
Abstract
Heart failure with preserved ejection fraction (HFpEF) is highly prevalent, and lacks effective treatment. The aberration of WNT pathway underlies many pathological processes including cardiac fibrosis and hypertrophy, while porcupine is an acyltransferase essential for the secretion of WNT ligands. In this study we investigated the role of WNT signaling pathway in HFpEF as well as whether blocking WNT signaling by a novel porcupine inhibitor CGX1321 alleviated HFpEF. We established two experimental HFpEF mouse models, namely the UNX/DOCA model and high fat diet/L-NAME (“two-hit”) model. The UNX/DOCA and “two-hit” mice were treated with CGX1321 (3 mg·kg−1·d−1) for 4 and 10 weeks, respectively. We showed that CGX1321 treatment significantly alleviated cardiac hypertrophy and fibrosis, thereby improving cardiac diastolic function and exercise performance in both models. Furthermore, both canonical and non-canonical WNT signaling pathways were activated, and most WNT proteins, especially WNT3a and WNT5a, were upregulated during the development of HEpEF in mice. CGX1321 treatment inhibited the secretion of WNT ligands and repressed both canonical and non-canonical WNT pathways, evidenced by the reduced phosphorylation of c-Jun and the nuclear translocation of β-catenin and NFATc3. In an in vitro HFpEF model, MCM and ISO-treated cardiomyocytes, knockdown of porcupine by siRNA leads to a similar inhibitory effect on WNT pathways, cardiomyocyte hypertrophy and cardiac fibroblast activation as CGX1321 did, whereas supplementation of WNT3a and WNT5a reversed the anti-hypertrophy and anti-fibrosis effect of CGX1321. We conclude that WNT signaling activation plays an essential role in the pathogenesis of HFpEF, and porcupine inhibitor CGX1321 exerts a therapeutic effect on HFpEF in mice by attenuating cardiac hypertrophy, alleviating cardiac fibrosis and improving cardiac diastolic function.
Similar content being viewed by others

Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) is a clinical syndrome in which patients have symptoms and signs of HF despite normal or nearly normal left ventricular ejection fraction (LVEF ≥ 50%) [1]. HFpEF accounted for over half of the HF patients and its prevalence continues to increase [2]. The 5-year all-cause mortality rate of HFpEF was over 50%, but the drugs for HFrEF such as β-blockers and angiotensin converting enzyme inhibitors (ACEi) showed limited effect on HFpEF [3,4,5]. Despite the recent progress that sodium-glucose cotransporter 2 inhibitors (SGLT2is) and/or angiotensin receptor-neprilysin inhibitors (ARNIs) reduced the hospitalization of HFpEF patients [6, 7], HFpEF was still considered to be the greatest unmet need in cardiovascular medicine today, and more efficacious therapies were urgently needed [8, 9].
HFpEF was a highly integrated and multisystem disorder [10]. It was characterized by impaired left ventricle (LV) diastolic function and increased LV stiffness which caused the subsequent rising LV filling pressure and led to symptoms of dyspnea, lung gas exchange abnormalities and impaired aerobic capacity [11]. Although the molecular mechanisms for HFpEF pathogenesis were still not fully understood, it was well accepted that cardiac fibrosis and hypertrophy were the essential pathological features of HFpEF and contributed to the increased LV stiffness and impaired cardiac diastolic function [12, 13].
WNT pathway was a fundamental cell signaling pathway, and its aberration underlies many human pathologies, such as cancer, tissue degeneration and fibrosis, including cardiac fibrosis [14]. In normal healthy adult heart WNT signaling was quiescent, but it was activated and played an essential role in the pathogenesis in cardiac fibrosis and hypertrophy [15, 16]. A recent study of human blood proteome analyses showed that the activation of WNT signaling was a proteomics signature in HFpEF [17]. This suggested a potential role of WNT signaling in the pathogenesis of HFpEF.
WNT signaling included canonical and non-canonical pathways. The activation of WNT signaling pathways required the binding of various WNT ligands to membrane receptors frizzled and LRP5/6, and subsequent engagement of Dishevelled-1 in the cytoplasm [18]. Porcupine, an acyltransferase located on the endoplasmic reticulum, was essential for the lipid modification and secretion of all WNT proteins. Blocking porcupine could inhibit both canonical and non-canonical WNT pathways [19, 20]. In the past several years, targeting porcupine to block WNT signaling pathways has been extensively studied to treat cancer, and small molecule porcupine inhibitors such as WNT974, ETC-159, RXC004 and CGX1321 have been tested in clinical trials [20].
As revealed by our previous studies, CGX1321, a novel porcupine inhibitor discovered by Curegenix, exerted robust anti-hypertrophy and anti-fibrosis effects in mouse models of myocardial hypertrophy by transverse aortic constriction and myocardial infarction by coronary artery ligation [21, 22]. CGX1321 has completed phase 1 dose-escalation clinical safety study and entered into dose-expansion and phase 1b clinical efficacy studies for the treatment of cancer as a monotherapy and in combination with pembrolizumab (Keytruda®) (clinical trial identifier: NCT02675946). Our present study aimed to investigate the role of WNT signaling in HFpEF, and tested whether porcupine inhibitor CGX1321 could exert a therapeutic effect on HFpEF in two established HFpEF animal models.
Materials and methods
Experimental animal models, maintenance and treatment
Two established mouse models were employed to induce HFpEF phenotypes. The UNX/DOCA mouse model was established as described previously [23]. In brief, surgery was performed on 10–12 week-old male C57/BL6J mice (SPF Biotechnology, Beijing, China). According to the published procedures, mice underwent a surgery of right unilateral nephrectomy (UNX), subcutaneously implanted a controlled release deoxycorticosterone acetate (DOCA) pellet (0.7 mg/day, M-121, Innovative Research of American, FL, USA) and substituted drinking water with 1% NaCl and 0.1% KCl. Control animals were subjected a sham operation, implanted a placebo pellet and received water without additional NaCl and KCl. All surgeries were performed under anesthesia and all efforts were made to minimize animal suffering. After 1 weeks post-DOCA pellet implantation, both the control and UNX/DOCA mice were randomly divided into two groups respectively to receive a vehicle or CGX1321 (3 mg·kg−1·d−1) treatment randomly for another 4 weeks. The “two hit” mouse model was also employed in this study. As early described, 10–12 week-old male C57BL/6 mice (SPF Biotechnology, Beijing, China), were subjected 60% high fat diet (H10060, Hua Fukang, Beijing, China) and drinking water with 0.5 g/L of L-NAME (N5751, Sigma Aldrich, St. Louis, MO, USA) for 15 weeks [24]. Control mice were fed with a standard diet and water without L-NAME. At 5 weeks, the control mice and “two hit” mice were randomly divided into 2 groups then treated with a supplementation of vehicle or CGX1321 randomly for another 10 weeks. All of these mice were maintained in a 12/12-h light/dark cycle environment with a 22 °C constant temperature. The protocols used in this study were approved by the Institutional Animal Care and Use Committee at Third Military Medical University and all studies were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
CGX1321
CGX1321 is a novel porcupine inhibitor discovered by Guangzhou Curegenix Co. Ltd. and Curegenix Inc. By targeting porcupine, CGX1321 inhibits the acylation and secretion of all WNT proteins. CGX1321 has completed phase 1 dose-escalation clinical safety study and entered into dose-expansion and phase 1b clinical efficacy studies for treatment of cancer as a monotherapy and in combination with pembrolizumab (Keytruda®) (clinical trial identifier: NCT02675946). In our earlier pre-clinical studies, a dosage of 3 mg·kg−1·d−1 of CGX1321 in vivo and 0.1 μM in vitro could significantly inhibit the target genes of WNT signaling and there was no significant toxicities shown in vital organs in mice [21, 22].
Echocardiography
Serial transthoracic echocardiographic and Doppler analyses were performed using the vevo2100 high-resolution ultrasound imaging system (VisualSonics Inc, Toronto, Canada) with an MS400 transducer (24–30 MHz). Briefly, mice were anesthetized with 1.5% isoflurane and 98.5% O2 and placed in the supine position on a temperature-controlled heating platform to maintain the body temperature at 37 °C. Indices of systolic function were obtained in conscious, gently restrained mice from M-mode scans at middle ventricular level. The indices of diastolic function were obtained in anesthetized mice from apical four-chamber view using pulsed-wave and tissue Doppler imaging [25]. Three consecutive values were measured using the software resident on the ultrasonography. A random number was allocated to every mouse to guarantee the echocardiographic manipulator was blinded to treatment.
Invasive hemodynamics
Invasive hemodynamic measurements were carried out as described previously [26]. Briefly, the animals were anesthetized with isoflurane (3%–4% for induction; 1.5% for maintenance mixed with 98.5% oxygen). Following tracheal intubation, mice were artificially ventilated by using a volume-controlled ventilator trach and then placed on a controlled heating pads to maintain the body temperature at 37 °C. The left external jugular vein was cannulated with a polyethylene catheter for fluid administration. A 1.4 F micro tip pressure-conductance micro-catheter (SPR-839, Millar Instruments, Houston, TX, USA) was inserted into the right carotid artery and advanced into the ascending aorta. When a stable mean arterial blood pressure (MAP) and heart rate (HR) were recorded, the catheter was continuously advanced into the left ventricle (LV). After 5-min stabilization, signals were continuously recorded at a sampling rate of 1000 samples/s using pressure-volume (P-V) conductance system connected to the Power Lab 16/30 data acquisition system (AD Instruments, Colorado Springs, CO, USA). Maximal slope of systolic pressure increment (dp/dtmax), diastolic pressure decrement (dp/dtmin), time constant of LV pressure decay (Tau W; according to the Weiss method), stroke volume (SV) and end-diastolic volume (EDV) were analyzed by the Lab Chart 7.0 Pro Software System. Tibia length (TL; mm) and heart weight (HW; mg) were measured after the hemodynamic experiments.
Tail cuff blood pressure recordings
Systolic blood pressure was measured noninvasively in conscious mice using the tail-cuff method and a CODA instrument. Animals were placed in individual holders on a temperature-controlled platform (37 °C), and recordings were performed under steady-state conditions. Before testing, all mice were trained to become accustomed to short-term restraint. Blood pressure was recorded for at least 4 consecutive days and readings were averaged from at least 8 measurements per session [24].
Exercise calorimetry
Mice were subjected to exercise capacity testing on a closed-chamber treadmill. After an initial adaptation period of 2 min at 5 m/s, testing velocity was increased to 14 m/s and with an acceleration of 2 m/s per minute. The exercise session ended at maximal exhaustion, which was defined as the inability to maintain running speed despite being in contact with the electric shock for more than 5 s, accordingly, maximal running distance and workload were calculated. All mice were trained to be accustomed to running in the treadmill for 3 days before tested [24].
Measurement of cardiac fibrosis and cardiac hypertrophy
The mouse heart tissues were harvested and fixed in 4% PFA/PBS solution at 4 °C for 24 h. For fibrosis analysis, cardiac sections were deparaffinized and stained with a modified Masson trichrome staining kit (G1345, Solarbio, Bejing, China) according to the manufacturer’s protocols. For the analysis of cardiac hypertrophy, cardiac sections were deparaffinized and stained with wheat germ agglutinin (W11261, Invitrogen Corporation, Waltham, MA, USA) at 37 °C in dark for 1 h. The digital images of the stained sections were captured, the fibrosis areas and cross section of cardiomyocytes were measured. Quantification of staining was performed by using Image-Pro Plus software in a blinded manner.
Quantitative real-time polymerase chain reaction
Total RNA of hearts or cells was isolated by using TRIzol reagent (15596-026, Invitrogen Corporation, Waltham, MA, USA) according to the manufacturer’s protocols. RNA sample (1 μg) was reversely transcripted into cDNA using the cDNA Synthesis Kit (489703001, Roche, Basel, Switzerland). PCR was conducted in a 10 µL reaction system. Reversed transcription was performed at 37 °C for 15 min, and cDNA was amplified for 39 cycles: 95 °C for 10 s, 58 °C for 20 s, and 72 °C for 10 s. Values were normalized to GAPDH to calculate the relative RNA expression levels. The primer sequences used to detect mRNA expression were listed in Supplementary Table S1, S2.
Western blot
Total protein and nuclear protein of tissues, NRVMs and NRVFs was extracted by using RIPA lysis buffer (P0013B, Beyotime, Shanghai, China), phosphatase inhibitor cocktail (P1081, Beyotime, Shanghai, China) and Nuclear and Cytoplasmic Protein Extraction Kit (P0027, Beyotime, Shanghai, China). Protein concentrations were quantified with the BCA Protein Assay Kit (23227, Thermo Fisher Scientific, Waltham, MA, USA). Protein samples were separated by using SDS-PAGE, transferred onto PVDF membranes (FL00010, Millipore, Billerica, MA, USA). Then the membranes were blocked for 1 h with TBS (Tris-buffered saline), containing 5% of nonfat dry milk. Next, the blots were probed with primary antibodies overnight at 4 °C in 5% BSA TBST. WNT3a (26744-1-AP), WNT5a (55184-1-AP), β-catenin (51067-2-AP), TGF-β (21898-1-AP), Frizzled2 (24272-1-AP), c-myc (10828-1-AP), NCX1 (28447-1-AP) and GAPDH (60004) were purchased from Proteintech (Wuhan, China). NFATc3 (PA57934) and cyclin D1 (701421) were purchased from Invitrogen (Waltham, MA, USA), p-c-Jun (SC-23948) and c-Jun (SC-23948) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), α-SMA (68463), Phospholamba (PLN) (14562 s) and Phospho-Phospholamban (Ser16/Thr17) (8496 s) were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). SERCA2 ATPase (EPR9393) and Histone H3 (ab176840) were purchased from Abcam (Cambridge, UK). Following incubation with primary antibodies, blots were rinsed and probed with secondary antibodies for 1 h at room temperature. Protein bands were visualized and quantified by using Image Lab analysis software (Bio-Rad Laboratories, Hercules, CA, USA).
Cell isolation and culture
Less than 24 h old neonatal rat mice were prepared for isolation of ventricular myocytes (NRVMs). As early described [27], the hearts of neonatal rat mice were removed after an isoflurane-induced anesthesia and then placed in pre-cold PBS to remove the blood, then the tissues were cut into pieces and digested by 1.25 mg/mL trypsin and 0.8 mg/mL collagenase II. Isolated cells were pre-plated for 1.5 h to separate cardiomyocytes and cardiac fibroblasts. The cardiomyocytes and cardiac fibroblasts were collected and cultured with Dulbecco’s modified Eagles’s medium (C11330500BT, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS,10099-141, Gibco, Grand Island, NY, USA) and 1% penicillin-streptomycin (C0222, Beyotime, Shanghai, China). The in vitro models used to simulate the micro-environment of HFpEF were established as early described [26]. The RAW264.7 murine macrophages were cultured with DMEM containing 10% FBS and 1% penicillin-streptomycin. After treatment with ultrapure LPS 200 ng/mL (L4391, Sigma Aldrich, St. Louis, MO, USA) for 6 h and ATP 2 mM (A6559, Sigma Aldrich, St. Louis, MO, USA) for 30 min the medium was collected as macrophage-conditioned medium (MCM). To mimic the micro-environment of HFpEF, the cardiomyocytes were cultured with MCM and isoproterenol (ISO) 20 μM for 48 h and cardiac fibroblasts were cultured with MCM for 48 h. To test the effect of CGX1321 on hypertrophy and fibrosis the cardiomyocytes and fibroblasts were cultured in medium with or without CGX1321 (0.1 μM).
Immunohistochemistry
The cardiomyocytes and cardiac fibroblasts grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 and incubated with 2.5% normal donkey serum. To detect the location of β-catenin and NFATc3 in cardiomyocytes, the cardiomyocytes were stained by β-catenin (51067-2-AP, Proteintech, Wuhan, China), NFATc3 (PA57934, Invitrogen, Waltham, MA, USA), cTnT (ab56357, Abcam, Cambridge, UK). To detect the activation of fibroblast, the fibroblasts were stained by α-SMA (A2547, Sigma Aldrich, St. Louis, MO, USA), fibronectin (66042-1-Ig, Proteintech, Wuhan, China) and Ki67 (9449, CST, Danvers, MA, USA), respectively. Then the cells were probed with Alexa Fluor 488 and Alexa Fluor 555 secondary antibody for 1 h in the dark at 37 °C. The nucleus was counterstained with the DAPI (C1006, Beyotime, Shanghai, China). Images were obtained using laser confocal microscopy and evaluated using the Olympus Fluoview FV300 version 3 C Acquisition Software.
ELISA
The serum of each mouse in different groups was collected and the circulating protein levels of ANP (E-EL-M0166c, Elabscience, Wuhan, China), BNP (E-EL-M0204c, Elabscience, Wuhan, China), WNT3a (EM0737, FineTest, Wuhan, China) and WNT5a (EM0497, FineTest, Wuhan, China) were detected according to the instructions of the ELISA kit.
Glucose tolerance test
The glucose tolerance test was performed as previous report [24]. Briefly, mice were fasted for 12 h and intraperitoneally injected with a bolus of D-glucose at 2 g/kg. Blood glucose levels were measured from the tip of the tail with a glucometer (Abbott, Chicago, IL,USA) at 0, 15, 30, 45, 60 and 90 min after injection.
Statistical analysis
All data were expressed as the mean±standard deviation (SD). GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA, USA) was used for statistical analyses. Differences between the multiple groups were evaluated by one-way ANOVA, followed by the post hoc Tukey test. Two groups were analyzed using an unpaired Student’s t test. P < 0.05 was considered statistically significant.
Results
Porcupine inhibitor CGX1321 exerted a therapeutic effect against HFpEF in UNX/DOCA mouse model
We detected the mRNA expression level of all WNT ligands in two HFpEF mouse models (UNX/DOCA and high fat diet/L-NAME “two-hit”), and found that the mRNA levels of WNT ligands were increased in both HFpEF models heart compared with control. Of all WNT ligands, WNT3a and WNT5a were most significantly elevated (Supplementary Fig. S1a). Consist with the result of mRNA, the protein expressions of WNT3a and WNT5a were also elevated in HFpEF mice heart tissues (Supplementary Fig. S1b1–b3). Besides, the circulating WNT3a and WNT5a levels in serum were also significantly increased (Supplementary Fig. S1c, d). These two ligands could stimulate both canonical and non-canonical WNT pathways which were associated with cardiac hypertrophy and fibrosis [28, 29].
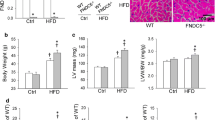
Then, we examined if CGX1321 could exert a therapeutic effect on UNX/DOCA induced HFpEF mice (Fig. 1a). After 5-week post-induction, CGX1321 did not change LVEF (%) (Fig. 1b2) and FS (%) (Fig. 1b3), while remarkably attenuating the increase of left ventricular posterior wall diameter (LVPWd) (Fig. 1b4), E/A ratio (Fig. 1b5) and E/E’ (Fig. 1b6) in HFpEF mice. Additionally, invasive hemodynamic measurements showed that CGX1321 treatment suppressed the decrease of end-diastolic volume (EDV) (Fig. 1c2), stroke volume (SV) (Fig. 1c3) and -dp/dtmin (Fig. 1c4) while significantly suppressed the increased tau value (Fig. 1c5) in HFpEF mice. These findings indicated that CGX1321 attenuated the diastolic dysfunction of HFpEF. While, there was no difference in dp/dtmax in four groups (Fig. 1c6) which reflected the systolic function of heart. Additionally, consistent with previous reports [30], UNX/DOCA could induce mild hypertension in mice. CGX1321 treatment did not affect blood pressure in different groups (Supplementary Fig. S2a, b). These suggested that CGX1321 attenuated the cardiac diastolic dysfunction in HFpEF independent of the blood pressure regulation.

CGX1321 exerted a therapeutic effect against HFpEF in UNX/DOCA mouse model. a A schematic representation of the experimental strategy. b Representative images of M-mode echocardiography, pulse wave Doppler and tissue Doppler tracings from different experimental groups at 5 weeks (b1), the calculated LV EF (%) (b2), FS% (b3), LVPWd (b4), mitral E/A ratio (b5) and E/E’ ratio (b6) in different groups (n = 8 mice per group). c Representative steady-state left ventricular pressure-volume (P-V) loops in different groups (c1), the calculated LV EDV (c2), stroke volume (SV) (c3), diastolic relaxation (-dp/dt, mmHg/s) (c4) Tau (Mirsky, ms) (c5), and dp/dtmax (mmHg/s) (c6) from the P-V loop analysis (n = 6 mice per group). n.s. not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
CGX1321 exerted a therapeutic effect against HFpEF in “two-hit” mouse model
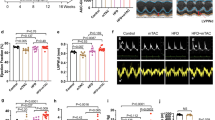
We also tested the therapeutic effect of CGX1321 on “two-hit” HFpEF mouse model (Fig. 2a). Consistent with previous reports [24], the “two-hit” HFpEF mice exhibited preserved LVEF (%) (Fig. 2b2) and FS (%) (Fig. 2b3), significantly increased LVPWd (Fig. 2b4), E/A ratio (Fig. 2b5) and E/E’ (Fig. 2b6), accompanied with increased tau (Fig. 2c5) and decreased EDV (Fig. 2c2), SV (Fig. 2c3) and -dp/dtmin (Fig. 2c4) at 15 weeks post-induction. CGX1321 administration significantly reversed those changes in “two hit” HFpEF mice (Fig. 2b4–b6, Fig. 2c2–c5) and did not influence the dp/dtmax (Fig. 2c6).

CGX1321 exerted a therapeutic effect against HFpEF in “two hit” mouse model. a A schematic representation of the experimental strategy. b Representative images of M-mode echocardiography, pulse wave Doppler and tissue Doppler tracings from different experimental groups at 15 weeks (b1). The calculated LV EF (%) (b2), FS% (b3), LVPWd (b4), mitral E/A ratio (b5) and E/E’ ratio in different groups (n = 8 mice per group). c Representative steady-state left ventricular pressure-volume (P-V) loops in different groups (c1), the calculated LV EDV (c2), stroke volume (SV) (c3), diastolic relaxation (-dp/dt, mmHg/s) (c4), Tau (Mirsky, ms) (c5) and dp/dtmax (mmHg/s) (c6) from the P-V loop analysis (n = 6 mice per group). n.s. not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Besides, consistent with previous reports [30], “two-hit” HFpEF mice were companied with hypertension, obesity and impaired glucose tolerance. Thus, we detected the blood pressure, body weight and glucose tolerance in different groups. The result showed that there were no differences between vehicle and CGX1321 treated groups (Supplementary Fig. S3a-e). These data indicated that the therapeutic effect of CGX1321 on HFpEF was not associated with blood pressure, obesity and glucose tolerance.
CGX1321 improved the exercise performance and attenuated the cardiac hypertrophy and fibrosis in HFpEF mice
We tested the exercise capacity of mice in different groups. Compared with the control ones, UNX/DOCA mice showed a decrement in running distance, which was restored by CGX1321 (Fig. 3a1-a2). Further researches showed that the UNX/DOCA HFpEF mouse model increased heart weight/tibia length ratio (Fig. 3b1–b2) and cardiomyocytes cross sectional areas (Fig. 3c1-c2) at 5-weeks post-induction, accompanied with increased interstitial and perivascular fibrosis (Fig. 3d1–d2). CGX1321 treatment decreased both heart weight/tibia length ratio and cardiomyocytes size in UNX/DOCA model (Fig. 3b-c). Such effect on hypertrophy was further corroborated by the decreases in the levels of hypertrophy biomarkers β-MHC and circulating ANP and BNP (Fig. 3g–i). Moreover, both interstitial and perivascular fibrosis degrees were reduced after CGX1321 treatment, as indicated by Masson trichrome staining (Fig. 3d) and the decreased mRNA levels of fibrotic biomarkers Col1a1 and Col3a1 (Fig. 3e, f). These data suggested that CGX1321 treatment reduced cardiac hypertrophy and fibrosis in UNX/DOCA HFpEF mice.

CGX1321 attenuated the cardiac hypertrophy and fibrosis in HFpEF mice. a The schematic representation of exercise capacity analysis in mice (a1) and running length in different groups (a2) (n = 8 mice per group). b Representative heart images (b1) and the ratio of heart weight normalized to tibia length (HW/TL, mg/mm) in different groups (b2) (n = 8 mice per group). c Representative images of wheat germ agglutinin (WGA) staining (c1) and the calculated cardiomyocyte cross-sectional area (c2) (n = 6 mice per group, scale bar = 20 μm). d Representative images of Masson’s trichrome staining (d1) and calculated fibrosis area (d2) (n = 6 mice per group). e–g The mRNA expression of Col1a1 (e), Col3a1 (f), β-MHC (g) in heart of different groups (n = 6 mice per group). h, i The ANP and BNP in serum of mice in different groups. (n = 6 mice per group). n.s. not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Additionally, these effects of CGX1321 on heart weight/tibia length ratio, cardiomyocyte hypertrophy, cardiac fibrosis and exercise capacity were also confirmed in the “two-hit” mouse HFpEF model (Supplementary Fig. S4a-f).
CGX1321 inhibited both canonical and non-canonical WNT signaling pathways and then exerted anti-hypertrophy and anti-fibrosis effect
Both canonical and non-canonical WNT pathways regulated cardiac hypertrophy and fibrosis [21, 31]. Activation of canonical and non-canonical WNT pathways were dependent on the phosphorylation of c-Jun and the nuclear translocation of β-catenin and NFATc3 [32]. Our data showed that the total c-Jun, β-catenin and NFATc3 proteins were comparable between different groups, while the nuclear translocation of β-catenin (canonical WNT signaling pathways), phosphorylation of c-Jun and the nuclear translocation of NFATc3 (non-canonical WNT signaling pathways) were obviously increased in HFpEF mice hearts (Fig. 4a1-a6). After the treatment of CGX1321 these changes were significantly reversed (Fig. 4a1-a6). Similar results were observed in mRNA levels of Axin2, an indicator of the activation of the canonical WNT/β-catenin pathway (Fig. 4b). Besides, the concentrations of WNT3a and WNT5a were also significantly decreased with the treatment of CGX1321 (Fig. 4e, f).

CGX1321 attenuated the activation of WNT signaling pathways and blocked the expression of hypertrophy and fibrosis associated protein in HFpEF mice. a Representative images of c-Jun, nucleus and total β-catenin and NFATc3 protein expression in heart tissue by Western-blot analysis (a1), densitometric analysis of different protein band in different experimental groups (a2–a6). GAPDH and Histone H3 were used as controls (n = 6 mice per group). b The expression of Axin2 in heart tissue (n = 6 mice per group). c Representative images of Frizzled2, Cyclin D1 and c-myc protein expression in heart tissue by Western-blot analysis (c1). Densitometric analysis of different protein band in different experimental groups (c2–c4). GAPDH was used as control (n = 6 mice per group). d Representative images of TGF-β and α-SMA protein expression in heart tissue by Western-blot analysis (d1). Densitometric analysis of different protein band in different experimental groups (d2, d3). GAPDH was used as control (n = 6 mice per group). e, f The WNT3a (e) and WNT5a (f) in serum of mice in different groups (n = 6 mice per group). n.s. not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The phosphorylation of c-Jun, nuclear translocations of β-catenin and NFATc3 could activate the expression of hypertrophy and fibrosis associated genes [21, 33]. We measured these genes regulated by WNT signaling pathways and found that the hypertrophy associated genes frizzled2, c-myc, Cyclin D1, and the fibrosis associated genes TGF-β and α-SMA were significantly increased in HFpEF mice, while the CGX1321 treatment reversed these changes (Fig. 4c, d).
These data indicated that both canonical and non-canonical WNT signaling pathways were activated in HFpEF mice hearts, while the treatment of CGX1321 obviously inhibited the secretion of WNT ligands and then effectively suppressed the activation of canonical and non-canonical WNT pathways, as well as the expression of hypertrophy and fibrosis associated genes.
CGX1321 attenuated cardiomyocyte hypertrophy by blocking WNT signaling pathways
We used an in vitro HFpEF models to further investigate the mechanism of CGX1321 in HFpEF (Fig. 5a) [26]. The results showed that cardiomyocytes treated with MCM and isoprenaline (ISO) showed a significant increase of cell surface areas and hypertrophy biomarker expression levels, which were significantly blocked by CGX1321 (Fig. 5b1-b3, 5f-h). MCM treatment also increased the phosphorylation of c-Jun, the nuclear translocation of β-catenin and NFATc3, and upregulated the mRNA level of Axin2 in cardiomyocytes. These effects were reversed by CGX1321 (Fig. 5c-d). Along with the WNT activation, frizzled2, Cyclin D1 and c-myc were also significantly elevated, which were attenuated by CGX1321 (Fig. 5e1-e4). However, when we added WNT3a and WNT5a in MCM, the anti-hypertrophy effect of CGX1321 was abolished (Supplementary Fig. S6a-b).

CGX1321 attenuated cardiomyocyte hypertrophy by blocking WNT signaling pathways. a A schematic representation of in-vitro experimental model for HFpEF. b Representative images of β-catenin (b1) and NFATc3 (b2) immuno-fluorescence images in NRVMs, quantification of the surface area of NRVMs (b3) (n = 6, scale bar= 20 μm). c The expression of Axin2 in heart tissue (n = 6). d Representative images of p-c-Jun, nucleus β-catenin, NFATc3 protein expression in NRVM (d1), densitometric analysis of different protein band in different experimental groups (d2–d4), c-Jun and Histone H3 were used as controls (n = 6). e Representative images of Frizzled2, Cyclin D1 and c-myc protein expression in heart tissue by Western-blot analysis (e1), densitometric analysis of different protein band in different experimental groups (e2–e4) GAPDH was used as control (n = 6). f–h The RNA expression of hypertrophy markers ANP (f), BNP (g) and β-MHC (h) (n = 6). n.s. not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
These results indicated that the effect of CGX1321 was dependent on repressing the secretion of WNT ligands (especially WNT3a and WNT5a) which subsequently inhibited the canonical/non-canonical WNT signaling pathways, and decreased the expression of hypertrophy associated protein.
CGX1321 attenuated cardiac fibroblast activation and proliferation by blocking WNT signaling pathways
Cardiac fibrosis was another essential pathological change in the development of HFpEF [34]. We found that MCM treatment (Fig. 6a) increased the number of α-SMA, fibronectin and Ki67 positive fibroblasts (Fig. 6b, c, Supplementary Fig. S6c). In accordance with the in vivo results, WNT signaling pathways were activated by MCM in fibroblasts as determined by the phosphorylation of c-Jun, nuclear translocation of NFATc3 and β-catenin, as well as the mRNA expression of Axin2 (Fig. 6d, e). In addition to the activation of WNT pathway, the protein levels of α-SMA and TGF-β (Fig. 6f), as well as the mRNA levels of Col1a1 and Col3a1 (Fig. 6g, h), were all obviously increased in MCM cultured fibroblast. All these effects were reversed by CGX1321. However, CGX1321’s effect was abolished when WNT3a and WNT5a were added in MCM (Supplementary Fig. S6a-c).

CGX1321 attenuated cardiac fibroblast activation by blocking WNT signaling pathways. a A schematic representation of in-vitro experimental model for HFpEF induced cardiac fibroblast activation. b, c Representative images of the activation of cardiac fibroblast in cultured NRVFs, indicated by α-SMA (b) and fibronectin (c) staining (n = 6, scale bar=50 μm). d Representative images of nucleus p-c-Jun, NFATc3 and β-catenin protein expression in NRVFs (d1), densitometric analysis of different protein band in different experimental groups (d2–d4), c-Jun and Histone H3 were used as controls (n = 6). e The mRNA expression of Axin 2 in different groups (n = 6). f Representative images of protein expression of TGF-β and α-SMA in NRVFs (f1), densitometric analysis of different protein band in different experimental groups (f2, f3) (GAPDH was used as control, n = 6). g, h The mRNA expression of Col1a1 and Col3a1 in NRVFs (n = 6). n.s. not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Knockdown of porcupine with siRNA led to inhibitory effect similar to CGX1321 on nuclear translocation of NFATc3 and β-catenin, as well as on cardiomyocyte hypertrophy and fibroblast activation (Supplementary Fig. S7a–d). These data suggested that CGX1321 by targeting porcupine exerted therapeutic effect on cardiac hypertrophy and fibrosis.
CGX1321 did not affect the expression and phosphorylation of cytoskeletal composition and Ca2+-handling proteins
In addition to cardiac fibrosis and hypertrophy, the expression and phosphorylation of cytoskeletal composition and Ca2+-handling proteins are also involved in the pathogenesis of HFpEF [35, 36]. We investigated if these alterations were also associated with CGX1321’s beneficial effects. We found that the total phospholamban (PLN) and TnI were comparable among different groups. While the sarcoplasmic reticulum Ca2+ ATPase (Serca2a) and p-PLN (Ser16/Thr17) were significantly decreased in HFpEF mouse hearts compared with control. On the other hand, the sodium/calcium exchanger (NCX) was increased in HFpEF mice hearts. All these changes were unaffected by CGX1321 treatment (Supplementary Fig. S8a). These indicated that the therapeutic effect of CGX1321 might not through regulating cytoskeletal and Ca2+-handling proteins.
Finally, we detected the WNT signaling pathways in control, TAC and two HFpEF models (UNX/DOCA and “two hit”), and found that canonical and non-canonical WNT signaling pathways were both activated in TAC and HFpEF models. In HFpEF the phosphorylation of c-Jun and nuclear translocation of β-catenin and NFATc3 were more obvious (Supplementary Fig. S9a1–a4). These results indicated that targeting WNT might be a new strategy to treat both HFrEF and HFpEF.
Discussion
By using two HFpEF mouse models, the present study demonstrated that WNT signaling was activated during the development of HFpEF. The blockade of the WNT pathways by the novel WNT porcupine inhibitor CGX1321 exerted a therapeutic effect against HFpEF. HFpEF was a complex clinical syndrome with a large pathophysiological heterogeneity. Due to the wide range of comorbidities and clinical presentations, the potential underlying etiology of HFpEF was diverse. We performed the experiments in two different HFpEF mouse models, UNX/DOCA and “two-hit”, with diverse etiologies and pathophysiologic features. We found that in both models, CGX1321 reduced the two cardinal pathologies-fibrosis and hypertrophy. Thus, the WNT signaling involvement and therapeutic effect of porcupine inhibitors CGX1321 might be generally applicable to HFpEF with various etiologies and pathophysiologic features.
Accumulating evidence in recent years showed that WNT signaling pathways played an important role in cardiac remodeling, especially cardiac hypertrophy and fibrosis [18, 29, 37, 38]. The activation of WNT signaling pathways increased the nuclear translocation of β-catenin (canonical WNT-β-catenin signal pathways), phosphorylation of c-Jun (non-canonical WNT-PCP signaling pathways), and the nuclear translocation of NFATc3 (non-canonical WNT-Ca2+ signaling pathways) [39]. The β-catenin, phosphorylated c-Jun and NFATc3 increased the expression of hypertrophy and fibrosis associated genes in cardiac remolding [21, 33]. Both canonical and non-canonical WNT signaling pathways were involved in the pathological progress of cardiac hypertrophy and fibrosis [18]. Thus, targeting canonical or non-canonical WNT signaling pathway alone might be not well enough to attenuate these pathological processes. Instead inhibiting both of them may obtain better therapeutic effect.
Porcupine, an O-acyltransferase located in the membrane of the endoplasmic reticulum, was necessary for the processing and secretion of WNT ligands [32]. CGX1321 was a novel porcupine inhibitor discovered by Guangzhou Curegenix Co. Ltd. In previously studies, we showed that CGX1321 could block the secretion of WNT ligands, inhibited both canonical and non-canonical WNT pathways in hearts, and exerted a therapeutic effect on cardiac hypertrophy and myocardial infarction [21, 22]. In present study, we found that the administration of CGX1321 on two HFpEF mouse models alleviated cardiac hypertrophy and fibrosis, thereby improving cardiac diastolic function. Further, in vitro HFpEF model found that either porcupine knocked down by siRNA or inhibited by CGX1321 exerted the same anti-hypertrophy and anti-fibrosis effects. Additionally, CGX1321 have accomplished the pre-clinical test, our unpublished data have also demonstrated the safety of CGX1321 in over seventy cancer patients treated in phase 1 and 1b clinical trials (clinical trial identifier: NCT02675946). Thus, the translational potential of porcupine inhibitors in treating HFpEF is encouraging.
Due to the complexity of pathogenesis of HFpEF, traditional medicine showed limited effect on it [40]. In previous study, we confirmed the therapeutic effect of CGX1321 in TAC mouse models which could be regarded as a hypertrophy induced HFrEF model. In present study, we also found that the activation of WNT signaling pathways in HFpEF and CGX1321 ameliorated the cardiac hypertrophy and fibrosis, and improved the diastolic function in two HFpEF mouse models. Further, we detected the WNT signaling in control, TAC model, UNX/DOCA and “two hit” and found that the activation of WNT signaling pathways was a common feature of HFrEF and HFpEF. Moreover, the activation of canonical and non-canonical WNT signaling pathways was more obviousl in HFpEF which was evidenced by the greater degree of phosphorylation of Jun and nuclear translocation of β-catenin and NFATc3. The recent proteomic analysis of human also confirmed these results [17]. All these findings indicated that the WNT signaling pathways might be a potential target to treat both HFrEF and HFpEF.
In conclusion, our study demonstrated that activation of WNT signaling played a key role in the pathogenesis of HFpEF. Blocking WNT signaling by a clinical-stage porcupine inhibitor CGX1321 exerted a therapeutic effect against the diastolic dysfunction of HFpEF by reducing cardiac hypertrophy and fibrosis (Fig. 7). Targeting porcupine to block the activation of WNT in treating HFpEF was mechanically different with the existing medicine such as SGLT2is and ARNIs. Thus, CGX1321 is expected to bring additional/synergistic efficacy to the clinical treatment for HFpEF.

WNT signaling blockade by novel porcupine inhibitor CGX1321 alleviates HFpEF. In the pathological progress of HFpEF, both canonical and non-canonical WNT signaling pathways were activated and involved in the cardiac hypertrophy and fibrosis. By targeting porcupine, CGX1321 inhibited the secretion of all WNT ligands and blocked the activation of both canonical and non-canonical WNT signaling pathways in HFpEF. Blocking WNT signaling pathways by CGX1321 exerted a therapeutic effect against the diastolic dysfunction of HFpEF by reducing cardiac hypertrophy and fibrosis.
References
Gladden JD, Chaanine AH, Redfield MM. Heart failure with preserved ejection fraction. Annu Rev Med. 2018;69:65–79.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596.
Pfeffer MA, Pitt B, McKinlay SM. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;371:181–2.
Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–67.
Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362:777–81.
Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451–61.
Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609–20.
Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, et al. Research priorities for heart failure with preserved ejection fraction: national heart, lung, and blood institute working group summary. Circulation. 2020;141:1001–26.
Cunningham J, Claggett B, O’Meara E, Prescott M, Pfeffer M, Shah S, et al. Effect of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFpEF. J Am Coll Cardiol. 2020;76:503–14.
Lam CSP, Voors AA, de Boer RA, Solomon SD, van Veldhuisen DJ. Heart failure with preserved ejection fraction: from mechanisms to therapies. Eur Heart J. 2018;39:2780–92.
Borlaug BA, Kane GC, Melenovsky V, Olson TP. Abnormal right ventricular-pulmonary artery coupling with exercise in heart failure with preserved ejection fraction. Eur Heart J. 2016;37:3293–302.
Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–9.
Díez J, Querejeta R, López B, González A, Larman M, Martínez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–7.
Goswami VG, Patel BD. Recent updates on Wnt signaling modulators: a patent review (2014-2020). Expert Opin Ther Pat. 2021;31:1009–43.
Bastakoty D, Saraswati S, Joshi P, Atkinson J, Feoktistov I, Liu J, et al. Temporary, systemic inhibition of the WNT/β-Catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen Med. 2016;2:2472–81.
Zhao Z, Liu H, Li Y, Tian J, Deng S. Wnt-C59 attenuates pressure overload-induced cardiac hypertrophy via interruption of Wnt pathway. Med Sci Monit. 2020;26:e923025.
Adamo L, Yu J, Rocha-Resende C, Javaheri A, Head RD, Mann DL. Proteomic signatures of heart failure in relation to left ventricular ejection fraction. J Am Coll Cardiol. 2020;76:1982–94.
Tao H, Yang JJ, Shi KH, Li J. Wnt signaling pathway in cardiac fibrosis: New insights and directions. Metabolism. 2016;65:30–40.
Yu J, Liao PJ, Xu W, Jones JR, Everman DB, Flanagan-Steet H, et al. Structural model of human PORCN illuminates disease-associated variants and drug-binding sites. J Cell Sci. 2021;134:624–36.
Shah K, Panchal S, Patel B. Porcupine inhibitors: Novel and emerging anti-cancer therapeutics targeting the Wnt signaling pathway. Pharmacol Res. 2021;167:105532.
Jiang J, Lan C, Li L, Yang D, Xia X, Liao Q, et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3459–67.
Yang D, Fu W, Li L, Xia X, Liao Q, Yue R, et al. Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin Sci (Lond). 2017;131:2919–32.
Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519–28.
Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–6.
Schnelle M, Catibog N, Zhang M, Nabeebaccus AA, Anderson G, Richards DA, et al. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol. 2018;114:20–8.
Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by beta-hydroxybutyrate mitigates HFpEF. Circ Res. 2021;128:232–45.
Li L, Fu W, Gong X, Chen Z, Tang L, Yang D, et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J. 2021;42:1415–30.
Działo E, Rudnik M, Koning RI, Czepiel M, Tkacz K, Baj-Krzyworzeka M, et al. WNT3a and WNT5a transported by exosomes activate WNT signaling pathways in human cardiac fibroblasts. Int J Mol Sci. 2019;20:1436–51.
Hermans KC, Blankesteijn WM. Wnt signaling in cardiac disease. Compr Physiol. 2015;5:1183–209.
Withaar C, Lam CSP, Schiattarella GG, de Boer RA, Meems LMG. Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J. 2021;42:4420–30.
Xiang FL, Fang M, Yutzey KE. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8:712.
Stylianidis V, Hermans KCM, Blankesteijn WM. Wnt signaling in cardiac remodeling and heart failure. Handb Exp Pharmacol. 2017;243:371–93.
Seo HH, Lee S, Lee CY, Lee J, Shin S, Song BW, et al. Multipoint targeting of TGF-beta/Wnt transactivation circuit with microRNA 384-5p for cardiac fibrosis. Cell Death Differ. 2019;26:1107–23.
Oatmen KE, Cull E, Spinale FG. Heart failure as interstitial cancer: emergence of a malignant fibroblast phenotype. Nat Rev Cardiol. 2020;17:523–31.
van der Velden J, Stienen GJM. Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol Rev. 2019;99:381–426.
Hegemann N, Primessnig U, Bode D, Wakula P, Beindorff N, Klopfleisch R, et al. Right-ventricular dysfunction in HFpEF is linked to altered cardiomyocyte Ca2+ homeostasis and myofilament sensitivity. ESC Heart Fail. 2021;8:3130–44.
Malekar P, Hagenmueller M, Anyanwu A, Buss S, Streit MR, Weiss CS, et al. Wnt signaling is critical for maladaptive cardiac hypertrophy and accelerates myocardial remodeling. Hypertension. 2010;55:939–45.
Bogdanova E, Beresneva O, Galkina O, Zubina I, Ivanova G, Parastaeva M, et al. Myocardial hypertrophy and fibrosis are associated with cardiomyocyte beta-catenin and TRPC6/Calcineurin/NFAT signaling in spontaneously hypertensive rats with 5/6 nephrectomy. Int J Mol Sci. 2021;22:4645–61.
Fu WB, Wang WE, Zeng CY. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharmacol Sin. 2019;40:9–12.
Gorter TM, van Veldhuisen DJ, Bauersachs J, Borlaug BA, Celutkiene J, Coats AJS, et al. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2018;20:16–37.
Acknowledgements
We thank for the Guangzhou Curegenix, Ltd. Co for providing CGX1321 and technical assistance. This work was supported by grants from the National Natural Science Foundation of China (81922005, U20A20344), National Key Research & Development Program of China (2018YFA0107403) and Chongqing Natural Science Foundation (cstc2020jcyj-jqX0016).
Author information
Authors and Affiliations
Contributions
WEW, CYZ and SZMA designed, supervised the project and edited the manuscript. HW, LXT and XMW performed most experiments. HW and LPL wrote and revised the manuscript. XKC, YJH, DZY, JLS, YS, ZSZ, LW, BJL conducted experiments. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that there are no competing interests associated with the manuscript. ZZ, LW and SMA are employees of Guangzhou Curegenix, Co. Ltd. and Curegenix, Inc.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, H., Tang, Lx., Wang, Xm. et al. Porcupine inhibitor CGX1321 alleviates heart failure with preserved ejection fraction in mice by blocking WNT signaling. Acta Pharmacol Sin 44, 1149–1160 (2023). https://doi.org/10.1038/s41401-022-01025-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01025-y
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Animal models of heart failure with preserved ejection fraction (HFpEF): from metabolic pathobiology to drug discovery
Acta Pharmacologica Sinica (2024)