nazha A, Narkhede M, Radivoyevitch T, Seastone DJ, Patel BJ, Gerds AT, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia. 2016;30:2214–20.
Sallman DA, Komrokji R, Vaupel C, Cluzeau T, Geyer SM, McGraw KL, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2016;30:666–73.
Sebaa A, Ades L, Baran-Marzack F, Mozziconacci MJ, Penther D, Dobbelstein S, et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer. 2012;51:1086–92.
Singhal D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW, et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia. 2019;33:2842–53.
Takahashi K, Patel K, Bueso-Ramos C, Zhang J, Gumbs C, Jabbour E, et al. Clinical implications of TP53 mutations in myelodysplastic syndromes treated with hypomethylating agents. Oncotarget. 2016;7:14172–87.
Weinberg OK, Siddon A, Madanat YF, Gagan J, Arber DA, Dal Cin P, et al. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022;6:2847–53.
Welch JS. Patterns of mutations in TP53 mutated AML. Best Pract Res Clin Haematol. 2018;31:379–83.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–5.
McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:513–27.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood. 2022. https://doi.org/10.1182/blood.2022015850.
Hiwase D, Hahn C, Tran ENH, Chhetri R, Baranwal A, Al-Kali A, et al. TP53 mutation in therapy-related myeloid neoplasm defines a distinct molecular subtype. Blood. 2023. https://doi.org/10.1182/blood.2022018236.
Shah MV, Chhetri R, Dholakia R, Kok CH, Gangat N, Alkhateeb HB, et al. Outcomes following venetoclax-based treatment in therapy-related myeloid neoplasms. Am J Hematol. 2022;97:1013–22.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54.
Sperling AS, Guerra VA, Kennedy JA, Yan Y, Hsu JI, Wang F, et al. Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood. 2022;140:1753–63.
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65.
Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–9.
Mrózek K, Eisfeld AK, Kohlschmidt J, Carroll AJ, Walker CJ, Nicolet D, et al. Complex karyotype in de novo acute myeloid leukemia: typical and atypical subtypes differ molecularly and clinically. Leukemia. 2019;33:1620–34.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N Engl J Med. 2011;364:2496–506.
Tashakori M, Kadia T, Loghavi S, Daver N, Kanagal-Shamanna R, Pierce S, et al. TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia. Blood. 2022;140:58–72.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl J Med. 2017;376:536–47.
Goel S, Hall J, Pradhan K, Hirsch C, Przychodzen B, Shastri A, et al. High prevalence and allele burden-independent prognostic importance of p53 mutations in an inner-city MDS/AML cohort. Leukemia. 2016;30:1793–5.
Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5:e331–e331.
Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–21.
Aguirre LE, Al Ali N, Jain AG, Chan O, Ball S, Kuykendall A, et al. Characterization of TP53-mutated myelodysplastic syndromes and impact of allelic status and concurrent cytogenetic abnormalities on survival outcomes. Blood. 2022;140:4034–5.
Kutyna MM, Kok CH, Lim Y, Tran ENH, Campbell D, Paton S, et al. A senescence stress secretome is a hallmark of therapy-related myeloid neoplasm stromal tissue occurring soon after cytotoxic exposure. Leukemia. 2022;36:2678–89.
Stengel A, Meggendorfer M, Walter W, Baer C, Nadarajah N, Hutter S, et al. Interplay of TP53 allelic state, blast count, and karyotype on survival of patients with AML and MDS. Blood. 2022;140:2073–4.
Baslan T, Morris JP, Zhao Z, Reyes J, Ho YJ, Tsanov KM, et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature. 2022;608:795–802.
Light N, Layeghifard M, Attery A, Subasri V, Zatzman M, Anderson ND, et al. Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis. Nat Commun. 2023;14:77.
Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35:1598–605.
Shah MV, Mangaonkar AA, Begna KH, Alkhateeb HB, Greipp P, Nanaa A, et al. Therapy-related clonal cytopenia as a precursor to therapy-related myeloid neoplasms. Blood Cancer J. 2022;12:106.
Sridharan A, Schinke CD, Georgiev G, Da Silva Ferreira M, Thiruthuvanathan V, MacArthur I, et al. Stem cell mutations can be detected in myeloma patients years before onset of secondary leukemias. Blood Adv. 2019;3:3962–7.
Acknowledgements
We are grateful to our patients, their families, Mayo Clinic Acute Leukemia and Myeloid Neoplasms Biobank, and South Australia Cancer Research Biobank (SACRB). Some of the data in this publication were produced in the Mayo Clinic Cytogenetics Core Laboratory, which in part, is supported by the Mayo Clinic Comprehensive Cancer Center Grant, funded by National Cancer Institute (P30CA15083). MVS was supported by Mayo Clinic Research Pipeline K2R Transition Award and Bridget Kiely Clinician Career Development in Transplant Research and Mayo Clinic, Rochester. SK was supported by a NHMRC Investigator Grant (GNT2007739). DT is supported by a CSL Centenary Fellowship, Medical Research Futures Fund, and Leukemia-Lymphoma Translational Research Program funding. DH was supported by a National Health and Medical Research Council (NHMRC)/Medical Research Future Fund (MRFF) Investigator Grant (MRF1195517), Cancer Australia, and Leukemia Foundation Australia.
Author information
Authors and Affiliations
Contributions
MVS designed the study, contributed the patient data, and edited the manuscript; ET and RC collated the data, analyzed the data, and edited the manuscript; SS, AB, and CS abstracted patient data; DL contributed to cytogenetic analysis; DC reviewed pathology; PG contributed to cytogenetics and chromosomal array analyses; CK performed statistical analysis and edited the manuscript; CH, ET, ALB, and HS contributed to variant annotation and edited the manuscript; DT edited manuscript; SK contributed DDR expertise and edited the manuscript; AA, HA, NG, MP, and AT contributed patients and edited manuscript. DH designed the study, contributed the patient data, analyzed the data, and wrote the manuscript. All authors agree to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
MVS—Research funding to the institution from AbbVie, Celgene, MRKR Therapeutics, and Astellas; AA—Research support to institution (Novartis, Astex); MP—Membership on an entity’s Board of Directors or advisory committees (Stemline Therapeutics) and Research funding (Kura Oncology); DH—Membership on an entity’s Board of Directors or advisory committees (AbbVie, Novartis). All other authors disclose no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Scientific category for submission: Myeloid Neoplasia.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, M.V., Tran, E.N.H., Shah, S. et al. TP53 mutation variant allele frequency of ≥10% is associated with poor prognosis in therapy-related myeloid neoplasms. Blood Cancer J. 13, 51 (2023). https://doi.org/10.1038/s41408-023-00821-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-023-00821-x
- Springer Nature Limited
This article is cited by
-
Mutational variant allele frequency profile as a biomarker of response to immune checkpoint blockade in non-small cell lung Cancer
Journal of Translational Medicine (2024)
-
TP53-mutated acute myeloid leukemia and myelodysplastic syndrome: biology, treatment challenges, and upcoming approaches
Annals of Hematology (2024)
-
Real world predictors of response and 24-month survival in high-grade TP53-mutated myeloid neoplasms
Blood Cancer Journal (2024)