
Abstract
Sulfur mustard (SM) is a highly toxic chemical warfare agent that causes acute lung injury (ALI) and/or acute respiratory distress syndrome (ARDS). There are no effective therapeutic treatments or antidotes available currently to counteract its toxic effects. Our previous study shows that bone marrow-derived mesenchymal stromal cells (BMSCs) could exert therapeutic effects against SM-induced lung injury. In this study, we explored the therapeutic potential of BMSC-derived exosomes (BMSC-Exs) against ALI and the underlying mechanisms. ALI was induced in mice by injection of SM (30 mg/kg, sc) at their medial and dorsal surfaces. BMSC-Exs (20 μg/kg in 200 μL PBS, iv) were injected for a 5-day period after SM exposure. We showed that BMSC-Exs administration caused a protective effect against pulmonary edema. Using a lung epithelial cell barrier model, BMSC-Exs (10, 20, 40 μg) dose-dependently inhibited SM-induced cell apoptosis and promoted the recovery of epithelial barrier function by facilitating the expression and relocalization of junction proteins (E-cadherin, claudin-1, occludin, and ZO-1). We further demonstrated that BMSC-Exs protected against apoptosis and promoted the restoration of barrier function against SM through upregulating G protein-coupled receptor family C group 5 type A (GPRC5A), a retinoic acid target gene predominately expressed in the epithelial cells of the lung. Knockdown of GPRC5A reduced the antiapoptotic and barrier regeneration abilities of BMSC-Exs and diminished their therapeutic effects in vitro and in vivo. BMSC-Exs-caused upregulation of GPRC5A promoted the expression of Bcl-2 and junction proteins via regulating the YAP pathway. In summary, BMSC-Exs treatment exerts protective effects against SM-induced ALI by promoting alveolar epithelial barrier repair and may be an alternative approach to stem cell-based therapy.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) is a serious and potentially fatal inflammatory disease characterized by damage to the alveolar epithelial–endothelial barrier that results in the accumulation of protein-rich edematous fluid in the alveoli, which in turn leads to acute respiratory failure [1]. ALI can be caused by various injurious stimuli, including sepsis, pneumonia (bacterial or viral), smoke inhalation, major trauma, and mechanical ventilation with high tidal volume, as well as exposure to and/or inhalation of industrial toxic compounds, such as phosgene, sulfur mustard, and various oxides of nitrogen (including nitric oxide, nitrogen dioxide, etc.) [2, 3]. Sulfur mustard (2,2-dichlorodiethyl sulfide, SM) is a highly toxic chemical warfare agent that can cause damage to the respiratory tract, eyes, skin, and multiple organ systems; however, most postexposure mortality and morbidity associated with SM are due to pulmonary toxicity [4]. Because there are currently no ideal treatments for SM-induced acute lung injury, improved therapies are needed to help prevent additional pulmonary damage [5, 6].
Increasing evidence has shown that mesenchymal stem cell (MSC)-based therapy is a promising new therapeutic approach for treating ALI [7,8,9]. We previously found that BMSCs exert protective effects against SM-induced ALI by alleviating inflammation and promoting tissue repair [10]. Although advances have been made in MSC transplantation to treat lung injury, concerns remain regarding tumor formation and long-term safety. New treatments that do not require the administration of live cells are therefore an increasingly attractive area of investigation [11]. There is growing evidence suggesting that the protective paracrine effects exerted by MSCs are largely mediated by the secretion of extracellular vesicles (EVs) [12, 13]. Since MSC-EVs may have protective and reparative properties similar to those of their source cells, their therapeutic potential for treating lung injuries and diseases is being actively explored.
Exosomes are EVs that are secreted via the fusion of multivesicular endosomes with the cell membrane and have been shown to act as regulators of cell-cell communication. Exosomes contain functional proteins, mRNAs, and microRNAs that can be transferred between cells and affect protein expression in the target cell [14, 15]. A number of studies have suggested that MSC-derived exosomes (MSC-Exs) ameliorate the causal factors of ALI and other inflammatory lung diseases in preclinical contexts both in vivo and ex vivo [16, 17]. However, the key factors in MSC-Exs and the mechanism(s) underlying their therapeutic effects are unclear and need to be examined. ALI is mainly characterized by the destruction of the alveolar and vascular membrane barrier and inflammatory response. Repairing barrier function to decrease alveolar epithelial–endothelial membrane permeability has been considered a treatment strategy for ALI [12]. Previous studies have suggested that transplanted MSCs restore barrier function [18]; however, the molecular mechanism by which this repair occurs is unclear.
In this study, we assessed the use of BMSC-derived exosomes (BMSC-Exs) to treat SM-induced ALI and investigated the mechanism of action. The antiapoptotic effects of BMSC-Exs and their regulatory effects on the expression and distribution of cell junction proteins in SM-induced lung injury models in vitro and in vivo were also examined. We found that BMSC-Exs promoted the repair of SM-induced lung epithelial tight junction dysfunction and inhibited apoptosis by regulating the GPRC5A–YAP axis. Our results provide an improved understanding of the use of BMSC-Exs to treat ALI.
Materials and methods
Cells and mice
Murine MLE-12 cells (ATCC, CRL-2110) were purchased from the American Type Culture Collection and cultured in DMEM/F12 medium (HyClone) containing 10% fetal bovine serum (FBS) (Invitrogen), penicillin, and streptomycin (Gibco) at 37 °C with 5% CO2. Human lung fibroblasts (HFL-1 cells) were purchased from the Chinese Academy of Sciences and cultured in minimal essential medium alpha (MEM-α) containing 10% FBS (Invitrogen) and penicillin/streptomycin (Gibco) at 37 °C with 5% CO2. Mycoplasma contamination tests were conducted on all cells.
Male ICR mice (25–30 g) were purchased from Sino-British SIPPR/BK Lab Animal, Ltd. (Shanghai, China), and GPRC5A-knockout mice (25–30 g) were obtained from the Model Animal Research Center of Nanjing University. All animals were maintained in a pathogen-free Animal Experimental Center and fed for 7 days before the experiments. All animals used in this study were treated according to the Guide for the Care and Use of Laboratory Animals, and the study procedures were approved by the Institutional Animal Care and Use Committee.
Exosome isolation and characterization
Exosomes were isolated and purified by ultracentrifugation as previously described [19]. FBS (Invitrogen) that was used for cell culture was first ultracentrifuged at 100,000×g for 16 h at 4 °C to remove bovine exosomes and protein aggregates. BMSCs and HFL-1 cells at passages 3–8 were cultured in serum-free medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C with 5% CO2 for 48 h before the medium was harvested. The conditioned medium was centrifuged at 2000×g for 10 min at 4 °C to remove cellular debris and then passed through a 0.22 μm filter (Millipore). The cleared supernatant was transferred to an ultracentrifuge tube underlaid with a 30% sucrose/D2O cushion (1.21 g/cm3) ultracentrifuged at 100,000×g for 70 min at 4 °C. Exosomes were collected from the bottom of the tube and concentrated using a 100 kDa molecular weight cutoff ultrafiltration membrane (Millipore) at 1000×g for 30 min at 4 °C. The final pellet was resuspended in 200 μL of PBS and stored at −80 °C until further use. The protein content was determined using a BCA protein assay kit (Pierce, ThermoFisher). The morphology of the extracted exosomes was observed using transmission electron microscopy (Tecnai G2 Spirit FEI, Philips). The size distribution was analyzed using a NanoSight LM10 system equipped with fast video capture and particle-tracking software.
Acute lung injury mouse model and treatment
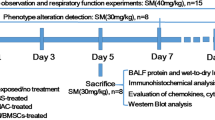
SM was diluted to the desired concentration (30 or 40 mg/kg) in a propanediol solution as described previously (Sigma, St. Louis, MO, USA) [10]. There were five randomized groups in this study: (i) the control group, (ii) the SM group, (iii) the SM + NAC group, (iv) the SM + BMSC-Ex group, and (v) the SM + HFL-Ex group. In the control group, the appropriate propanediol solution was injected into the mice. In the SM-exposed group, SM (30 mg/kg) was subcutaneously injected into the medial and dorsal surfaces of the mouse skin as previously described. Since N-acetylcysteine (NAC) has been reported to be the lead candidate for the treatment of SM-induced pulmonary toxicity, it was chosen as a positive control in our study [20]. The mice in the SM + NAC group were administered the same treatment as mice in the SM group and were then treated with NAC by gavage (200 mg/kg) once per day. Mice in the SM + BMSC-Ex and SM + HFL-Ex groups were treated with BMSC-Exs/HFL-Exs (20 mg/kg, resuspended in 200 μL of PBS) via tail vein injection on the first and third days after SM exposure. The mice were either evaluated or sacrificed at the indicated times after SM exposure, and lung tissues were collected for lung injury assessment and immunohistochemical and immunofluorescence analyses.
Histopathological staining
Lungs were fixed in 4% paraformaldehyde for 24 h. After the samples were dehydrated, cleared, immersed in wax, and embedded, the lung sections (4 μm) were stained with hematoxylin–eosin (H&E) for histological examination. Histological evaluation was performed by a pathologist who was blinded to the study groups [21]. The severity of lung injury was quantified and blindly assessed by an experienced pathologist based on images from 10 randomly selected high-power fields. For each section, edema, alveolar and interstitial inflammation, alveolar and interstitial hemorrhage, atelectasis, necrosis, and hyaline membrane formation were each scored using a 0- to 4-point scale according to the scoring system as described previously.
Measurement of BALF protein content and the wet-to-dry lung weight ratio
The thoracic cavity was opened after anesthetic administration, and the trachea was isolated and fixed with forceps. A tube was then introduced into the trachea and secured using a 2/0 suture. The right lung was lavaged three times with 800 µL of PBS, and the lavage fluid was collected in centrifuge tubes. The collected BALF was immediately pooled and centrifuged (1500×g, 15 min). Total protein in the cell-free BALF was quantified using a BCA protein assay kit (Thermo Scientific) according to the manufacturer’s instructions.
To determine the wet-to-dry lung weight ratio, the left lung was excised, and the wet weight was measured. The lung was then placed into an oven at 72 °C for 72 h to obtain the dry weight.
Immunohistochemical analysis
Lung tissue sections were deparaffinized, dehydrated, and subjected to antigen retrieval by microwave treatment in boiling 0.01 M citrate buffer (pH 6.0) for 20 min. Endogenous peroxidase activity was blocked by treatment with 3% hydrogen peroxide for 10 min. The sections were incubated with goat serum to block nonspecific binding, followed by overnight incubation at 4 °C with 50 µL of anti-Ki-67 antibody (ab15580, Abcam). The sections were then incubated with a secondary antibody for 1 h and horseradish peroxidase (HRP)-streptavidin for 30 min at 37 °C, and DAB was the substrate. The proliferative index was defined as the number of positively stained nuclei.
For immunofluorescence staining of junction proteins, the sections were blocked and then incubated with primary antibodies against occludin (40-4700, Invitrogen), claudin-1 (71-7800, Invitrogen), or E-cadherin (#3195, CST) for 1 h at room temperature, after which the sections were washed three times using PBS, incubated with Alexa Fluor 488- or 555-conjugated secondary antibodies for 30 min at room temperature, washed, and mounted with 4′,6-diamidino-2-phenyl-indole (DAPI, Roche) for 15 min to label the nuclei. The sections were examined with an automated microscope (Lionheart FX, BioTek).
Transwell monolayer preparation and permeability measurement
MLE-12 cells were grown as monolayers in 6.5-mm-diameter transwell filter inserts with a pore size of 3.0 µm (Corning Life Sciences, Lowell, MA, USA) and placed in 24-well tissue culture plates. The cells were cultured for 96 h, and then the consistency, confluence, and integrity of the monolayer were examined as previously described [22,23,24]. Transepithelial resistance (TER) was measured as previously described using an epithelial voltohmmeter under open-circuit conditions (Millicell ERS-2, Millipore) to determine the paracellular permeability of the epithelial cells. The average TER was 250 Ω·cm2 after subtraction of a blank that included the filter and fluid resistance. To facilitate comparisons between experiments, the TERs of all monolayers were normalized to those of control monolayers in the same experiment.
Cell viability assay
Cell viability was determined using the Cell Counting Kit-8 (CCK-8, CK04, Dojindo Laboratories) assay according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates at 4 × 104 cells/cm2, incubated with SM (300 μM) for 30 min, and then treated with BMSC-Exs or NAC for 24 h. The CCK-8 assay was performed by adding 110 μL of fresh medium containing 10 μL of CCK-8 solution to the cells and incubating them for 1–4 h at 37 °C. The absorbance of the solution at 450 nm was measured using a microplate reader (BioTek).
Apoptosis analysis
SM-treated MLE-12 cells were incubated with NAC or BMSC-Exs for 24 h. The treated cells were then harvested and subjected to annexin V/propidium iodide (PI) double-staining (BD) and analyzed by flow cytometry (Beckman CytoFlex) according to the manufacturer’s protocol as previously described [25]. Briefly, both floating and attached cells were pooled, washed, and resuspended in binding buffer. Fluorescein isothiocyanate (FITC)-conjugated annexin V and PI were then added to the suspended cells at a ratio of 1:60, and the mixture was incubated in the dark for 15 min. Flow cytometric analysis was then performed on the gated cells.
JC-1 and Hoechst 33342 staining
SM-treated MLE-12 cells were incubated with NAC or BMSC-Exs for 24 h and then stained with JC-1 (Beyotime) or Hoechst 33342 (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, the medium was replaced with 1 mL of fresh medium containing JC-1 (5 mg/mL) or Hoechst 33342 (50 mg/mL) and incubated at 37 °C for 20 min. After being washed twice with PBS, the cells were examined under an automated microscope (Lionheart FX, BioTek). Images were analyzed using Gene 5 software. Mitochondrial membrane potential depolarization was indicated by an increase in the green/red fluorescence intensity ratio, and apoptotic cells were identified by nuclear condensation and/or fragmentation.
Quantitative real-time PCR
Total RNA was extracted from MLE-12 cells with TRIzol reagent (Takara), and cDNA was synthesized using PrimeScript RT Master Mix (TaKaRa) according to the manufacturer’s instructions. Quantitative real-time PCR (RT-qPCR) was used to compare the relative expression levels of target genes using SYBR Premix Ex Taq (TaKaRa). All reactions were performed in triplicate, and the mRNA level of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous reference control. The primer sequences used in this study were as follows:
E-cadherin forward primer: 5′-CAGGTCTCCTCATGGCTTTGC-3′, reverse primer: 5′-CTTCCGAAAAGAAGGCTGTCC-3′; occludin forward primer: 5′-ACTGGGTCAGGGAATATCCA-3′, reverse primer: 5′-TCAGCAGCAGCCATGTACTC-3′; claudin-1 forward primer: 5′-TCAGGTCTGGCGACATTAGT-3′, reverse primer: 5′-TCAGCAGCAGCCATGTACTC-3′; ZO-1 forward primer: 5′-ACTCCCACTTCCCCAAAAAC-3′, reverse primer: 5′-CCACAGCTGAAGGACTCACA-3′; mouse GAPDH forward primer: 5′-AACATCTACAAGCCCAACAACAAGG-3′, reverse primer: 5′-GGTTCTGCAATCACATCTTCAAAGTC-3′.
The data were analyzed relative to the controls. All assays were performed on an ABI 7300 system (Applied Biosystems).
Confocal microscopy
Cells were seeded onto coverslips or Transwell filter inserts, incubated with SM (300 μM) for 30 min, and then treated with BMSC-Exs or NAC for 24 h. The cells were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 at room temperature, blocked in 5% BSA, and incubated with primary antibodies against ZO-1 (61-7300, Invitrogen), occludin (40-4700, Invitrogen), claudin-1 (71-7800, Invitrogen), E-cadherin (#3195, CST) and YAP (#14074, CST) at the indicated dilutions at room temperature for 1 h. After three washes with PBS, the cells were incubated with Alexa Fluor 488- or 555-conjugated secondary antibodies for 1 h. The cells were washed three times with PBS, and the nuclei were stained with DAPI for 10 min. Images were obtained using a confocal laser scanning microscope (Zeiss LSM710 Meta, Carl Zeiss) and analyzed using ZEN Light Edition software (Carl Zeiss).
Transient siRNA transfection
For MLE-12 cell transfection, FECT reagent (Dharmacon) was used to transfect GPRC5A siRNA (L-057339-00-0010, Dharmacon) or nontargeting control siRNA (D-001810-10-05, Dharmacon) according to the manufacturer’s instructions. Briefly, cells were seeded in 24-well plates or Transwell filter inserts for 12 h, transfected with 100 nM GPRC5A siRNA/nontargeting (NT) siRNA mixed with Opti-MEM (Invitrogen) and incubated at 37 °C for 72 h to ensure effective gene knockdown. Each siRNA transfection was performed in triplicate. Knockdown levels were monitored by Western blotting at 72 h post transfection.
Western blotting
Western blotting was performed as previously described [25]. Cells were washed with cold PBS and then lysed in radioimmunoprecipitation assay (RIPA) buffer (Pierce). Total protein concentrations were measured by BCA protein assay kits, and equal amounts of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk in 1× Tris-buffered saline (TBS) with 1% Tween-20 (SCR) for 2 h at room temperature (RT). Then, the membranes were incubated with the indicated primary antibodies against cleaved PARP (#94885, CST), cleaved caspase-3 (#9661, CST), cleaved caspase-9 (#9509, CST), Bcl-2 (#3498, CST), ZO-1 (61-7300, Invitrogen), occludin (40-4700, Invitrogen), claudin-1 (71-7800, Invitrogen), E-cadherin (#3195, CST), YAP (#14074, CST), phospho-YAP (#13008, CST), GPRC5A (#12968, CST), β-tubulin (#2146, CST) and GAPDH (#2118, CST) at 4 °C for 16 h. After being rinsed, each membrane was incubated with horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were detected with ECL Plus enhanced chemiluminescence Western blotting detection reagents (PerkinElmer Life Sciences). The protein bands were scanned and quantified based on optical densities using ImageJ software (version 1.34s) and normalized to GAPDH.
Statistical analysis
All statistical analyses were performed using SPSS 21.0 software. Experimental data are reported as the mean ± standard deviation. Significance between two groups was determined with the two-tailed Student’s t-test, and significance between multiple groups was determined by ANOVA. Statistical significance of P < 0.05 is indicated by one (*) asterisk, and significance of P < 0.01 and P < 0.001 is indicated by two (**) and three (***) asterisks, respectively, in the figure.
Results
BMSC-Exs ameliorated SM-induced acute lung injury
To assess the therapeutic potential of BMSC-Exs, mice were given 20 mg/kg exosomes by tail vein administration 24 h after injection of SM, and detailed information regarding their general health was recorded for 2 weeks. Beginning on day 3, the SM-exposed mice suffered significant loss of weight and appetite, a gradual increase in mortality was observed until the fifth day, and no mice died after the eighth day. The administration of BMSC-Exs alleviated these SM-induced effects. The general health and survival rates were significantly improved by BMSC-Ex injection (Fig. 1a and Supplementary Fig. S2a). H&E staining confirmed the presence of epithelial necrosis, massive exudate and inflammatory cell infiltration, pulmonary edema, substantial thickening of the alveolar septum and hemorrhage after SM administration, while BMSC-Ex administration significantly decreased tissue lesions and perivascular infiltrates, and the observation of mild bleeding and edema indicated a significant protective effect of BMSC-Exs (Fig. 1b and Supplementary Fig. S2b). In addition, the BALF (bronchoalveolar lavage fluid) protein levels and W/D (wet-to-dry weight) ratio, which are important indicators of exudation associated with pulmonary edema, decreased at 120 h postinjection in animals that were administered BMSC-Exs, confirming that BMSC-Exs reduced the acute, extensive lung injury caused by SM (Fig. 1c, d and Supplementary Fig. S2c, d).

a Survival curves of SM-injured mice treated with BMSC-Exs (20 mg/kg) by tail vein injection. BMSC-Ex administration protected recipient animals from lung failure. (n = 15, **P < 0.01 vs the SM group). b Representative photographs of H&E-stained lung sections from SM-exposed mice after NAC or BMSC-Ex treatment. Original magnification: ×200. The BMSC-Ex group exhibited obvious protection against SM-induced lung tissue damage, as revealed by the relatively normal alveolar cavity, mucosal epithelium and airways and minimal inflammatory cell infiltration, and septal thickening in the mice (left panel). Comparison of pathological lung injury scores in SM-exposed mice. ** represents P < 0.01 compared with the SM group (right panel). c, d Comparisons of BALF protein levels (c) and wet-to-dry lung weight ratios (d) in SM-exposed mice. * represents P < 0.05, ** represents P < 0.01 compared with the SM group.
BMSC-Exs reduced apoptosis in SM-injured lung epithelial cells
SM-induced cytotoxicity was analyzed in the MLE-12 cell line, which is a murine lung epithelial cell line that has been widely used to study acute lung injury and pulmonary fibrosis [24, 26]. SM-induced cytotoxicity was assessed by measuring cell viability using a CCK-8 assay after BMSC-Ex or NAC treatment. As shown in Fig. 2a, viability was reduced in SM-injured cells and increased in BMSC-Ex-treated cells in a dose-dependent manner. The SM-induced decrease in viability was also significantly alleviated in NAC-treated cells. These results indicate that BMSC-Exs alleviated SM-induced cytotoxicity and promoted recovery in MLE-12 cells. The decrease in cell viability may be associated with apoptosis or necrosis. We found that SM exposure significantly induced apoptosis and that at 24 h after treatment, the number of apoptotic cells decreased in the BMSC-Ex group compared with the SM group (Fig. 2b and Supplementary Fig. S3a). Hoechst 33342 staining confirmed that there were fewer apoptotic cells in the BMSC-Ex group than in the SM group (Fig. 2c). We previously reported that SM accumulates in the mitochondria of living cells and that mitochondrial injury can promote apoptosis [27]. Thus, JC-1 staining was performed to measure changes in mitochondrial membrane potential in MLE-12 cells treated with or without BMSC-Exs. As shown in Fig. 2d, the ratio of green to red fluorescence was significantly decreased in cells treated with BMSC-Exs, indicating a reversal of SM-induced perturbation of mitochondrial membrane potential. Apoptosis is usually associated with caspase activation, and we found that in the BMSC-Ex group, Bcl-2 levels increased, while cleaved PARP, cleaved caspase-3, and cleaved caspase-9 levels decreased, compared to those in the SM group (Fig. 2e). To illustrate the effects of BMSC-Exs, human lung fibroblast (HFL-1)-derived exosomes (HFL-Exs) were used as controls. Compared with those in the SM group, HFL-Ex treatment had little effect on cell viability and apoptosis, indicating that HFL-Exs exerted no significant protective effects (Supplementary Fig. S3). Taken together, these results indicate that BMSC-Ex treatment reduced SM-induced lung epithelial cell apoptosis.

MLE-12 cells were left untreated (Ctrl) or treated with SM (300 μM) for 30 min and then treated with BMSC-Exs (10, 20, or 40 μg) or NAC (100 μM) for 24 h. a Cell viability was measured by CCK-8 assays. b The induction of apoptosis in MLE-12 cells was determined by Annexin V/PI flow cytometry. The quantification of apoptotic cells is presented as the percent of total cells. c Hoechst 33342 staining of apoptotic SM-injured MLE-12 cells cultured with PBS, NAC, or BMSC-Exs (20 μg) for 24 h. Original magnification: ×200. d Mitochondrial membrane potentials in the PBS, NAC, or BMSC-Ex (20 μg) groups. Increases in green fluorescence indicate perturbed membrane potentials. Original magnification: ×200. e Western blot analysis of apoptosis-related proteins in MLE-12 cells treated with PBS, NAC, or BMSC-Exs (20 μg) for 24 h. The levels of Bcl-2, cleaved PARP, cleaved caspase-3, and cleaved caspase-9 were quantitated by densitometric analysis using ImageJ software and normalized to GAPDH. The data represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001.
BMSC-Exs alleviated SM-induced epithelial barrier damage
ALI is characterized by damage to the barrier functions of the alveolar epithelium and vascular endothelial cells, resulting in pulmonary edema [2, 12]. The pathophysiological changes in SM-induced ALI resulted in a decline in alveolar epithelial barrier function (Fig. 1b). To determine whether SM directly affects alveolar epithelial cells, MLE-12 monolayers were established in vitro. As shown in Fig. 3a, SM exposure caused TER, which is a sensitive measure of epithelial barrier function, to fall by 51% in monolayers in the SM group compared with the control group, indicating that barrier function was disrupted by SM. Both NAC and BMSC-Ex treatment inhibited the SM-induced reduction in TER (Fig. 3a). The TER increased by 30% in the BMSC-Ex-treated group compared with the SM group, showing that BMSC-Exs promoted barrier function repair. Adherens and tight junctions are important structures that maintain the alveolar membrane barrier and the permeability of pulmonary epithelial cells [28]; thus, the distribution of E-cadherin, claudin-1, occludin, and ZO-1 was examined. As shown in Fig. 3b, SM significantly induced the endocytosis of E-cadherin, claudin-1, and occludin, leading to tight junction protein damage or discontinuous distribution. Compared with the SM and NAC, BMSC-Ex treatment reduced barrier loss and the internalization of E-cadherin, claudin-1, and occludin (Fig. 3b). Combined with the in vivo data, these in vitro data indicate that BMSC-Exs alleviate SM-induced epithelial barrier damage.

MLE-12 cell monolayers were pretreated with or without SM (300 μM) for 30 min in the basal chamber, after which the cells were transferred to fresh media containing PBS, NAC, or BMSC-Ex (20 μg) and cultured for 24 h. a TER was measured using an epithelial voltohmmeter under open-circuit conditions. TER data were normalized to those of identically handled control monolayers. b Localization and distribution of adherens and tight junction proteins. MLE-12 cell monolayers were fixed and subjected to immunofluorescence analysis to detect claudin-1, occludin, ZO-1, and E-cadherin (green). The nuclei were stained with DAPI (blue). Images were obtained using a confocal microscope (scale bar = 50 μm). c Claudin-1, occludin, ZO-1, and E-cadherin mRNA levels were measured by RT-qPCR. d Claudin-1, occludin, ZO-1, and E-cadherin protein levels were measured by Western blotting, quantitated by densitometric analysis using ImageJ software, and normalized to GAPDH. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001.
To investigate whether BMSC-Ex treatment regulates adherens or tight junction expression, RT-qPCR analysis of SM-injured MLE-12 cells treated with or without BMSC-Exs was performed. The data showed that SM exposure inhibited the expression of E-cadherin (3.6-fold) and had no significant effect on the expression of claudin-1, occludin, or ZO-1. However, both adherens and tight junction mRNA levels were upregulated in the SM + BMSC-Ex group compared with the SM group (Fig. 3c), indicating that BMSC-Exs promoted adherens and tight junction mRNA expression. Furthermore, the protein levels of E-cadherin, claudin-1, occludin, and ZO-1 were examined after BMSC-Ex treatment by Western blotting, and the results were consistent with the mRNA results, showing that BMSC-Ex treatment promoted the expression of these proteins (Fig. 3d). Interestingly, while SM did not affect the mRNA levels of claudin-1 or occludin, their protein levels were decreased, which may be due to the internalization (white arrows) and degradation of these proteins, as shown in Fig. 3b. Taken together, these results show that BMSC-Exs promoted the repair of adherens and tight junction integrity in SM-induced acute lung injury.
BMSC-Exs protected lung epithelial cells from apoptosis and epithelial barrier damage by regulating GPRC5A in vitro and in vivo
To further elucidate the molecular mechanism of the antiapoptotic and barrier-regenerating properties of BMSC-Exs, we performed a gene microarray analysis of MLE-12 cells treated with or without BMSC-Exs after exposure to SM. A total of 107 genes, including 26 upregulated and 81 downregulated genes, were affected by BMSC-Exs (>1.5-fold change, adjusted P-value < 0.05, data not shown). Among the 10 genes with the most significant changes in BMSC-Ex-treated MLE-12 cells, GPRC5A expression was significantly upregulated (Fig. 4a). This upregulation was confirmed by measuring GPRC5A protein levels by WB, which showed that GPRC5A may participate in BMSC-Ex-mediated repair of lung damage (Fig. 4b).

a, b MLE-12 cells were untreated (Ctrl) or treated with SM (300 μM) for 30 min and then treated with PBS or BMSC-Exs (20 μg) for 24 h. a Variations in the top 10 differentially expressed genes as determined by gene microarray analysis. The values represent the log2-fold change in expression in the BMSC-Ex treatment group compared to that in the PBS treatment group after SM exposure. b The GPRC5A protein level was measured by Western blotting, quantitated by densitometric analysis using ImageJ software, and normalized to GAPDH. c–h MLE-12 cells were transfected with GPRC5A-specific siRNA (100 nM) or nontargeting (NT) siRNA (100 nM) for 72 h. GPRC5A siRNA- or NT siRNA-transfected cells were exposed to SM (300 μM) for 30 min and then treated with BMSC-Exs (20 μg) for 24 h. Cell viability was analyzed by CCK-8 assays (c), and the induction of apoptosis was determined by Annexin V/PI flow cytometry (d). Hoechst 33342 staining of apoptotic SM-injured MLE-12 cells (e) and mitochondrial membrane potential detection using JC-1 staining (f) are shown. Original magnification: ×200. g TER levels were measured and normalized to those of the control monolayers. h The localization and distribution of claudin-1, occludin, ZO-1, and E-cadherin (green) in MLE-12 cell monolayers were examined using immunofluorescence, and images were obtained using a confocal microscope (scale bar = 50 μm). The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001.
GPRC5A is predominately expressed in type I and II lung epithelial cells, and previous studies have shown that GPRC5A has an important role in controlling susceptibility to ALI [29]. To investigate the role of GPRC5A in BMSC-Ex-mediated antiapoptotic effects and cellular injury repair, we knocked down GPRC5A in MLE-12 cells with siRNA (depletion confirmed by Western blotting (Supplementary Fig. S4a). GPRC5A knockdown reversed both the promotion of cell viability (Fig. 4c) and the reduction in MLE-12 apoptosis mediated by BMSC-Exs (Fig. 4d and Supplementary Fig. S4b, e, f). Thus, GPRC5A knockdown abrogated BMSC-Ex-mediated inhibition of SM-induced cytotoxicity. The BMSC-Ex-mediated increase in TER and alleviation of epithelial barrier damage were abolished by GPRC5A knockdown in vitro (Fig. 4g, h), indicating that GPRC5A has an important role in epithelial barrier regulation.
In addition, GPRC5A knockout attenuated the therapeutic effects of BMSC-Exs on SM-induced ARDS, as shown by the in vivo reversal of BMSC-Ex-mediated inhibition of tissue lesions, perivascular infiltrates, and inflammatory infiltration (Fig. 5a). The expression of Ki67, a cellular marker of proliferation, was inhibited (Fig. 5b), pneumocyte apoptosis was promoted (Fig. 5c), and epithelial barrier repair was abolished in the GPRC5A−/− groups (Fig. 5d). GPRC5A knockout also reversed the decrease in BALF protein levels and the W/D ratio in BMSC-Ex-treated mice (Fig. 5e, f). Taken together, the in vitro and in vivo data showed that GPRC5A knockdown delayed BMSC-Ex-induced recovery from an acute lung injury, demonstrating that GPRC5A is a key player in BMSC-Ex-mediated lung repair and protection.

Wild-type (GPRC5A+/+) and GPRC5A-ko (GPRC5A−/−) mice were treated with or without BMSC-Exs for 5 days after SM exposure (n = 3). a Representative optical and H&E-stained images of lung tissues from SM-exposed mice after BMSC-Ex treatment (left panel). Original magnification: ×200. Comparisons of pathological lung injury scores in SM-exposed mice (right panel). b Ki67-stained lung sections from SM-injured mice after PBS or BMSC-Ex treatment (×200). c TUNEL staining of apoptotic cells in SM-injured mouse lung sections after PBS or BMSC-Ex treatment (×200). d Expression and distribution of claudin-1 (red), occludin (green), and E-cadherin (green) in SM-injured mouse lung sections after PBS or BMSC-Ex treatment (×200). e, f Comparisons of BALF protein levels (e) and wet-to-dry lung weight ratios (f) in SM-injured mouse lung sections after PBS or BMSC-Ex treatment. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001.
BMSC-Exs promoted the repair of lung epithelial cells by activating the YAP pathway via GPRC5A
The Hippo/YAP kinase cascade is critical for maintaining vascular barrier integrity and formation, as well as for regenerating the alveolar epithelium [30, 31]. To investigate whether BMSC-Ex-mediated cell regeneration and barrier repair require the YAP pathway, YAP phosphorylation was measured after GPRC5A knockdown. GPRC5A depletion abolished the BMSC-Ex-mediated increases in Bcl-2, claudin-1, occludin, ZO-1, and E-cadherin expression levels (Fig. 6a). SM induced YAP phosphorylation, while BMSC-Ex treatment significantly inhibited YAP signaling pathway activation (Fig. 6b). This inhibition was lost after GPRC5A knockdown, indicating that GPRC5A may regulate YAP phosphorylation. The total YAP protein level was upregulated in the BMSC-Ex-treated group, and this increase was reversed by GPRC5A knockdown (Fig. 6b). YAP nuclear localization was also examined, and we observed that GPRC5A knockdown inhibited the BMSC-Ex-mediated promotion of the nuclear translocation of YAP (Fig. 6c), which may promote the expression of tight junctions and antiapoptotic genes. GPRC5A knockout also reversed the BMSC-Ex-mediated increase in E-cadherin, claudin-1, occludin, ZO-1, Bcl-2, and YAP expression in vivo, and the inhibition of YAP phosphorylation was abolished by GPRC5A knockout (Fig. 6d, e), which was consistent with the in vitro results. Taken together, these results indicate that BMSC-Exs promote lung epithelial cell protection and barrier repair by regulating GPRC5A-mediated regulation of the YAP pathway.

a–c GPRC5A siRNA- or NT siRNA-transfected MLE-12 cells were exposed to SM (300 μM) for 30 min and then treated with BMSC-Exs (20 μg) for 24 h. a Western blot analysis of apoptosis-related proteins (ai) and junction proteins (aii) in MLE-12 cells treated with or without BMSC-Exs. The levels of Bcl-2, claudin-1, occludin, ZO-1, and E-cadherin were quantitated by densitometric analysis using ImageJ software and normalized to GAPDH. b The total protein level of YAP and YAP phosphorylation was examined by Western blotting. GAPDH served as a loading control. The relative quantification of the detected signals was determined using ImageJ software and normalized to GAPDH. c Expression and localization of YAP. MLE-12 cells were fixed after being treated with or without BMSC-Exs for 24 h and were subjected to immunofluorescence analysis to detect YAP (green). The nuclei were stained with DAPI (blue). Images were obtained using a confocal microscope (scale bar = 50 μm). The fluorescence intensity of YAP was processed and quantified using ZEN Light Edition software (right panel). d, e Wild-type (GPRC5A+/+) and GPRC5A-ko (GPRC5A−/−) mice treated with or without BMSC-Exs for 5 days after SM exposure (n = 3). The protein levels of Bcl-2 (dii), claudin-1, occludin, ZO-1, E-cadherin (di), and total and phosphorylated YAP (e) were measured by Western blotting. Tubulin served as a loading control. The relative quantification of the detected signals was determined using ImageJ software and normalized to Tubulin. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001.
Discussion
SM is a highly toxic and vesicant chemical warfare agent that remains a significant threat to public security due to its availability, stable chemical properties, and ease of production and storage. While SM exposure can cause damage to the respiratory tract, eyes, skin, and multiple organ systems, lung injury is the major determinant of morbidity and mortality [32, 33]. Despite extensive research into ALI and acute respiratory distress syndrome (ARDS) caused by different risk factors, treatment is still limited to supportive measures, as no effective pharmacotherapy exists.
In recent years, MSCs have been shown to greatly reduce inflammation and promote the regeneration of injured lung tissue in preclinical ALI models. MSCs secrete soluble factors, such as growth factors, anti-inflammatory cytokines, and antimicrobial peptides, that stabilize the alveolocapillary barrier, enhance alveolar fluid clearance, and decrease infection [13, 34, 35]. Our previous work demonstrated the systemic therapeutic effects of BMSCs in SM-induced lung injury in mice, including anti-inflammatory, immunomodulatory, and reparative effects [10]. Yan reported that in rats exposed to SM, the significant therapeutic effects of human MSCs were due to immunoregulation and functional improvements in the hemopoietic microenvironment [36]. All of these results suggest that MSC administration after SM exposure may ameliorate pulmonary toxicity by facilitating the delivery of MSC-Exs. It has recently been established that the therapeutic effect of MSCs is largely due to the transfer of exosomes/microvesicles, suggesting that MSC-derived exosomes may be critical to reversing lung injury [37]. Thus, in this study, we sought to understand the potential role and mechanism of BMSC-Exs in the treatment of ALI.
Using a previously established murine model of SM-induced ALI, we investigated BMSC-Ex doses to determine their efficacy in treating lung injury. Our data showed that a single administration of BMSC-Exs generated an effective therapeutic effect. Treatment with BMSC-Exs rescued mice from SM-induced lung failure similar to that of treatment with NAC, which is an appealing antioxidant therapy for both the acute and chronic phases of vesicant-induced pathology [6, 32]. In our study, with increasing exosome doses, the therapeutic effects became more obvious, suggesting that this treatment is indeed a promising therapeutic candidate for acute manifestations of vesicant injury. The acute phases of SM-induced lung injury are characterized by thickening of the alveolar septal walls, perivascular edema, lung parenchymal congestion, hemorrhage, and epithelial cell apoptosis, and analyses of histopathology, BALF protein levels, the W/D ratio, cell viability, and cell apoptosis performed in this study all confirmed that BMSC-Exs repaired lung injury induced by SM toxicity.
Although DNA alkylation, inflammatory responses, oxidative stress, and proteolytic enzyme activation induced by SM are associated with its toxicity and SM exposure leads to dysregulated cellular metabolism, causing apoptosis or necrosis [38, 39], the exact mechanism of SM toxicity has not yet been fully elucidated. In our study, SM-induced MLE-12 cells exhibited increased apoptosis. Perturbations in mitochondrial membrane potential, caspase-3/9 apoptotic pathway activation, and decreased Bcl-2 expression, which are indicative of apoptosis triggered through mitochondrial signaling pathways, were also observed. After the administration of BMSC-Exs, SM-induced apoptosis was significantly inhibited, and epithelial cell viability was enhanced. Consequently, these results suggest that SM induces epithelial cell injury mainly by promoting apoptosis, and in this process, BMSC-Exs act as apoptosis inhibitors.
Damage to the alveolar epithelial–endothelial barrier is one of the most significant pathological changes in ALI and results in increased pulmonary vascular membrane permeability [28, 40]. Adherens and tight junctions are important structures that maintain the alveolar membrane barrier and the permeability of pulmonary epithelial cells. The primary causes of increased permeability are direct damage to alveolar epithelial cells and decreased expression of junction proteins [41, 42]. Using an in vitro pulmonary epithelial barrier model, we found that SM increased barrier permeability and changed the cell membrane localization and protein levels of adherens and tight junction proteins. Treatment with BMSC-Exs significantly improved the expression and membrane localization of junction proteins, leading to the repair of barrier function. It is interesting to note that while the mRNA levels of these genes were not affected by SM treatment, their protein levels decreased significantly. Previous studies have shown that ventilator-induced lung injury leads to the endocytosis and degradation of both E-cadherin and occludin via RhoA activation [28] and that myosin II regulatory light chain kinase (MLCK) activation induces the internalization of tight junctions via cytoskeletal and caveolar-mediated endocytosis, resulting in a loss of the intestinal epithelial barrier [23]. There is increasing evidence that junction proteins are mainly degraded by the ubiquitination pathway after endocytosis [43,44,45]. In this study, we showed that SM induced E-cadherin, occludin, and claudin-1 endocytosis, suggesting that endocytosis and degradation of these molecules are important causes of barrier dysfunction.
Exosomes may mimic the function of their parental MSCs by transferring components such as lipids, multiple RNA species, various proteins, and even organelles such as mitochondria to recipient cells through endocytosis [46]. Various strategies for exogenously loading isolated exosomes with specific proteins and nucleic acids have been investigated [47], including freeze-thaw cycles and electroporation. To date, more than 850 unique gene products and 150 miRNAs have been identified in MSC-derived exosomes. Moreover, MSC-derived exosomes have been genetically modified with certain proteins and miRNAs using transgenic MSCs to identify their functional components. However, optimized conditions are required for genetically modified EVs to acquire additional effective functional properties. In addition, information regarding the quality control of EV composition and the safety of these exosomes is needed before EVs can be used in clinical applications [48, 49].
BMSC-Exs may function via a single component or by the synergistic actions of multiple components; however, BMSC-Exs can ultimately regulate signaling pathways and/or gene expression in recipient cells [50]. We found that GPRC5A, a retinoic acid target gene that is predominately expressed in the lung epithelium [51], has a key role in promoting the repair of SM-induced lung epithelial barrier dysfunction. SM exposure downregulated GPRC5A expression, while BMSC-Ex treatment significantly upregulated GPRC5A expression. BMSC-Ex-induced repair of epithelial barrier function was significantly inhibited by GPRC5A knockdown in vitro and in vivo. Although GPRC5A has been confirmed to be a critical factor in the regulation of ALI and lung tumorigenesis [29, 52,53,54], the present study is the first report of its role in regulating lung epithelial cell apoptosis and the expression of tight junction proteins. We also found that GPRC5A promoted the expression of Bcl-2 and junction proteins by activating the Hippo pathway effector YAP. Previous work on cancer cell adaptation to hypoxia showed that GPRC5A enables hypoxic cell survival by activating YAP and its antiapoptotic target gene Bcl-2L1 [55]. The Hippo/YAP pathway has also been shown to participate in regulating vascular barrier maturation and blood–brain barrier restoration [30, 31, 56]. These findings demonstrate that the GPRC5A–YAP pathway may be critical in the modulation of the pulmonary epithelial barrier and the pulmonary capillary endothelial barrier.
In summary, the administration of exosomes derived from murine bone marrow mesenchymal stromal cells rescues SM-induced acute lung injury in vivo and in vitro. BMSC-Exs exert antiapoptotic effects, promote the restoration of epithelial barrier function, and alleviate extensive lung damage. The antiapoptotic and barrier-regenerating effects may be mediated by the upregulation of GPRC5A expression in recipient cells, which activates the YAP pathway, leading to the promotion of Bcl-2 and junction protein expression and relocalization. Thus, the administration of BMSC-Exs may be an alternative approach to stem cell-based therapy for acute lung injury and represents a novel therapeutic strategy for restoring biological barrier function.
References
Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 2016;140:345–50.
Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18.
Spadaro S, Park M, Turrini C, Tunstall T, Thwaites R, Mauri T, et al. Biomarkers for acute respiratory distress syndrome and prospects for personalised medicine. J Inflamm. 2019;16:1.
Ghabili K, Agutter PS, Ghanei M, Ansarin K, Panahi Y, Shoja MM. Sulfur mustard toxicity: history, chemistry, pharmacokinetics, and pharmacodynamics. Crit Rev Toxicol. 2011;41:384–403.
Sun M, Yang Y, Meng W, Xu Q, Lin F, Chen Y, et al. Advanced biotherapy for the treatment of sulfur mustard poisoning. Chem-Biol Interact. 2018;286:111–8.
Weinberger B, Malaviya R, Sunil VR, Venosa A, Heck DE, Laskin JD, et al. Mustard vesicant-induced lung injury: advances in therapy. Toxicol Appl Pharmacol. 2016;305:1–11.
Nejad-Moghaddam A, Ajdari S, Tahmasbpour E, Goodarzi H, Panahi Y, Ghanei M. Adipose-derived mesenchymal stem cells for treatment of airway injuries in a patient after long-term exposure to sulfur mustard. Cell J. 2017;19:117–26.
Simonson OE, Mougiakakos D, Heldring N, Bassi G, Johansson HJ, Dalen M, et al. In vivo effects of mesenchymal stromal cells in two patients with severe acute respiratory distress syndrome. Stem Cells Transl Med. 2015;4:1199–213.
Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med. 2019;7:154–62.
Feng Y, Xu Q, Yang Y, Shi W, Meng W, Zhang H, et al. The therapeutic effects of bone marrow-derived mesenchymal stromal cells in the acute lung injury induced by sulfur mustard. Stem Cell Res Ther. 2019;10:90.
Lee HY, Hong IS. Double-edged sword of mesenchymal stem cells: cancer-promoting versus therapeutic potential. Cancer Sci. 2017;108:1939–46.
Abraham A, Krasnodembskaya A. Mesenchymal stem cell-derived extracellular vesicles for the treatment of acute respiratory distress syndrome. Stem Cells Transl Med. 2020;9:28–38.
Monsel A, Zhu YG, Gudapati V, Lim H, Lee JW. Mesenchymal stem cell derived secretome and extracellular vesicles for acute lung injury and other inflammatory lung diseases. Expert Opin Biol Ther. 2016;16:859–71.
Li X, Liu L, Yang J, Yu Y, Chai J, Wang L, et al. Exosome derived from human umbilical cord mesenchymal stem cell mediates miR-181c attenuating burn-induced excessive inflammation. EBioMedicine. 2016;8:72–82.
Zhang B, Shi Y, Gong A, Pan Z, Shi H, Yang H, et al. HucMSC exosome-delivered 14-3-3zeta orchestrates self-control of the Wnt response via modulation of YAP during cutaneous regeneration. Stem Cells. 2016;34:2485–500.
Hao Q, Zhu YG, Monsel A, Gennai S, Lee T, Xu F, et al. Study of bone marrow and embryonic stem cell-derived human mesenchymal stem cells for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells Transl Med. 2015;4:832–40.
Park J, Kim S, Lim H, Liu A, Hu S, Lee J, et al. Therapeutic effects of human mesenchymal stem cell microvesicles in an ex vivo perfused human lung injured with severe E. coli pneumonia. Thorax. 2019;74:43–50.
Han J, Liu Y, Liu H, Li Y. Genetically modified mesenchymal stem cell therapy for acute respiratory distress syndrome. Stem Cell Res Ther. 2019;10:386.
Yan Y, Jiang W, Tan Y, Zou S, Zhang H, Mao F, et al. HucMSC exosome-derived GPX1 is required for the recovery of hepatic oxidant injury. Mol Ther. 2017;25:465–79.
Sawyer TW. N-acetylcysteine as a treatment for sulphur mustard poisoning. Free Radic Biol Med. 2020;161:305–20.
Dong L, He HL, Lu XM, Yang Y, Qiu HB. Modulation of FLT3 signaling targets conventional dendritic cells to attenuate acute lung injury. APMIS. 2012;120:808–18.
Bachler G, Losert S, Umehara Y, von Goetz N, Rodriguez-Lorenzo L, Petri-Fink A, et al. Translocation of gold nanoparticles across the lung epithelial tissue barrier: combining in vitro and in silico methods to substitute in vivo experiments. Part Fibre Toxicol. 2015;12:18.
Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, et al. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology. 2007;132:2383–94.
Shen CH, Lin JY, Chang YL, Wu SY, Peng CK, Wu CP, et al. Inhibition of NKCC1 modulates alveolar fluid clearance and inflammation in ischemia-reperfusion lung injury via TRAF6-mediated pathways. Front Immunol. 2018;9:2049.
Xu Q, Shi W, Lv P, Meng W, Mao G, Gong C, et al. Critical role of caveolin-1 in aflatoxin B1-induced hepatotoxicity via the regulation of oxidation and autophagy. Cell Death Dis. 2020;11:6.
Soni S, Wilson MR, O’Dea KP, Yoshida M, Katbeh U, Woods SJ, et al. Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax. 2016;71:1020–9.
Meng W, Sun M, Xu Q, Cen J, Cao Y, Li Z, et al. Development of a series of fluorescent probes for the early diagnostic imaging of sulfur mustard poisoning. ACS Sens. 2019;4:2794–801.
Gu C, Liu M, Zhao T, Wang D, Wang Y. Protective role of p120-catenin in maintaining the integrity of adherens and tight junctions in ventilator-induced lung injury. Respir Res. 2015;16:58.
Liao Y, Song H, Xu D, Jiao H, Yao F, Liu J, et al. Gprc5a-deficiency confers susceptibility to endotoxin-induced acute lung injury via NF-kappaB pathway. Cell Cycle. 2015;14:1403–12.
Kim J, Kim YH, Park DY, Bae H, Lee DH, Kim KH, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest. 2017;127:3441–61.
LaCanna R, Liccardo D, Zhang P, Tragesser L, Wang Y, Cao T, et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J Clin Invest. 2019;129:2107–22.
Etemad L, Moshiri M, Balali-Mood M. Advances in treatment of acute sulfur mustard poisoning—a critical review. Crit Rev Toxicol. 2019;49:191–214.
Wolfe GA, Petteys SM, Phelps JF, Wasmund JB, Plackett TP. Sulfur mustard exposure: review of acute, subacute, and long-term effects and their management. J Spec Oper Med. 2019;19:81–6.
Lee JW, Fang X, Krasnodembskaya A, Howard JP, Matthay MA. Concise review: mesenchymal stem cells for acute lung injury: role of paracrine soluble factors. Stem Cells. 2011;29:913–9.
Zheng G, Huang L, Tong H, Shu Q, Hu Y, Ge M, et al. Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: a randomized, placebo-controlled pilot study. Respir Res. 2014;15:39.
Yan X, Shu Y, He J, Zhao J, Jia L, Xie J, et al. Therapeutic effects of human umbilical cord mesenchymal stromal cells in Sprague-Dawley rats with percutaneous exposure to sulfur mustard. Stem Cells Dev. 2019;28:69–80.
Witwer KW, Van Balkom BWM, Bruno S, Choo A, Dominici M, Gimona M, et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J Extracell Vesicles. 2019;8:1609206.
Heinrich A, Balszuweit F, Thiermann H, Kehe K. Rapid simultaneous determination of apoptosis, necrosis, and viability in sulfur mustard exposed HaCaT cell cultures. Toxicol Lett. 2009;191:260–7.
Lang S, Popp T, Kriegs CS, Schmidt A, Balszuweit F, Menacher G, et al. Anti-apoptotic and moderate anti-inflammatory effects of berberine in sulfur mustard exposed keratinocytes. Toxicol Lett. 2018;293:2–8.
Janga H, Cassidy L, Wang F, Spengler D, Oestern-Fitschen S, Krause MF, et al. Site-specific and endothelial-mediated dysfunction of the alveolar-capillary barrier in response to lipopolysaccharides. J Cell Mol Med. 2018;22:982–98.
Higuita-Castro N, Nelson MT, Shukla V, Agudelo-Garcia PA, Zhang W, Duarte-Sanmiguel SM, et al. Using a novel microfabricated model of the alveolar-capillary barrier to investigate the effect of matrix structure on atelectrauma. Sci Rep. 2017;7:11623.
Short KR, Kasper J, van der Aa S, Andeweg AC, Zaaraoui-Boutahar F, Goeijenbier M, et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur Respir J. 2016;47:954–66.
Cai J, Culley MK, Zhao Y, Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell. 2018;9:754–69.
Stamatovic SM, Johnson AM, Sladojevic N, Keep RF, Andjelkovic AV. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. 2017;1397:54–65.
Sumi A, Hayes P, D’Angelo A, Colombelli J, Salbreux G, Dierkes K, et al. Adherens junction length during tissue contraction is controlled by the mechanosensitive activity of actomyosin and junctional recycling. Dev Cell. 2018;47:453–63 e3.
Gonda A, Kabagwira J, Senthil GN, Wall NR. Internalization of exosomes through receptor-mediated endocytosis. Mol Cancer Res. 2019;17:337–47.
Kotmakçı M, Bozok, Çetintaş V. Extracellular vesicles as natural nanosized delivery systems for small-molecule drugs and genetic material: steps towards the future nanomedicines. J Pharm Pharm Sci. 2015;18:396–413.
Park K-S, Bandeira E, Shelke GV, Lässer C, Lötvall J. Enhancement of therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther. 2019;10:288.
Mendt M, Rezvani K, Shpall E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transpl. 2019;54:789–92.
Harrell CR, Jovicic N, Djonov V, Volarevic V. Therapeutic use of mesenchymal stem cell-derived exosomes: from basic science to clinics. Pharmaceutics. 2020;12:474.
Tao Q, Fujimoto J, Men T, Ye X, Deng J, Lacroix L, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99:1668–82.
Wang J, Farris AB, Xu K, Wang P, Zhang X, Duong DM, et al. GPRC5A suppresses protein synthesis at the endoplasmic reticulum to prevent radiation-induced lung tumorigenesis. Nat Commun. 2016;7:11795.
Huang Z, Wang S, Liu Y, Fan L, Zeng Y, Han H, et al. GPRC5A reduction contributes to pollutant benzo[a]pyrene injury via aggravating murine fibrosis, leading to poor prognosis of IIP patients. Sci Total Environ. 2020;739:139923.
Kantrowitz J, Sinjab A, Xu L, McDowell TL, Sivakumar S, Lang W, et al. Genome-wide gene expression changes in the normal-appearing airway during the evolution of smoking-associated lung adenocarcinoma. Cancer Prev Res. 2018;11:237–48.
Greenhough A, Bagley C, Heesom KJ, Gurevich DB, Gay D, Bond M, et al. Cancer cell adaptation to hypoxia involves a HIF-GPRC5A-YAP axis. EMBO Mol Med. 2018;10:e8699.
Cao Z, Xu T, Tong X, Wang Y, Zhang D, Gao D, et al. Maternal yes-associated protein participates in porcine bastocyst development via modulation of trophectoderm epithelium barrier function. Cells. 2019;8:1606.
Acknowledgements
We are grateful to You-heng Wei (State Key Laboratory of Genetic Engineering, Institute of Genetics, Fudan University, Shanghai, China) for providing technical assistance. This work was funded by the National Natural Science Foundation of China (81671858 and 81871521) and the Natural Science Foundation of Shanghai (20ZR1470300).
Author information
Authors and Affiliations
Contributions
GCM, CCG, and ZW performed the experiments, collected data, and prepared the manuscript. KX, QQX, and YL conceived the study and wrote the main manuscript. GCM, ZPP, and MXS checked the data and performed data analysis. XWH and WQM participated in the design of the study and helped draft the manuscript. JFC and QQX helped perform data analysis and prepare the images. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Mao, Gc., Gong, Cc., Wang, Z. et al. BMSC-derived exosomes ameliorate sulfur mustard-induced acute lung injury by regulating the GPRC5A–YAP axis. Acta Pharmacol Sin 42, 2082–2093 (2021). https://doi.org/10.1038/s41401-021-00625-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00625-4
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Translational medicine for acute lung injury
Journal of Translational Medicine (2024)
-
The efficacy of extracellular vesicles for acute lung injury in preclinical animal models: a meta-analysis
BMC Pulmonary Medicine (2024)
-
The dual role of mesenchymal stem cells in apoptosis regulation
Cell Death & Disease (2024)
-
Stem Cell Extracellular Vesicles as Anti-SARS-CoV-2 Immunomodulatory Therapeutics: A Systematic Review of Clinical and Preclinical Studies
Stem Cell Reviews and Reports (2024)
-
MiR-146a-5p delivered by hucMSC extracellular vesicles modulates the inflammatory response to sulfur mustard-induced acute lung injury
Stem Cell Research & Therapy (2023)