
Abstract
Endoplasmic reticulum (ER) homeostasis is regulated by ER-resident E3 ubiquitin ligase Hrd1, which has been implicated in inflammatory bowel disease (IBD). Ginsenoside Rb1 (GRb1) is the major ginsenoside in ginseng with multiple pharmacological activities. In this study we investigated the role of Hrd1 in IBD and its regulation by GRb1. Two mouse colitis models were established to mimic human IBD: drinking water containing dextran sodium sulfate (DSS) as well as intra-colonic infusion of 2, 4, 6-trinitrobenzene sulfonic acid (TNBS). Colitis mice were treated with GRb1 (20, 40 mg·kg−1·d−1, ig) or a positive control drug sulfasalazine (500 mg·kg−1·d−1, ig) for 7 days. The model mice showed typical colitis symptoms and pathological changes in colon tissue. In addition to significant inflammatory responses and cell apoptosis in colon tissue, colon epithelial expression of Hrd1 was significantly decreased, the expression of ER stress markers GRP78, PERK, CHOP, and caspase 12 was increased, and the expression of Fas was increased (Fas was removed by Hrd1-induced ubiquitination). These changes were partially, or completely, reversed by GRb1 administration, whereas injection of Hrd1 inhibitor LS102 (50 mg·kg−1· d−1, ip, for 6 days) exacerbated colitis symptoms in colitis mice. GRb1 administration not only normalized Hrd1 expression at both the mRNA and protein levels, but also alleviated the ER stress response, Fas-related apoptosis, and other colitis symptoms. In intestinal cell line IEC-6, the expression of Hrd1 was significantly decreased by LPS treatment, but was normalized by GRb1 (200 μM). GRb1 alleviated LPS-induced ER stress and cell apoptosis in IEC-6 cells, and GRb1 action was inhibited by knockdown of Hrd1 using small interfering RNA. In summary, these results reveal a pathological role of Hrd1 in colitis, and provide a novel insight into alternative treatment of colitis using GRb1 activating Hrd1 signaling pathway.
Similar content being viewed by others


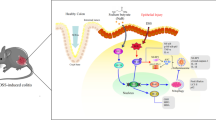
Introduction
Inflammatory bowel disease (IBD), a group of autoimmune diseases characterized by chronic, recurrent inflammation, and ulceration in one or more parts of the gastrointestinal tract, carries an increased risk of developing colorectal cancer [1]. Both Crohn’s disease (CD) and ulcerative colitis (UC), the two primary clinically defined forms of IBD, can lead to severe symptoms in patients and a consequent social burden. It is estimated that more than 3.7 million people worldwide are affected by IBD; both the prevalence and morbidity of IBD are rapidly increasing [2]. According to statistical data collected from 2006 to 2016, in China, the prevalence of UC is 11.6/100,000, while that of CD is 1.4/100,000 [3].
IBD is caused by a complex series of interactions between the environment and immune system [4, 5]. Multiple signaling pathways, such as the nuclear factor-κB (NF-κB) pathway, interleukin (IL)-22 cascade, and Nod-like receptor pathway [6,7,8], are involved in these interactions and maintain intestinal balance. Medications that modulate immune balance have been the main means of IBD therapy until now [9]. Treatment options for IBD have been developed over recent years, and more novel biological therapies, such as antitumor necrosis factor α (TNF-α) antibodies and the activation of IL-22, are now utilized alongside classic therapies [10, 11]. Therefore, exploring potential IBD therapeutic targets to supplement existing therapies is very worthwhile.
The endoplasmic reticulum (ER)-resident E3 ubiquitin ligase Hrd1 has received much attention because of its regulatory role in immune cell function. Hrd1 has been identified as a positive regulator of T-cell immunity [12]. Hrd1 also protects peripheral B cells from activation-induced cell death through degradation of the death receptor Fas [13]. Hrd1 promotes dendritic cells to activate CD4+ T cells during inflammatory processes [14]. Hrd1 is also involved in many autoimmune diseases, such as multiple sclerosis, type-1 diabetes, and rheumatoid arthritis. In multiple sclerosis patients, Hrd1 expression in CD4+ T cells is increased, which promotes CD4+ T-cell autoimmunity [12]. In type-1 diabetes, Hrd1 promotes proinsulin degradation, after which the degradation products are processed into autoantigens that are recognized by CD8+ T cells [15]. In synovial cells from rheumatoid arthritis patients, Hrd1 expression is increased, which can suppress synovial cell apoptosis [16]. In contrast with the role of Hrd1 in the above mentioned diseases, the expression of Hrd1 in the inflamed intestine of patients with IBD is significantly downregulated [17]. The specific function and mechanisms of Hrd1 in intestinal epithelial cells in IBD are only partially understood.
In IBD, the imbalance of intestinal homeostasis triggers ER stress, and excessive ER stress in turn promotes IBD development [18,19,20]. In ER stress, Hrd1 can help to induce the degradation of misfolded proteins through ER-associated degradation (ERAD). During ERAD, misfolded proteins bind chaperones such as BiP, which are then retrotranslocated into the cytosol to be ubiquitinated by Hrd1 [21]. Hrd1-mediated degradation of misfolded proteins plays a protective role against a variety of diseases, such as pathological cardiac hypertrophy, diabetic retinopathy, and lung epithelial damage caused by tobacco smoking [22,23,24]. Therefore, activation of Hrd1 may be a potential target to treat IBD.
In recent years, herbal medicines have been widely used by IBD patients as complementary and alternative medicines. The ginsenoside Rb1 (GRb1), mainly derived from the stem, root, and flower bud of Panax ginseng, has received much attention because of its anti-inflammatory, antioxidant, and neuroprotective activities [25,26,27]. In particular, GRb1 has demonstrated anti-inflammatory effects in colitis [28]. The potential mechanisms through which GRb1 treatment ameliorates colitis are partially understood. In our study, mouse models of colitis induced by both dextran sulfate sodium (DSS) and 2,4,6-trinitrobenzene sulfonic acid (TNBS) were used to investigate the effects of GRb1 activation on Hrd1 signaling pathways in colitis.
Materials and methods
Reagents
GRb1 (purity ≥ 98%) was obtained from Chengdu Must Biotechnology Co., Ltd. (Chengdu, China). Sulfasalazine (SASP) was obtained from Tianjin Kingyork Group Co., Ltd. (Tianjin, China). Antibodies against Hrd1(13473-1-AP), CHOP(15204-1-AP), Fas (13098-1-AP), caspase 8(13423-1-AP), caspase 3 (19677-1-AP), cleaved-caspase 3 (cl-caspase 3) (19677-1-AP), and iNOS (14142-1-AP) were purchased from Proteintech Co., Ltd. (Wuhan, China). Antibodies against caspase 12 (WL03000), GRP78 (WL02372), PERK (WL03378), NF-κB-p65 (66535-1-Ig), and phosphorylated NF-κB-p65 (p-NF-κB-p65) (WL02169) were obtained from Wanleibio Co., Ltd. (Shenyang, China). Antibody against COX2 (12282) was purchased from Cell Signal Co., Ltd. (Boston, MA, USA). A Cell Counting Kit-8 (CCK-8) (WLA074b) was obtained from Wanleibio Co., Ltd. (Shenyang, China). Chemicals were obtained from Sigma–Aldrich (St. Louis, MO, USA), unless otherwise indicated.
Animals
Sixty male C57BL/6 mice (6–8 weeks old, weighing 18–20 g) were obtained from the Laboratory Animal Center of Dalian Medical University (certificate of conformity: No. SYXK (Liao) 2018-0007). The experimental protocol was approved by the Dalian Medical University Animal Care and Ethics Committee. The animals were acclimatized to laboratory conditions (23 °C, a 12/12-h light/dark cycle, 50% humidity, and ad libitum access to food and water) for 2 weeks prior to the experiments. The animal protocol was designed to minimize pain and discomfort to the animals. Mice were housed one per cage and deprived of food for 12 h before the experiments. All mice were euthanized by barbiturate overdose (intravenous injection, 150-mg/kg pentobarbital sodium) for intestinal tissue collection. All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal studies were reported in compliance with the ARRIVE guidelines [29]. All applicable institutional and/or national guidelines for the care and use of animals were followed.
Cell culture
IEC-6 rat intestinal epithelial cells were purchased from the Cell Bank of the Shanghai Institute (Shanghai, China). The cells used in this study were evaluated before the experiments were conducted, and no significant interspecies variations in Hrd1 signaling that might have affected the results of the current study were observed by preliminary experiments. The cells were maintained at 37 °C in a 5% CO2 environment. The culture medium consisted of DMEM (Invitrogen, Waltham, MA, USA) with 4.5-mg/mL glucose, 50-U/mL penicillin, 50-U/mL streptomycin, 4-mM glutamine, 25-mM HEPES, and 10% fetal bovine serum (Invitrogen).
Experimental design
For the experiment in the DSS-induced colitis model, mice were randomly divided into five groups of six mice each. The mice were treated as follows: group I, sham-operated control mice administered saline by gavage; group II, mice given colitis; and groups III–V, mice treated with SASP (500-mg/kg body weight dissolved in saline, administered by gavage), low-dose GRb1 (20-mg/kg body weight dissolved in saline, administered by gavage), and high-dose GRb1 (40-mg/kg body weight dissolved in saline, administered by gavage), respectively, at 1 day after colitis induction with DSS. SASP is an anti-inflammatory drug widely used for the clinical treatment of diseases such as IBD; therefore, it was used as a positive control for the effects of GRb1 on colitis. Mice in groups III–V were administered SASP or GRb1 by gavage once a day for 7 successive days. Mice in groups II–V were given drinking water containing DSS (4% w/v) dissolved in autoclaved distilled water to induce colitis symptoms. Mice in the control group were given autoclaved distilled water but otherwise treated in the same way as the other mice. No mice showed abnormalities or became moribund during the study. Food intake and the body weight of the mice were recorded once a day. On the seventh day, distal colon samples were harvested from mice in groups I–V for biochemical studies. Mice in the Hrd1 inhibition group were intraperitoneally injected with 50-mg/kg LS102 per day for 6 consecutive days with the experimental designs described above.
For the experiment in the TNBS-induced colitis model, mice were randomly divided into five groups of six mice each. The mice were treated as follows: group I, sham-operated control mice administered saline by gavage; group II, mice in the colitis group; and groups III–V, mice treated with SASP (500-mg/kg body weight dissolved in saline administered by gavage), low-dose GRb1 (20-mg/kg body weight dissolved in saline administered by gavage), and high-dose GRb1 (40-mg/kg body weight dissolved in saline administered by gavage), respectively, at 1 day after colitis induction. Mice in groups III–V were administered SASP and GRb1 by gavage once a day for seven successive days. Colitis was induced with TNBS as described previously [30]. A catheter was inserted through the anus to approximately the level of the splenic flexure under urethane anesthesia. The colon was then infused with 0.1 mL of TNBS dissolved in ethanol (50% v/v) at a dose of 125 mg/kg. The mice were allowed to eat and drink ad libitum from 1 h after the operation. On the seventh day, distal colon samples were harvested from mice in groups I–V for biochemical studies.
Assessment of disease activity index
Mouse body weights were monitored daily. Disease activity indexes were assessed on the last day by an observer blinded to the treatments as previously described; scores for weight loss, stool bleeding, and stool consistency were added [31]. The loss of body weight was calculated as the percent difference between the original body weight (day 1) and the body weight on a particular day and scored as follows: 0: no change; 1: ≤5%; 2: 6%–10%; 3: 11%–20%; and 4: ≥20%. The stool bleed score was assessed according to the following criteria: 0: none; 2: trace visible blood in stool; and 4: total rectal bleeding. Stool consistency scores were determined as follows: 0: normal; 1: soft but still formed; 2: very soft; 3: half diarrhea; 4: and diarrhea.
Isolation of colonic epithelial cells
The distal colons of the euthanized mice were isolated and immediately rinsed with ice-cold phosphate-buffered saline (PBS) to clear the luminal contents. The distal colons were then opened longitudinally along the mesenteric border. Tissue was then cut into ~2-mm long pieces that were then submerged in 40 mL of ice-cold PBS with 5 mM EDTA in a 50-mL Falcon tube. The pieces of tissue in PBS-EDTA were then incubated at 37 °C with gentle rocking for 30 min. Following incubation, the colonic tissue was vigorously shaken to disperse colonic crypts and the surface epithelium into solution. The supernatant was then loaded into 1.5-mL microcentrifuge tubes and spun at 1 × 103 g for 5 min to pellet the suspended cells [32]. The isolated epithelial cells were used in downstream applications.
Hematoxylin-eosin staining and immunohistochemical and immunofluorescent analyses
The colon tissues were fixed with 4% paraformaldehyde at 4 °C. The samples were dehydrated, embedded in paraffin, and sectioned into 3-µm thick transverse sections. For hematoxylin-eosin staining, the sections were dewaxed, dehydrated, and stained with hematoxylin and eosin. After being washed with distilled water and dehydrated, the sections were treated with xylene. For immunohistochemical staining, the sections were dewaxed, dehydrated, and washed with distilled water three times. The sections were then treated with 3% H2O2 for 10 min, washed in PBS containing Tween-20 (PBST), treated with 5% BSA, and then incubated with rabbit anti-Hrd1 (1:100 dilution) overnight at 4 °C. After three washes with PBST, the sections were incubated with secondary antibody for 30 min at 37 °C. After being rinsed, the sections were incubated with diaminobenzidine. After counterstaining with hematoxylin, washing with distilled water and dehydration, the sections were treated with xylene. For immunofluorescent staining, the cells were washed three times with PBS, fixed with paraformaldehyde, permeabilized in 0.1% Triton X-100 (Amresco, 100219-910) in PBS, and stained with anti-Hrd1 (Bioss, bs-0679R) at a dilution of 1:100 in 1% BSA (Solarbio, A8020) overnight at 4 °C. The cells were then incubated with secondary antibody (Proteintech, SA00013-2) for 60 min. Glass slides were used to mount the cells on coverslips with Prolong Gold antifade reagent with DAPI (Invitrogen, P36941). The surface areas of single cells were determined by measuring Hrd1 staining with a quantitative digital image analysis system (Leica, version 4.5).
Enzyme-linked immunosorbent assay (ELISA)
Levels of myeloperoxidase (MPO) and the proinflammatory cytokines IL-1β, IL-6, and TNF-α were determined using ELISA kits (Shang Hai Lengton Bioscience Co., Ltd., China) according to the manufacturer’s instructions.
Western blot analysis
Colon segments were isolated from mice and immediately stored in liquid nitrogen. The total protein was isolated from colonic epithelial cells using a total protein extraction kit (P0012 Beyotime Biotechnology, China). Proteins were transferred to nitrocellulose filters and probed with the corresponding antibodies overnight at 4 °C with gentle shaking. Bands were detected and quantified using a Bio-Rad ChemiDoc XRS+ imaging system (Bio-Rad, Hercules, CA, USA).
Cell transfection
IEC-6 cells were transfected using Lipofectamine 2000 (Invitrogen) with Hrd1-targeted or control small interfering RNA (siRNA) oligos (GenePharma, China) according to the manufacturer’s instructions (Takara Biotechnology (Dalian) Co., Ltd.). The siRNA sequence targeting Hrd1 was 5′-GCACACAUUUCCGCUCUUUTTAAAGAGCGGAAAUGUGUGCTT-3′. The efficiency of gene silencing was confirmed by Western blotting.
CCK-8 viability assay
The effects of GRb1 on the viability of IEC-6 cells were measured by the CCK-8 assay. Briefly, a total of 5 × 103 cells in logarithmic growth phase in 100 μL of DMEM with 10% FBS were plated in each well of a 96-well plate. Cells were treated with GRb1 at the indicated concentrations for the indicated durations. Thereafter,10 μL of CCK-8 solution was added to the cells, which were incubated for 4 h at 37 °C. The absorbance at 450 nm was measured using an automated microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) to evaluate cell viability.
Electron microscopy
Transmission electron microscopy was used to examine the morphology of the tight junctions between epithelial cells. The colon tissue samples were exposed to 3% glutaraldehyde in 0.2-mol/L PBS for at least 2 h at 4 °C. After rinsing three times with sucrose-PBS, the samples were fixed in 1% osmic acid for 2 h at 4 °C, dehydrated through a graded series of acetone solutions and embedded in EPON epoxyresin. Ultrathin sections were prepared with an ultramicrotome and stained with uranyl acetate and lead citrate. Images were collected on a JEM-2000EX electron microscope (JEM, Japan) with an AMT 2K digital camera.
Annexin V assay
Apoptosis was determined by flow cytometry analysis (BD Pharmingen, Heidelberg, Germany). After the different treatments, IEC-6 cells were harvested, lysed in 0.25% cold trypsin without EDTA, washed with PBS, and stained with fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide (PI) according to the manufacturer’s protocol (Annexin V-FITC Apoptosis Detection Kit, BD Pharmingen, USA). Apoptotic cells (Annexin V-positive and PI-negative) were then quantified by flow cytometry using the BD analysis program.
Real-time PCR
Total RNA was isolated from colonic epithelial cells using RNAiso Plus (9108, Takara) and reverse-transcribed to single-stranded cDNA using a reverse transcription system (KR116, Tiangen). Real-time PCR was performed using SuperReal PreMix Plus (SYBR Green, FP205, Tiangen) with an Applied Biosystems StepOnePlus Real-Time PCR System, according to the manufacturer’s instructions. The primers used to detect the expression of Hrd1 target genes were as follows: forward, 5′-CCCGCTTTGTTGCACTCTTC-3′; reverse, 5′-GCAGTGAAAGAGCCAGGAGA-3′. Separate real-time PCR using primers for the detection of GAPDH was used as a control. The primers used to detect the expression of Hrd1 target genes were as follows: forward, 5′-GCATTGTGGAAGGGCTCATG-3′; reverse, 5′-CAGGGATGATGTTCTGGGCA-3′.
Statistical analysis
The animal experiments, in vitro experiments, and data analysis were conducted according to a single-blind study design. Data were compared between three or more groups using one-way ANOVA and between two groups using Student’s t test. Data are expressed as the mean ± standard deviation. Data were normally distributed, and each group showed similar variances. Further evaluations were carried out using the Kruskal–Wallis rank sum test. All experiments were repeated at least three times, and a P value < 0.05 was used to indicate statistical significance.
Results
Effect of GRb1 on colitis symptoms
The chemical structure of GRb1 is shown in Fig. S1a. The effects of GRb1 on the proliferation of IEC-6 cells were analyzed to determine the effective concentration of GRb1 with no toxicity for subsequent experiments. Exposure to 50, 100, 150, and 200-μM GRb1 for 12 h did not affect the viability of IEC-6 cells (Fig. S1b). Gavage administration of GRb1 to the mice for 7 consecutive days had no effect on body weight (Fig. S1c), food intake (Fig. S1d), or cytokine profiles in the colon tissue (TNF-α, IL-1β, and IL-6; Fig. S1e–g). Gavage administration of GRb1 to the mice had no significant effect on the expression of Hrd1 (Fig. S1h). Based on these preliminary experiments and previous reports [33, 34], maximum GRb1 treatment doses of 40 mg/kg for in vivo experiments and 200 μM for in vitro experiments were chosen.
Colitis symptoms, including weight loss, decreased food intake, an increased disease activity index, and an increased colon weight-to-length ratio (Fig. 1a–e), were observed in the DSS-induced colitis mouse model. These results confirmed successful establishment of the colitis model. HE staining showed a variety of colitis symptoms, including a damaged villous structure (green ring), the infiltration of inflammatory cells (black arrow), submucosal edema (blue ring), and muscle fiber separation (blue arrow) (Fig. 1f). Both GRb1 (20 and 40 mg/kg) and SASP reversed the visible pathological changes in the colitis model after 7 days of treatment. Electron microscopy was used to assess cell connections (Fig. S2). A wide gap (yellow ring, red arrow) between cells in the DSS group was seen. After GRb1 treatment, the gap between cells decreased in size (yellow ring, red arrow), and button-like (yellow ring, blue arrow) and thread-like (yellow ring, green arrow) cell connections were also found, which suggests the restoration of cell junctions. These results indicate that GRb1 significantly ameliorated colitis symptoms.

Colitis symptoms were recorded after 7 days of GRb1 treatment. Effects of GRb1 on a body weight, b food intake, c disease activity index, and d, e colon weight-to-length ratio. f Hematoxylin-eosin staining of mouse colonic tissue. Scale bars, 100 μm (upper panel) and 50 μm (lower panel). The green ring indicates damage to the villous structure; the black arrow indicates the infiltration of inflammatory cells; the blue ring indicates submucosal edema; the blue arrow indicates muscle fiber separation. Data are expressed as the mean ± SD. **P < 0.01 compared with the sham group; ##P < 0.01 compared with the DSS-induced colitis group; n = 6 samples.
Effect of GRb1 on inflammation and apoptosis in a colitis model
Imbalance between pro- and anti-inflammatory cytokines is known to be a major contributor to the development of IBD, leading to a proinflammatory cytokine profile in IBD [35]. In this study, compared to control mice, colitis model mice had higher MPO activity and higher levels of the proinflammatory cytokines IL-1β, IL-6, and TNF-α (Fig. 2a–d). The levels of inflammation-related proteins (iNOS, COX2, and p-NF-κB-p65) and the apoptosis-related protein cl-caspase 3 were significantly increased in colitis mice (Fig. 2e–h). However, these changes were mostly normalized by GRb1 treatment for 7 days (Fig. 2). These results suggest that GRb1 had anti-inflammatory and antiapoptotic activities in the colitis model mice.

Expression levels of the colonic cytokines a MPO, b IL-1β, c IL-6, and d TNF-α were determined by ELISA. Western blot analysis of the inflammation-related proteins e iNOS, f COX2, and g p-NF-κB-p65 (calculated as p-NF-κB-p65/NF-κB-p65) and the apoptosis-related protein h cleaved-caspase 3 (cl-caspase 3), calculated as cl-caspase 3/caspase 3. Data are expressed as the mean ± SD. Values in the sham group were set to 100%, and other values are given relative to those in the sham group, **P < 0.01 compared with the sham group; ##P < 0.01 compared with the DSS control group; n = 3 samples for Western blot experiments; n = 6 samples for other experiments.
Effect of GRb1 on the ER stress response in colitis model mice
The ER stress markers GRP78, PERK, CHOP, and caspase 12 in the colitis model were measured by Western blotting. The expression of all four markers was significantly increased in the colitis model, but these changes were partially or completely reversed by GRb1 (Fig. 3a–d).

Western blot analysis of the ER stress marker proteins a GRP78, b PERK, c CHOP, and d caspase 12. e Western blot analysis of Hrd1. f Expression of Hrd1 determined by real-time PCR. g Immunohistochemical staining of Hrd1 in the colonic epithelium. Scale bars, 200, 100, and 50 μm. Data are expressed as the mean ± SD; **P < 0.01 compared with the sham group; ##P < 0.01 compared with the DSS control group; n = 3 samples for Western blot experiments; n = 6 samples for other experiments.
As Hrd1 plays an important role in ER stress, we further studied whether the changes in Hrd1 were related to ER stress inhibition by GRb1. The levels of Hrd1 were significantly decreased in colitis model mice (Fig. 3e–g) but partially or completely normalized in mice treated with GRb1. These results suggest that GRb1 alleviates colitis through reducing the ER stress response; the changes in Hrd1 expression were consistent with the involvement of Hrd1 in the anti-inflammatory effect of GRb1 in the ER.
Effect of GRb1 on Hrd1 expression and the inhibition of ER stress
In vivo, the Hrd1 inhibitor LS102 was used to test the relationship between Hrd1 and the GRb1 treatment-induced effects on colitis. As shown in Fig. 4a, Hrd1 expression was significantly inhibited by LS102, and LS102 significantly exacerbated colitis symptoms. Colitis symptoms, including increased disease activity (Fig. 4b), increased colon weight-to-length ratio (Fig. 4c, e), and weight loss (Fig. 4d), were reversed by GRb1, and this abrogation was significantly inhibited by the Hrd1 inhibitor LS102. These results were also confirmed by HE staining (Fig. 4f). The colitis symptoms, including a damaged villous structure (green ring), the infiltration of inflammatory cells (black arrow), and submucosal edema (blue ring), were not alleviated by GRb1 in the presence of LS102. These results show that the inhibition of Hrd1 could exacerbate colitis and that Hrd1 may be involved in the mechanism of GRb1 treatment-induced effects on colitis.

a Hrd1 expression in the presence of the Hrd1 inhibitor LS102. Effects of GRb1 on disease activity (b), colon weight/length (c, e), weight loss (d). f Hematoxylin-eosin staining analysis of the effects of GRb1 on colitis. Scale bars, 100 μm. The green ring indicates damage to the villous structure; the black arrow indicates the infiltration of inflammatory cells; the blue ring indicates submucosal edema. g Effects of GRb1 on ER stress marker protein expression in the absence and presence of siRNA targeting Hrd1. h Expression of Hrd1 in the absence and presence of siRNA targeting Hrd1. i Hrd1 protein expression in IEC-6 cells incubated with 0.1–1μM BFA (an ER stress inducer). Values in the corresponding sham or normal control (NC) group were set to 100%, and other values are given relative to the control values. **P < 0.01 compared with the sham or NC group; ##P < 0.01 compared with the DSS/LPS control group or as indicated; n = 3 samples for Western blot experiments; n = 6 samples for other experiments.
The effect of GRb1 on Hrd1 expression was also evaluated in vitro using IEC-6 cells incubated with LPS. Expression of the Hrd1 protein was inhibited by 0.1–1 μg/mL LPS in a dose-dependent manner (Fig. S3a). As determined by CCK-8 analysis, the viability of IEC-6 cells was also reduced by LPS treatment (Fig. S3b). In IEC-6 cells incubated with 1 μg/mL LPS, expression of the Hrd1 protein was also decreased in a time-dependent manner (Fig. S3c). In IEC-6 cells incubated with 1 μg/mL LPS, the decrease in Hrd1 protein expression was abrogated by 50–200 μM GRb1 in a dose-dependent manner and normalized by 200 μM GRb1 (Fig. S3d, e). Treatment with 50–200 μM GRb1 abrogated the reduction in IEC-6 cell viability upon incubation with 1 μg/mL LPS in a dose-dependent manner (Fig. S3f). These findings suggest that GRb1 can stimulate Hrd1 signaling pathways to protect against LPS-induced cell damage.
Further experiments were carried out to study the effects of GRb1-activated Hrd1 signaling pathways on ER stress. The ER stress marker proteins CHOP and caspase 12 were significantly increased in the presence of LPS, but GRb1 inhibited these increases (Fig. 4g). The GRb1-induced reduction in ER stress marker proteins was inhibited by siRNA targeting Hrd1. In addition, Hrd1 expression was significantly inhibited by siRNA targeting Hrd1 (Fig. 4h). Brefeldin A (BFA) was also used to induce ER stress to test the effects of Hrd1 signaling pathways in IEC-6 cells. In IEC-6 cells incubated with BFA at different concentrations, the expression level of Hrd1 was reduced, but this reduction did not occur in a dose-dependent manner (Fig. 4i). These findings indicate that GRb1 can stimulate Hrd1 signaling pathways, resulting in the inhibition of ER stress.
Effect of GRb1 on apoptosis and the relationship of GRb1 to Hrd1 and the Fas receptor
The effect of GRb1 on the apoptosis of LPS-treated IEC-6 cells was determined by flow cytometry (Fig. 5). The apoptosis rate in the LPS-treated cells was significantly increased compared with that in the sham group, but this increase was mostly abrogated by GRb1 (Fig. 5a). LPS significantly increased the expression of both Fas and its downstream activation target, caspase 8 (Fig. 5b–d). The increase in Fas expression resulting from LPS treatment was inhibited by siRNA targeting Hrd1 (Fig. 5e). These results indicate that Hrd1 downregulated Fas, thereby inhibiting intestinal epithelial cell apoptosis. The antiapoptotic effect of GRb1 was also significantly inhibited by siRNA targeting Hrd1 (Fig. 5f). It appears that GRb1 can inhibit Fas expression by activating Hrd1, exerting an antiapoptotic effect, in intestinal epithelial cells.

a Effect of GRb1 (200 μM) on IEC-6 cell apoptosis, as determined by flow cytometry. Effect of GRb1 on the expression of b Fas and c caspase 8 in vivo. Effect of GRb1 (200 μM) on the expression of Fas in LPS-treated IEC-6 cells in the absence (d) and presence (e) of siRNA targeting Hrd1. f Effects of GRb1 (200 μM) on IEC-6 cell apoptosis in the presence of siRNA targeting Hrd1. Data are expressed as the mean ± SD. **P < 0.01 compared with the corresponding control group; ##P < 0.01 compared with the DSS control group. n = 3 samples for Western blot experiments, n = 6 samples for other experiments.
Effect of GRb1 on TNBS-induced colitis in mice
To validate the protective effects of GRb1 against colitis, another colitis model induced by TNBS was established. Compared with the sham group, the TNBS-treated group had a lower body weight, lower food intake, higher colon weight-to-length ratio, higher MPO activity, and higher proinflammatory cytokine (IL-1β, IL-6, and TNF-α) levels. In agreement with the results from our DSS-induced colitis model, GRb1 significantly alleviated the above colitis symptoms (Fig. 6). These results were also confirmed by HE staining (Fig. 6e). The colitis symptoms, including a damaged villous structure (green ring), the infiltration of inflammatory cells (black arrow), and submucosal edema (blue ring), were alleviated by GRb1.

Effects of GRb1 on a body weight, b food intake, and c colon weight-to-length ratio in colitis mice; d the gross morphology of colon tissue, and e hematoxylin-eosin staining of mouse colonic tissue. Scale bars, 100 μm. The green ring indicates damage to the villous structure; the black arrow indicates the infiltration of inflammatory cells; the blue ring indicates submucosal edema. Expression levels of colonic f MPO, g IL-1β, h IL-6, and i TNF-α were determined by ELISA. Data are expressed as the mean ± SD. **P < 0.01 compared with the sham group; ##P < 0.01 compared with the TNBS-induced colitis group; n = 6 samples.
Expression of the ER stress-related proteins CHOP and GRP78, the apoptosis marker protein cl-caspase 3, and the inflammation marker protein p-NF-κB-p65 was significantly increased by TNBS treatment, and the expression of all was reduced by GRb1 treatment. In addition to the changes in colitis symptoms, the expression of Hrd1 was significantly decreased in TNBS-induced colitis, and this decrease was inhibited by GRb1 treatment (Fig. 7). Overall, these results suggest that GRb1 alleviates TNBS-induced colitis and that modulation of the Hrd1 signaling pathways is involved in this effect.

Western blot analysis of a Hrd1, b CHOP, c GRP78, d cleaved-caspase 3 (cl-caspase 3) (calculated as cl-caspase 3/caspase 3), and e p-NF-κB-p65 (calculated as p-NF-κB-p65/NF-κB-p65). Data are expressed as the mean ± SD. **P < 0.01 compared with the sham group (n = 3 mice); ##P < 0.01 compared with the TNBS-induced colitis group; n = 3 mice.
Discussion
In colitis model mice, intestinal epithelial Hrd1 production was significantly decreased compared with that in the normal control mice. Gavage administration of GRb1 significantly alleviated colitis symptoms in both DSS- and TNBS-induced colitis mouse models. The DSS-induced colitis model is a classic colitis model initiated by intestinal barrier dysfunction. In the TNBS-induced colitis model, inflammation persists for a longer time and can transform from acute inflammation into chronic inflammation [36]. GRb1 significantly alleviated ER stress in the colitis models. In vivo and in vitro blockade of Hrd1 significantly inhibited the effects of GRb1 treatment on colitis symptoms. Overall, these results suggest that GRb1 activated epithelial Hrd1-ER stress signaling pathways to exert a protective role against colitis in the models.
GRb1, one of the major active ingredients extracted from ginseng, has been shown to target multiple signaling pathways, such as the Rho/ROCK, PI3K/mTOR, and NF-κB signaling pathways, suggesting that it has effects against autophagy, inflammation, and apoptosis [37, 38]. A previous report in the TNF-α-induced LS174T cell line suggested the potential use of GRb1 for the treatment of colitis by targeting the PXR/NF-κB interaction [28]. In this study, we demonstrated that GRb1 could activate the intestinal epithelial Hrd1 signaling pathway to inhibit ER stress, resulting in the alleviation of colitis in a mouse model. These findings provide another potential mechanism of action by which GRb1 alleviates colitis.
Hrd1, the central regulator of ERAD, participates in the ER stress response and catalyzes the degradation of misfolded and unfolded proteins [39, 40]. Decreased expression of Hrd1 has been found in the intestines of IBD patients [17]. Disruption of the ER stress balance causes intestinal epithelial cell apoptosis, impairs innate immunity, and activates proinflammatory responses in the colon [41, 42]. In both DSS- and TNBS-induced mouse colitis models and in the LPS-induced model of epithelial cell inflammation, Hrd1 expression was reduced. This reduction in Hrd1 was accompanied by increases in the ER stress-response markers GRP78 and PERK and the ER stress-dependent apoptotic proteins CHOP and caspase 12.
Decreased epithelial Hrd1 expression may result in an increase in Fas-related apoptosis. Hrd1 also carries out a variety of important functions independent of ERAD. Hrd1 can ubiquitinate denatured non-ERAD protein substrates, such as PTEN, eIF2α, and S100A8, to target them for removal and degradation [43,44,45]. Fas/CD95, a well-known cell-death receptor, localizes on the surface of various cells and interacts with its ligand, FasL/CD95L, to trigger apoptosis through an alternative pathway, the mitochondrial apoptosis-triggering pathway. In B cells, Fas, as one of the non-ERAD substrates of Hrd1, has received attention. Hrd1 inhibits cell apoptosis by ubiquitination of the death receptor Fas in B cells independent of ERAD [13]. The effects of Hrd1 on autoimmune disease development in B cells may differ from its effects in epithelial cells. In B cells, Hrd1 can promote disease development, but our results showed that Hrd1 exerted a protective effect against chemically induced colitis in intestinal epithelial cells. Our findings show that intestinal epithelial Fas levels were significantly increased in the colitis model, whereas Hrd1 levels were significantly decreased. GRb1-induced Hrd1 activation significantly reduced Fas expression and intestinal cell apoptosis, but these effects were attenuated by the inhibition of Hrd1. These results indicate that the Hrd1 and Fas signaling pathways are also involved in the regulation of intestinal epithelial cell apoptosis. Further study on the detailed functional role of Hrd1/Fas in epithelial cells and immune cells in IBD is needed.
The active forms of absorbed GRb1 in the intestine, as well as the specific mechanisms by which they activate Hrd1, should be investigated by future research. In vivo, when β-glucosidase-producing bacterial strains were used to biotransform GRb1, the GRb1 was converted into Rd, gypenoside XVII, F2, compound K, and Rg3. The GRb1 was converted in the following sequence: Rb1 → Rd → F2→ compound K [46]. Compound K, the main metabolite of aprotopanaxadiol-type ginsenoside produced by intestinal bacteria after oral administration of ginseng extract, is thought to be the major form of protopanaxadiol saponin absorbed from the intestine [47, 48]. In addition, compound K promotes the recovery of colitis by suppressing activation of the NF-κB pathway [49]. In summary, GRb1 appears to activate intestinal epithelial Hrd1 signaling pathways, thereby alleviating colitis. The findings of this study provide some understanding of the pathological mechanisms of IBD and suggest the development of GRb1 as a natural product to treat IBD. In the clinic, GRb1 has been shown to reduce the degree of oxidative stress and inflammation in patients with chronic kidney disease and to improve glycemic status in men during recovery from resistance training [50, 51]. The dose of GRb1 used in the clinic is ~10 mg/kg, and a maximum dose of 40 mg/kg GRb1 was used in our mouse study, which suggests the safety of GRb1 to treat IBD in the future. Moreover, GRb1 may also have potential for the treatment of other autoimmune diseases accompanied by decreased Hrd1 expression.
References
Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229–44.
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2011;142:46–54.e42.
Na Z, Xiuqin Z, Xifei H, Ru C, Yanqiu M. Bibliometric analysis of quality of life in 473 patients with inflammatory bowel disease from 2006 to 2016 in China. Chin J Mod Nurs. 2018;24:309–13.
Chiodini RJ, Dowd SE, Galandiuk S, Davis B, Glassing A. The predominant site of bacterial translocation across the intestinal mucosal barrier occurs at the advancing disease margin in Crohn’s disease. Microbiology. 2016;162:1608.
Theresa A, Osborne LC, Saenz SA, Dmytro K, Ziegler CGK, Mullican SE, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504:153.
Lin JC, Wu JQ, Wang F, Tang FY, Sun J, Xu B, et al. QingBai decoction regulates intestinal permeability of dextran sulphate sodium-induced colitis through the modulation of notch and NF-κB signalling. Cell Prolif. 2019;52:e12547.
Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 2018;53:465–74.
Ruiz PA, Morón B, Becker HM, Lang S, Atrott K, Spalinger MR, et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut. 2017;66:1216–24.
Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53 Suppl 5:V1–16.
Hibi T, Fujiyama Y. Biological therapies for inflammatory bowel disease. J Gastroenterol. 2002;37:43.
Sugimoto S, Naganuma M, Kiyohara H, Arai M, Ono K, Mori K, et al. Clinical efficacy and safety of oral Qing-Dai in patients with ulcerative colitis: a single-center open-label prospective study. Digestion. 2016;93:193.
Xu Y, Zhao F, Qiu Q, Chen K, Wei J, Kong Q, et al. The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat Commun. 2016;7:12073.
Kong S, Yang Y, Xu Y, Wang Y, Zhang Y, Melo-Cardenas J, et al. Endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls B-cell immunity through degradation of the death receptor CD95/Fas. Proc Natl Acad Sci U S A. 2016;113:10394–9.
Yang H, Qiu Q, Gao B, Kong S, Lin Z, Fang D. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med. 2014;211:2467–79.
Hoelen H, Zaldumbide A, van Leeuwen WF, Torfs EC, Engelse MA, Hassan C, et al. Proteasomal degradation of proinsulin requires Derlin-2, HRD1 and p97. PLoS One. 2015;10:e0128206.
Amano T, Yamasaki S, Yagishita N, Tsuchimochi K, Shin H, Kawahara K, et al. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003;17:2436–49.
Sun S, Lourie R, Cohen SB, Ji Y, Goodrich JK, Poole AC, et al. Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol Biol Cell. 2015;27:483–90.
Kaser A, Lee A, Franke A, Glickman J, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56.
Tréton X, Pédruzzi E, Cazals–Hatem D, Grodet A, Panis Y, Groyer A, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology. 2011;141:1024–35.
Ma X, Dai Z, Sun K, Zhang Y, Chen J, Yang Y, et al. Intestinal epithelial cell endoplasmic reticulum stress and inflammatory bowel disease pathogenesis: an update review. Front Immunol. 2017;8:1271.
Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require herp. Mol Cell. 2007;28:544–54.
Doroudgar S, Völkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, et al. Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res. 2015;117:536–46.
Shruthi K, Reddy SS, Reddy GB. Ubiquitin-proteasome system and ER stress in the retina of diabetic rats. Arch Biochem Biophys. 2017;627:10–20.
Tan SX, Jiang DX, Hu RC, Dai AG, Gan GX, Fu DY, et al. Endoplasmic reticulum stress induces HRD1 to protect alveolar type II epithelial cells from apoptosis induced by cigarette smoke extract. Cell Physiol Biochem. 2017;43:1337–45.
Tan S, Yu W, Lin Z, Chen Q, Shi J, Dong Y, et al. Anti-inflammatory effect of ginsenoside Rb1 contributes to the recovery of gastrointestinal motility in the rat model of postoperative ileus. Biol Pharm Bull. 2014;37:1788–94.
Oh SJ, Kim K, Lim CJ. Protective properties of ginsenoside Rb1 against UV-B radiation-induced oxidative stress in human dermal keratinocytes. Die Pharmazie. 2015;70:381–7.
Zhang YL, Liu Y, Kang XP, Dou CY, Zhuo RG, Huang SQ, et al. Ginsenoside Rb1 confers neuroprotection via promotion of glutamate transporters in a mouse model of Parkinson’s disease. Neuropharmacology. 2017;131:223–37.
Zhang J, Cao L, Wang H, Cheng X, Wang L, Zhu L, et al. Ginsenosides regulate PXR/NF-kappaB signaling and attenuate dextran sulfate sodium-induced colitis. Drug Metab Dispos. 2015;43:1181–9.
Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharm. 2010;160:1577–9.
Xiong Y, Shi L, Wang L, Zhou Z, Wang C, Lin Y, et al. Activation of sirtuin 1 by catalpol-induced down-regulation of microRNA-132 attenuates endoplasmic reticulum stress in colitis. Pharm Res. 2017;123:73–82.
Sann H, Erichsen J, Hessmann M, Pahl A, Hoffmeyer A. Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. 2013;92:708–18.
Rottgen TS, Nickerson AJ, Minor EA, Stewart AB, Harold AD, Rajendran VM. Dextran sulfate sodium-induced chronic colitis attenuates Ca2+-activated Cl− secretion in murine colon by downregulating TMEM16A. Am J Physiol Cell Physiol. 2018;315:C10–20.
Zhou P, Lu S, Luo Y, Wang S, Yang K, Zhai Y, et al. Attenuation of TNF-alpha-induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-kappaB, JNK and p38 signaling pathways. Front Pharmacol. 2017;8:464.
Bo Q, Lan Z, Zhiqun Z, Jiangqiong O, Hui H. Effects of ginsenosides-Rb1 on exercise-induced oxidative stress in forced swimming mice. Pharmacogn Mag. 2014;10:458–63.
Das UN. Inflammatory bowel disease as a disorder of an imbalance between pro- and anti-inflammatory molecules and deficiency of resolution bioactive lipids. Lipids Health Dis. 2016;15:11.
Lixuan S, Nan G, Bing C, Weixin L, Min J. Comparison of DSS, TNBS and OXZ-induced colitis in animal models. Chin J Gastroen Hepatol. 2013;22:1221–4.
Yang T, Miao Y, Zhang T, Mu N, Ruan L, Duan J, et al. Ginsenoside Rb1 inhibits autophagy through regulation of Rho/ROCK and PI3K/mTOR pathways in a pressure-overload heart failure rat model. J Pharm Pharmacol. 2018;70:830–8.
Wang J, Qiao L, Li Y, Yang G. Ginsenoside Rb1 attenuates intestinal ischemia-reperfusion-induced liver injury by inhibiting NF-kappaB activation. Exp Mol Med. 2008;40:686–98.
Xu Y, Melo-Cardenas J, Zhang Y, Gau I, Wei J, Montauti E, et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight. 2019;4:e121887.
Dibdiakova K, Saksonova S, Pilchova I, Klacanova K, Tatarkova Z, Racay P. Both thapsigargin- and tunicamycin-induced endoplasmic reticulum stress increases expression of Hrd1 in IRE1-dependent fashion. Neurol Res. 2019;41:177–88.
Cao SS. Epithelial ER stress in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2016;22:984–93.
Adolph TE, Niederreiter L, Blumberg RS, Kaser A. IBD is a disorder of defective autophagy and innate immunity: endoplasmic reticulum stress and inflammation. Dig Dis. 2012;30:341.
Liu L, Long H, Wu Y, Li H, Dong L, Zhong JL, et al. HRD1-mediated PTEN degradation promotes cell proliferation and hepatocellular carcinoma progression. Cell Signal. 2018;50:90–9.
Huang Y, Sun Y, Cao Y, Sun H, Li M, You H, et al. HRD1 prevents apoptosis in renal tubular epithelial cells by mediating eIF2α ubiquitylation and degradation. Cell Death Dis. 2017;8:3202.
Wang Y, Guo A, Liang X, Li M, Shi M, Li Y, et al. HRD1 sensitizes breast cancer cells to Tamoxifen by promoting S100A8 degradation. Oncotarget. 2017;8:23564–74.
Cheng LQ, Kim MK, Lee JW, Lee YJ, Yang DC. Conversion of major ginsenoside Rb1 to ginsenoside F2 by Caulobacter leidyia. Biotechnol Lett. 2006;28:1121–7.
Kim DY, Park MW, Yuan HD, Lee HJ, Kim SH, Chung SH. Compound K induces apoptosis via CAMK-IV/AMPK pathways in HT-29 colon cancer cells. J Agric Food Chem. 2009;57:10573–8.
Lee ES, Choi JS, Kim MS, You HJ, Ji GE, Kang YH. Ginsenoside metabolite compound K differentially antagonizing tumor necrosis factor-alpha-induced monocyte-endothelial trafficking. Chem Biol Interact. 2011;194:13–22.
Li J, Zhong W, Wang W, Hu S, Yuan J, Zhang B, et al. Ginsenoside metabolite compound K promotes recovery of dextran sulfate sodium-induced colitis and inhibits inflammatory responses by suppressing NF-kappaB activation. PLoS One. 2014;9:e87810.
Xu X, Lu Q, Wu J, Li Y, Sun J. Impact of extended ginsenoside Rb1 on early chronic kidney disease: a randomized, placebo-controlled study. Inflammopharmacology. 2017;25:33–40.
Chang WH, Tsai YL, Huang CY, Hsieh CC, Chaunchaiyakul R, Fang Y, et al. Null effect of ginsenoside Rb1 on improving glycemic status in men during a resistance training recovery. J Int Soc Sports Nutr. 2015;12:34.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81600440); basic research projects of Liaoning Province Universities (Grant No. LQ2017042); Dalian High Level Talent Support program (No. 2017RQ017); and National Key Research and Development Project of China (2017YFD050160203).
Author information
Authors and Affiliations
Contributions
DPC and JYW designed the research; JYD, KJX, WL, LLL, FY, XSF, YJX, LW, ZJZ, CYL, and WDZ performed the experiments; JYD, KJX, and WL analyzed the data; KJX and WL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Dong, Jy., Xia, Kj., Liang, W. et al. Ginsenoside Rb1 alleviates colitis in mice via activation of endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 signaling pathway. Acta Pharmacol Sin 42, 1461–1471 (2021). https://doi.org/10.1038/s41401-020-00561-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00561-9
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Ginseng-derived nanoparticles alleviate inflammatory bowel disease via the TLR4/MAPK and p62/Nrf2/Keap1 pathways
Journal of Nanobiotechnology (2024)
-
Casting Light on the Janus-Faced HMG-CoA Reductase Degradation Protein 1: A Comprehensive Review of Its Dualistic Impact on Apoptosis in Various Diseases
Molecular Neurobiology (2024)
-
miR-18a-5p shuttled by mesenchymal stem cell-derived extracellular vesicles alleviates early brain injury following subarachnoid hemorrhage through blockade of the ENC1/p62 axis
Cell and Tissue Research (2023)
-
Therapeutic material basis and underling mechanisms of Shaoyao Decoction-exerted alleviation effects of colitis based on GPX4-regulated ferroptosis in epithelial cells
Chinese Medicine (2022)