
Abstract
The etiopathogenesis of late-onset Alzheimer’s disease (AD) is increasingly recognized as the result of the combination of the aging process, toxic proteins, brain dysmetabolism, and genetic risks. Although the role of mitochondrial dysfunction in the pathogenesis of AD has been well-appreciated, the interaction between mitochondrial function and genetic variability in promoting dementia is still poorly understood. In this study, by tissue-specific transcriptome-wide association study (TWAS) and further meta-analysis, we examined the genetic association between mitochondrial solute carrier family (SLC25) genes and AD in three independent cohorts and identified three AD-susceptibility genes, including SLC25A10, SLC25A17, and SLC25A22. Integrative analysis using neuroimaging data and hippocampal TWAS-predicted gene expression of the three susceptibility genes showed an inverse correlation of SLC25A22 with hippocampal atrophy rate in AD patients, which outweighed the impacts of sex, age, and apolipoprotein E4 (ApoE4). Furthermore, SLC25A22 downregulation demonstrated an association with AD onset, as compared with the other two transcriptome-wide significant genes. Pathway and network analysis related hippocampal SLC25A22 downregulation to defects in neuronal function and development, echoing the enrichment of SLC25A22 expression in human glutamatergic neurons. The most parsimonious interpretation of the results is that we have identified AD-susceptibility genes in the SLC25 family through the prediction of hippocampal gene expression. Moreover, our findings mechanistically yield insight into the mitochondrial cascade hypothesis of AD and pave the way for the future development of diagnostic tools for the early prevention of AD from a perspective of precision medicine by targeting the mitochondria-related genes.
Similar content being viewed by others
Introduction
Late-onset Alzheimer’s disease (AD) is aheterogeneous neurodegenerative disorder symptomatically defined by gradual cognitive decline [1, 2]. Despite a consensus that hippocampal dysfunction constitutes a pivotal biological basis for AD-related cognitive deficits as well as being a critical predictor of AD risk [2,3,4,5,6], the detailed mechanisms of hippocampal vulnerability in this neurodegenerative disorder thus far remain elusive. The etiopathogenesis of AD is proposed to be a perplexing combination of aging factors, protein aggregates, brain dysmetabolism, and genetic risks [7,8,9]. In addition to the deleterious influence of AD-associated toxic molecules, including amyloid beta (Aβ) and pathological tau, basic and clinical studies have accentuated a mitochondrial pathway of hippocampal pathology and thus the resulting cognitive deficits in AD [9,10,11,12,13]. Although the polygenic nature of AD pathogenesis has been increasingly recognized [14,15,16,17], the interaction between mitochondrial function and genetic variability in the development of AD remains understudied.
Mitochondrial solute carriers, also known as solute carrier family 25 (SLC25) spanning from SLC25A1 to SLC25A53, constitute a mitochondria-specific sub-family of solute carriers (SLCs) that transports a plethora of substrates across the mitochondrial membrane [18, 19]. The substrate repertoire of SLC25 includes a collection of molecules encompassing inorganic ions, nucleotides, amino acids, and enzyme cofactors, as well as Krebs cycle metabolites [19,20,21,22]. The SLC25 family composed of a collection of members typically feature three homologous repeats and structure into three distinct regions: the cytoplasmic gate, the substrate binding site, and the matrix gate [23]. Depending on the concentrations of transported substrates, counter-substrates, competing substrates, or interactions with allosteric inhibitors or activators, the substrate binding site of SLC25 family members opens to the intermembrane space in the cytoplasmic state (c-state) and rotates to the matrix side in the matrix state (m-state) to regulate substrate transport direction [24, 25]. Most of the SLC25 family isoforms exhibit widespread expression across various tissues, whereas some isoforms demonstrate tissue-specific expression patterns to accommodate cell- and tissue-specific functions such as the abundance of UCP1 carrier SLC25A7 in the brown adipose tissue, and UCP4 carrier SLC25A27 in the brain [18]. The importance of these SLC25 substrates to mitochondrial biology and the abundance of a full spectrum of SLC25 family members in the brain, including the hippocampus, together establish a pivotal role of SLC25 in brain physiology and further underscore a causal relationship between SLC25 mutations and multiple neurological disorders such as thiamine metabolism dysfunction syndrome 4, combined D-2- and L-2-hydroxyglutaric aciduria, mitochondrial phosphate carrier deficiency, early infantile epileptic encephalopathy, and many others [26,27,28,29,30,31]. The growing supportive evidence of hippocampal mitochondrial dysfunction in AD has thus raised a critical and yet-understudied scientific question of whether the gene traits of mitochondrial SLCs may predispose hippocampal lesions, promoting the development of this neurodegenerative disorder.
In recent decades, genetic variants have been associated with AD by genome-wide association studies (GWAS) [14,15,16,17], shedding light on the diagnosis, prevention, and management of AD from a perspective of precision medicine. Transforming this idea into reality, however, is stalled by obstacles in interpreting the influence(s) of GWAS-identified single nucleotide polymorphisms (SNPs) on transcriptomics and subsequently, proteomic landscapes. Owing to the recent progress in deep- and machine-learning gene expression imputation methods, tissue-specific transcriptome-wide association study (TWAS) is emerging as a critical supplement to GWAS in bridging the disconnect between gene expression and disease traits. By using the reference panels for gene expression in different tissues, TWAS incorporates regulatory weights of genetic variants to impute gene expression in specific tissues for the prediction of gene-trait association [32,33,34]. This method offers opportunities to gain insights into the biological consequences of gene polymorphisms and enables our examination of SLC25-associated traits in AD.
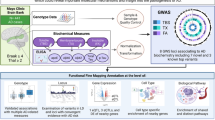
In the current study, we aimed to examine the association between regulatory variants of SLC25 family genes with AD risk and hippocampal pathology. We adopted whole-genome sequencing (WGS) GWAS summary statistics from two large-scale discovery cohorts and individual WGS data from the validation cohort, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. In reference to the Genotype-Tissue Expression (GTEx) Consortium atlas of genetic regulatory effects [35], we performed hippocampal TWAS [36] for AD-associated genes in the SLC25 family and identified three transcriptome-wide significant genes including SLC25A10, SLC25A17, and SLC25A22 among the SLC25 family. In further examination of the gene-trait relationship among the three studied genes, downregulation of SLC25A22 was associated with accelerated hippocampal atrophy in AD patients and increased hazard of dementia. Lastly, functional annotation mounted the effects of SLC25A22 regulatory variants to multiple neuronal development- and function-related pathways. Collectively, in this study, we have identified AD-susceptibility genes in the SLC25 family by TWAS-predicted hippocampal gene expression and further gene-trait association analysis. The results add supportive evidence to the mitochondrial pathway of AD and have positive impacts on future precision medicine targeting mitochondria-related SLC25 genes, especially SLC25A22, for the diagnosis and early prevention of AD in a subset of patients.
Materials and Methods
Participants and genetic data sources
Data from the participants were obtained from multiple sources, including two discovery cohorts: cohort 1 GWAS summary statistics downloaded from the Alzheimer disease (AD) GWAS Catalog on 11/27/2023 for study “GCST013197” with 90,338 AD cases and 1,036,225 nonAD controls [14], as well as cohort 2 freely-accessible GWAS summary data for Alzheimer’s disease cohorts with 71,880 AD cases and 383,378 nonAD controls [37]. Original data of the validation cohort, including WGS data, neuroimaging data, and patient information used in the preparation of this article, were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). For up-to-date information, see www.adni-info.org.
TWAS analysis using GWAS summary statistics
GWAS summary statistics of the two discovery cohorts were analyzed by transcriptome-wide association study (TWAS) to establish the connection between phenotypic associations of SNPs and gene expression levels. In this study, we employed the TIGAR-V2 tool for conducting TWAS analysis [36]. Bayesian DPR cis-eQTL weights for the hippocampus and reference LD covariance for chromosomes 1-22 based on GTEx V8 data were obtained from SYNAPSE (project SynID: syn16804286). TWAS analysis of summary statistics for cohort 1 directly employed the z score, while for cohort 2, SNP z scores were converted from odds ratio (OR), the lower bound of the 95% confidence interval (CI), and standard error (SE) using the following equation:
We conducted TWAS summary analysis on a genomic region spanning 1 million base pairs around the gene (± 1 million base pairs flanking the gene region) and set the weight threshold at 0. For dichotomous phenotypes, logistic regression was selected. We chose the statistical method employed by S-PrediXcan for the burden Z test to complement the TWAS individual analysis method.
TWAS analysis for individual genotyping data
The original WGS data from the ADNI cohort underwent an initial liftover process, transitioning from human genome version 19 (hg19) to Genome Reference Consortium Human Build 38 (GRCh38). We subsequently conducted TWAS analysis utilizing the PrediXcan software [38]. Consistent with the TWAS summary statistics analysis, we here used hippocampal Bayesian DPR cis-eQTL weights from chromosomes 1-22 as a reference database to calculate the effect size of genes. In alignment with the analysis of TWAS summary statistics, Bayesian DPR eQTL weights were employed as the reference database for gene expression predictions. Disease association analysis was executed on a cohort comprised of 229 individuals with AD and 246 nonAD healthy controls. Logistic regression was selected for dichotomous univariate phenotypes.
Neuroimaging association analysis
Individuals from 65 to 95 years of age, who had multiple magnetic resonance imaging (MRI)-based hippocampal volumetric measurements within a 2-year period of the last visit, with an interval between two MRI scans of at least 9 months in the ADNI cohort were included in calculations of the relationship between genetic regulation of susceptibility genes and the atrophy rate of the left, right, and total hippocampus (in percentage). Individuals with hippocampal MRI scans that failed to pass the quality check on their last visit were removed from the study. Hippocampal measurement data from the University of California, San Francisco (UCSF)-cross-sectional FreeSurfer 5.1 was used for the analysis [39,40,41,42,43,44,45]. The annualized hippocampal atrophy rate was calculated using the following equation:
Weighted least square regression (WLSR) analysis was used to investigate the correlation between hippocampal atrophy rate and the effect sizes of the regulatory variants of the susceptibility genes. Graphs were generated using R studio.
Functional annotation of the identified genes
QIAGEN Ingenuity Pathway Analysis (QIAGEN IPA) web service was used to examine the canonical pathways and core analysis of variants (loss and gain) related to the identified genes. Comparison analysis of genes with opposite regulation (z score) between subjects with SLC25A22 up- and down-regulation was performed to identify the differential pathways related to disease and functions also using Qiagen IPA. Hippocampal gene network analysis to examine the involvement of the identified genes with opposite regulation (z score) between subjects with SLC25A22 up- and downregulation in cohesive gene clusters was conducted by using the HumanBase (https://hb.flatironinstitute.org) [46].
Human single-cell RNA sequencing data
To investigate the expression patterns of SLC25A18 and SLC25A22, we leveraged open-access human RNA-Seq data available through the Cell Types Database on ALLEN BRAIN MAP. Datasets, including M1 10X genomics, Multiple Cortical Areas-smart-seq, and MTG 10X Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) studies from the “Human Multiple Cortical Areas” protocol, were obtained from the specified website (https://portal.brain-map.org/atlases-and-data/rnaseq). Data generation was supported by multiple awards, including the Brain Initiative Cell Census Network (BICCN) award U01MH114812 from the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke, and by the Allen Institute for Brain Science [47, 48]. The expression profiles of SLC25A18 and SLC25A22, represented by trimmed mean values, were retrieved from the three mentioned databases and subsequently visualized using R Studio.
Statistical analysis and meta-analysis
All data were analyzed using SPSS statistical software (version 29.0.0.0; IBM, Armonk, NY) unless otherwise indicated. The two-tailed Student’s t-test was employed to compare means between the two groups. Samples were normal distribution and of similar variance between the groups that are being statistically compared. Chi-squared and Fisher’s exact probability tests were used for the analysis of qualitative data differences. The meta-analysis of TWAS statistics from the two discovery cohorts and the ADNI cohort was performed with the use of the weighted Fisher’s method (wFisher) in metapro R package [49]. The p values from the TWAS analysis, corresponding sample sizes, and effect direction were input for this meta-analytical approach for the calculation of combined p values. Genes with a combined p value of less than 0.05 in the meta-analysis in the studied three cohorts were considered transcriptome-wide significant genes. The transcriptome-wide significant genes with a combined p value of less than 0.01 were considered as top candidate genes for further gene-trait analysis. Weighted least squares regression (WLSR) was used to examine the correlation between hippocampal atrophy rate and magnitude of genetic regulation. Partial least squares regression (PLSR) analysis was used to predict the influence of critical co-variables, including age (younger than 80 vs older than 80), sex (female vs male), and ApoE4 status (carrier vs non-carrier) as well as genetic regulation on the development of hippocampal lesions in small-scale cohort (n < 200) [50, 51]. Multivariable Cox proportional hazards models were used to examine the time-event relationship between the time point of event and covariates, including genetic regulation of susceptibility genes (downregulated vs. upregulated), age (age at the initial visit), sex (female vs. male), and ApoE4 status (carrier vs. noncarrier). In this time-event analysis, an event was defined as a global clinical dementia rating (CDR) score of 1 or higher within the follow-up period of up to ten years. The time point (year) of the CDR score of 1 was noted as “time of event”. Subjects with a CDR score of 0 or 0.5 by the end of the ten-year observation period were noted as “no event”. Statistical significance was set at p < 0.05 (two-sided).
Results
Identification of AD-associated SLC25 family genes with hippocampal TWAS analysis
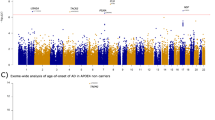
To integrate the WGS data with tissue-specific reference transcriptomic data for gene-based association studies, we performed hippocampal TWAS using Bayesian Dirichlet process regression (DPR) expression quantitative trait loci (eQTL) weights from the GTEx V8 reference [36]. A schematic workflow description of the TWAS analysis and further phenotype association is shown in Fig. 1. Our discovery cohorts consisted of two extensive datasets: the “GCST013197” study from the GWAS Catalog, encompassing 90,338 AD cases and 1,036,225 nonAD controls, and a freely-accessible Alzheimer’s disease cohort with 71,880 AD cases and 383,378 nonAD controls [14, 37]. GWAS summary statistics of the two discovery cohorts were subjected to hippocampal TWAS using the TIGAR-V2 tool to predict the effects of regulatory variants on gene expression in the hippocampus in reference to hippocampal SNP-expression associations. The TWAS summary-burden Z test results, quantified with the S-Predixcan calculation method, indicated that among the genes with predicted values, forty-one genes out of the fifty-three SLC25 family members were identified in the two discovery cohorts. Given the availability of neuroimaging data for further gene-trait analysis, we chose the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort for validation. The individual WGS data from the ADNI cohort (Table 1) underwent TWAS individual analysis using Predixcan software, employing the same reference as the mentioned TWAS summary analysis, to predict the regulation of genes, including the SLC25 family genes. We next utilized the weighted Fisher’s method (wFisher) to perform meta-analysis based on the TWAS statistics from two discovery cohorts and the ADNI cohort as validation to enhance statistical power for the combined p values [49]. At the nominal threshold of p value at 0.01, three AD-associated genes, SLC25A10, SLC25A17, and SLC25A22 were determined as candidate genes for further gene-to-disease trait association analysis (Fig. 2 and Table 2). The results showed that the upregulation of SLC25A10, which encodes the mitochondrial dicarboxylate carrier [52], and SLC25A17, which encodes the peroxisomal transporter for multiple nucleotides [53], are positively associated with AD (Fig. 2 and Table 2). In contrast, the genetic regulation of SLC25A22, which encodes the mitochondrial glutamate carrier [54], is in a negative relationship with AD (Fig. 2 and Table 2). In contrast, no disease association between genetic regulation of SLC25A10, SLC25A17, or SLC25A22 was determined in the cerebellum by TWAS statistics and meta-analysis at the nominal threshold of p value at 0.01 (Supplementary Table 1). These findings corroborate previous observations of brain region-related transcriptomic difference in humans, including brain aging and AD subjects [55,56,57].

GWAS: genome-wide association studies, WGS whole genome sequencing, ADNI the Alzheimer’s Disease Neuroimaging Initiative, eQTL expression quantitative trait loci, DPR Dirichlet process regression. GReX genetically regulated gene expression. wFisher weight Fisher method.

Each cohort is grouped based on the gene p value at a threshold of 0.05. Discovery cohort 1, n = 90,338 AD cases, n = 1,036,225 nonAD controls; discovery cohort 2, n = 71,880 AD cases, n = 383,378 nonAD controls; ADNI cohort, n = 229 AD cases, n = 246 nonAD controls. Genes including SLC25A10, SLC25A17, and SLC25A22 with a combined p value less than 0.01 via meta-analysis were selected for phenotype association study.
Association of susceptibility SLC25 family genes with hippocampal atrophy in AD
Hippocampal atrophy is a characteristic pathology of AD and accelerated volumetric loss of the hippocampus correlates with cognitive deficits associated with disease progression [58, 59]. To this end, it would be of great interest to examine the relationship between transcriptome-wide significant genes—including SLC25A10, SLC25A17, and SLC25A22—and hippocampal atrophy to establish the gene-trait association. The annualized rate of hippocampal atrophy was calculated in subjects with multiple MRI scans and MRI-based hippocampal volumetric measurements within 2 years of the last visit at an interval of at least 9 months between two MRI scans in the ADNI cohort (Table 3). In contrast to the lack of association between genetic regulation of SLC25A10 (Supplementary Fig. 1A & B) or SLC25A17 (Supplementary Fig. 2A & B) and annualized hippocampal loss, subjects with downregulation of hippocampal SLC25A22 demonstrated increased annualized loss of the left, right, and total (Fig. 3A) hippocampal volume as compared to those with SLC25A22 upregulation. Further weighted least square regression (WLSR) analysis showed a negative correlation between hippocampal atrophy rate and the magnitude of SLC25A22 genetic regulation (Fig. 3B). In the cohort of AD patients only, accelerated hippocampal atrophy was also associated with hippocampal SLC25A22 downregulation (Fig. 3C). Such an inverse relationship between hippocampal gene expression and hippocampal atrophy rate was not detected with SLC25A10 (Supplementary Fig. 1C) or SLC25A17 (Supplementary Fig. 2C) in the AD cohort. Next, to predict the contribution of the transcriptome-wide significant genes to the development of hippocampal atrophy accompanying AD, we performed partial least square regression (PLSR) analysis, which is a frequently-used statistical tool in neuroimaging studies of small-scale cohorts (N < 200) to reliably identify relevant variables and estimate their impacts [50, 51]. In addition to the tested genes, aging, sex, and ApoE4 status, which are strong risk factors associated with brain atrophy [60,61,62], were also included as critical co-variables in the analysis. By setting hippocampal atrophy rate as the dependent variable, the first three latent factors explained ~80% of variance, and SLC25A22 expression exhibited top variable importance in the projection (VIP), outweighing ApoE4 and age (Fig. 3D) and demonstrated, by its weight, a strong negative association with annualized hippocampal loss (Fig. 3E & F) in all three conditions. However, neither SLC25A10 (Supplementary Fig. 1D–F) nor SLC25A17 (Supplementary Fig. 2D-F) exhibited strong influence on hippocampal atrophy in AD patients by PLSR analysis. Collectively, these findings indicate the role of hippocampal SLC25A22 regulation with pathological characteristic of AD and further support an association of SLC25A22 with the development of AD.

A Comparison of hippocampal atrophy rate between SLC25A22 down- and up-regulated subjects in the AD and nonAD combined cohort. Two-tail student t-test. SLC25A22 downregulated n = 38, SLC25A22 upregulated n = 222. * p < 0.05, ** p < 0.01. Data is represented as mean ± 95% CI. B Weighted least square regression (WLSR) analysis for the correlation between hippocampal atrophy rate and the effect size of SLC25A22 genetic regulation. SLC25A22 downregulated n = 38, SLC25A22 upregulated n = 222. * p < 0.05, ** p < 0.01. C Comparison of hippocampal atrophy rate between SLC25A22 down-and up-regulated subjects in AD patients. Two-tail student t-test. SLC25A22 downregulated n = 17, SLC25A22 upregulated n = 79. * p < 0.05. Data is represented as mean ± 95% CI. D–F Partial least square regression (PLSR) analysis in the AD cohort. The annualized hippocampal atrophy rate was set as the dependent variable. SLC25A22, age, sex, and ApoE4 status were input as covariables in the analysis. D Variable importance in the projection (VIP) of SLC25A22, age, sex, and ApoE4. E, F GGraphics of latent factors 1 and 3 (E) as well as 2 and 3 (F).
Association of susceptibility SLC25 family genes with AD risk
To examine whether genetic regulation of hippocampal SLC25A10, SLC25A17, and SLC25A22 may promote AD development, a total of 124 subjects in the ADNI cohort with the diagnosis of nonAD and the global clinical dementia rating (CDR) score of 0 at the initial visit as well as yearly CDR evaluations were included in the study. The demographic information of the selected subjects is presented in Table 4. Within the follow-up period of up to ten years, the studied subjects with a change in global CDR score from 0 to 1 or greater were considered “at risk of dementia (event)”. Multivariable Cox proportional hazards regression was performed for the time-event relationship. After adjustment for ApoE4 status, sex, and age, hippocampal SLC25A22 downregulation was significantly associated with a faster development of dementia, demonstrated by a hazard ratio (HR) of 3.078 (95% CI:1.035 to 9.154) (Fig. 4A). In contrast, an effect on the speed of dementia onset was not obtained with genetic regulation of SLC25A10 (Fig. 4B) or SLC25A17 (Fig. 4C). Therefore, among the three susceptibility genes associated with AD, downregulation of hippocampal SLC25A22 demonstrates an association with an increased probability of dementia.

Cox proportional hazards regression models for the association of dementia onset with genetic regulation of hippocampal SLC25A22 (A), SLC25A10 (B), and SLC25A17 (C) within the follow-up period of up to ten years. Sex, ApoE4 status, and age were included as covariates in multivariable Cox regression. * p < 0.05. A total of 124 subjects were included for the analysis. HR: hazard ratio.
Functional annotation of hippocampal gene expression associated with genetic SLC25A22 regulation
To interpret the biological significance of the SLC25A22 gene trait and examine the pattern of transcriptomic regulation in subjects with opposite SLC25A22 regulation, the tested cohort was clustered into two subsets: SLC25A22 down- and up-regulated groups, respectively. After comparison of the mean value of all the genes predicted by hippocampal TWAS (Fig. 5A), a total of 622 genes with reversed z score direction between the two groups were identified (Fig. 5B). Genes with predicted values in the SLC25A22 down- and upregulated groups were subjected to variant loss/gain analysis using Qiagen Ingenuity Pathway Analysis (IPA) software for canonical pathway analysis. Among the downregulated signaling pathways, multiple pathways related to biological processes in the brain, including “glutaminergic receptor signaling pathway”, “dopamine-DARPP32 feedback in cAMP signaling”, “synaptogenesis signaling pathway”, “orexin signaling pathway”, “serotonin receptor signaling”, and “GABAergic receptor signaling pathway” as well as “acetylcholine receptor signaling pathway” were noted. “Senescence pathway” and “corticotropin releasing hormone signaling” topped the upregulated pathways (Fig. 5C). In sharp contrast, “synaptogenesis signaling pathway” and “GABAergic signaling pathway”, which were suppressed in the SLC25A22 downregulated group, were predicted to be enhanced in subjects with upregulated SLC25A22 (Fig. 5D). Furthermore, comparison pathway analysis using the identified 622 genes with reversed z score direction between the SLC25A22 down- and up-regulated groups demonstrated inverted regulation of several pathways related to neuronal function and development, with “formation of cellular protrusions”, “neuritogenesis”, and “development of neural cells” at the top (Fig. 5E). To examine the impact(s) of differentially-regulated genes on biological functions in the context of hippocampal tissues, the 622 genes with reversed z score direction between the SLC25A22 down- and up-regulated groups were mounted to the hippocampal network in the HumanBase online website (https://hb.flatironinstitute.org) [46] and clustered in multiple modules of biological processes such as “double-strand DNA stability” (M1), “developmental growth” (M2), “lipid metabolism and inflammation” (M3), “cytosolic transport” (M4), and “regulation of transporter activity” (M5) (Fig. 5F and Supplementary Table 2). Those findings support functional and developmental defects of hippocampal neurons in subjects with SLC25A22 downregulation.

A Regulation of hippocampal genes in SLC25A22 down- and up-regulated subjects. B Genes with reverse z score directions in SLC25A22 down- and up-regulated subjects. C, D Canonical pathway analysis of genes in SLC25A22 downregulated C and upregulated D subjects. E Comparison analysis of genes with opposite z score directions between SLC25A22 down- and up-regulated subjects. F Hippocampal network analysis of genes with opposite z score directions between SLC25A22 down- and up-regulated subjects in cohesive gene clusters. M1: double-strand DNA stability, M2: developmental growth, M3: lipid metabolism and inflammation, M4: cytosolic transport, M5: regulation of transporter activity.
Selective abundance of SLC25A22 gene expression in human glutamatergic neurons
Previous studies have determined two types of mitochondrial glutamate carriers, including SLC25A22 and SLC25A18 located in the inner mitochondrial membrane both designated for glutamate transport [54]. The high degree of biological homology in both their sequences and functions [54] thus raise an interesting question about whether genetic downregulation of SLC25A22 could be, at least in part, compensated by the unaffected expression of SLC25A18. To this end, we sought to examine expression patterns of SLC25A22 and SLC25A18 in the human brain at the cell-type resolution using the Allen brain map online tools (https://portal.brain-map.org/atlases-and-data/rnaseq) [47, 48]. In searching for SLC25A22 and SLC25A18 expression in the Human M1 10X single-cell sequencing data, we noticed that SLC25A22 in the brain is predominantly expressed by glutamatergic neurons, which differs from the abundance of SLC25A18 expression in astrocytes (Fig. 6A). Similar patterns of SLC25A22 and SLC25A18 expression in the human brain were determined in the SMART-SEQ (Fig. 6B) and Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) - Spatial transcriptomics - MERFISH studies (Fig. 6C). The distinct expression patterns of SLC25A22 and SLC25A18 indicate glutamatergic neuronal vulnerability to genetic downregulation of SLC25A22 and further correlate with our functional annotation of hippocampal genes showing deficits in neuronal function and development in subjects with genetic downregulation of SLC25A22. Of note, a study done in rats has determined higher expression of SLC25A22 in astrocytes than in neurons [63]. Furthermore, although mRNA levels do not always correlate with protein expression, the results of selective abundance of SLC25A22 mRNA in human neurons together with reported enrichment of SLC25A22 in mouse neurons [64, 65] seem to implicate a species-related difference in SLC25A22 expression.

A–C Heatmaps of SLC25A22 and SLC25A18 expression in different brain cell types of single cell sequencing data from A M1 10X GENOMICS (2020), B MULTIPLE CORTICAL AREAS-SMART-SEQ (2019), C MTG 10X Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) (2022). OPC: Oligodendrocyte progenitor cell.
Discussion
In contrast to the Mendelian inheritance pattern of familial Alzheimer’s disease (AD), the contribution of genetic components to the etiopathogenesis of late-onset Alzheimer’s disease (AD), especially in patients without a clear history of transmission, remains unclear [66]. In recent years, several powerful tools including tissue-specific transcriptome-wide association studies (TWAS) and Mendelian randomization (MR) have been developed to assist in the functional interpretation of genome-wide association studies (GWAS), which disentangle the association of genetic variants within genetically complex diseases such as late-onset AD [32, 67, 68]. The mitochondrial solute carrier family (SLC25), consisting of 53 members, constitutes crucial transporters for the exchange of a plethora of small molecules between mitochondria and the cytosol and/or other cytosolic organelles [19, 22]. Consistent with their importance to mitochondrial function, defects within the SLC25 members have been linked to mitochondrial dysfunction in cancers as well as in a diverse spread of neuro-developmental and degenerative disorders [22, 69,70,71]; however, the genetic variability of SLC25s and their phenotypic consequences in AD have never been comprehensively investigated. In this study, we conducted hippocampal TWAS analysis to predict gene expression of the SLC25 family and their association(s) with AD using GWAS summary data from two extensive cohorts of AD patients and individual WGS data from the ADNI cohort. The findings revealed a significant association between the genetic regulation of SLC25A10, SLC25A17, and SLC25A22, and the risk of developing AD. Further neuroimaging studies indicated a strong reverse relationship between cis-regulation of SLC25A22 and hippocampal atrophy rate. Notably, our partial least square regression (PLSR) analysis using the ADNI cohort weighed SLC25A22 over well-characterized risk factors for AD and AD-associated hippocampal atrophy such as sex (female), aging, and ApoE4 status [60,61,62, 72, 73] in disrupting hippocampal volumetric integrity. Therefore, these results support SLC25A22 as a susceptibility gene for AD and further connect SLC25A22 downregulation to hippocampal vulnerability in a subset of AD patients. It should be noted that in addition to the hippocampus, other brain regions such as the amygdala, cingulate gyrus, temporal and frontal cortexes, and others are also affected in AD [74]. Taking into consideration the abundance of SLC25A22 in glutamatergic neurons, SLC25A22 downregulation may also be associated with accelerated decay with disease progression in these brain regions, which warrants further investigation.
In view of our results, it would be of paramount interest to explore the potential mechanisms underlying the association of SLC25A22 with AD. SLC25A22-encoded protein [SLC25A22, also known as glutamate carrier 1 (GC1)] together with SLC25A18 [also known as mitochondrial glutamate carrier 2 (GC2)] are the two carriers for the mitochondrial transport of glutamate. Our analysis of the single-cell sequencing data from the Human M1 10X single-cell sequencing, SMART-SQ and SEA-AD data indicates an abundance of SLC25A22 expression in glutamatergic neurons and selective SLC25A18 expression in astrocytes. These findings not only suggest the limited capacity of SLC25A18 to compensate for neuronal loss of SLC25A22, but also raise a critical scientific question regarding the yet-underappreciated role of SLC25A22 in neuronal glutamate metabolism. It is well-established that glutamate, the primary excitatory neurotransmitter, is predominantly cleared by astrocytes after its release into the synaptic cleft [75]. In recent years, the role of neurons in glutamate metabolism has gained increasing attention, and experimental evidence suggests that part of the glutamate released from synapses can be sent back to neurons, probably via glutamate transporter 1 encoded by SLC1A2 [76]. In addition to its return into synaptic vesicles for reuse, the reabsorbed glutamate also can be used by mitochondria to fuel oxidative phosphorylation (OXPHOS) for ATP generation [77]. Previous studies have highlighted a key role of mitochondrial glutamate/aspartate antiporter, SLC25A12, in importing glutamate into mitochondria to complete the malate-aspartate shuttle (MAS) and energize mitochondria in neurons as well as other types of cells in the brain [78,79,80]. Additionally, in light of its function as a mitochondrial glutamate/H+ symporter, a significant contribution of SLC25A22 to mitochondrial glutamate transport and subsequent bioenergetic regulation of neurons is emerging [65, 81], which supports our hypothesis that lowered expression of SLC25A22, even to a small degree, compromises neuronal and mitochondrial glutamate metabolism, which causes energy deficiency and cytosolic glutamate overburden and predisposition to AD. Intriguingly, a recent study reported that activated expression of neuron-enriched mitochondrial proteins, including SLC25A22 in astrocytes promotes the conversion of astrocytes to neurons in vitro [64]. These interesting findings not only underscore the significance of SLC25A22 in the homeostatic regulation of neuronal function but also implicate the potentially deleterious impact of SLC25A22 downregulation on neuronal regeneration in neurodegenerative disorders, including AD. It should be noted that consistent with the importance of SLC25A22 to mitochondrial and neuronal fitness, exonic mutations in SLC25A22 have been linked to multiple developmental disorders such as thiamine metabolism dysfunction syndrome 4, combined D-2- and L-2-hydroxyglutaric aciduria, mitochondrial phosphate carrier deficiency, and early infantile epileptic encephalopathy [26,27,28,29,30,31]. In our current study, in contrast to exonic mutations in SLC25A22 that are associated with lethal developmental disorders, the identified AD-associated mutations are located in the non-coding regions of SLC25A22. Although the current study on intronic mutations is still in an early stage, emerging evidence highlights the potential impacts of mutations in the noncoding regions on gene expression or splicing through various pathways, such as the short-distance regulation of the efficacy of promoters and/or enhancers, and long-distance modulation of intron-promoter 3D interactions [82, 83]. Therefore, it is possible that the AD-associated SLC25A22 polymorphism has a relatively weak but persistent deleterious impact on gene expression, culminating in severe pathological consequences with age.
Subjects with SLC25A22 downregulation also demonstrated altered transcriptomic architecture with changes in the genetic regulation of a collection of genes including ABLIM1, which encodes actin-binding LIM protein 1, ABRA, which encodes actin-binding Rho-activating protein, VAMP5, which encodes vesicle-associated membrane protein 5, STX3, which encodes syntaxin 3, NLGN1, which encodes neuroligin 1, and many others. These genes were mounted to pathways related to neuronal function and development such as “formation of cellular protrusions”, “neuritogenesis”, and “development of neural cells”, as well as “dendritic growth/branching” and “shape change of neurons”. Although data from a larger population is needed to confidently establish the link between SLC25A22 and all or some of these genes, current results seem to add further supportive evidence to the deleterious influence of SLC22A22 downregulation on neuronal development and function. Indeed, it should be remembered that the TWAS-predicted effects of regulatory variants indicate the genetic regulation of selected gene(s) but may not fully represent actual gene expression and, to a further extent, actual protein expression in each individual [34]. To this end, the impact of genetic SLC25A22 due to regulatory variants on mitochondrial function and neuronal glutamate metabolism requires further in-depth investigation and validation.
Lastly, this study indicates that mitochondrial factors may independently contribute to AD. Previous studies on mitochondrial dysfunction in brain aging and AD have overwhelmingly focused on mitochondrial dysfunction in response to the aging process, AD-associated pathological molecules, and systemic factors with disease progression [7, 9,10,11,12,13, 84], thus underscoring the contributing role of mitochondrial dysfunction to AD development. However, a key scientific question about whether mitochondria play a proactive role in the etiopathogenesis of AD or act as passive responders to AD-associated toxic proteins remains unaddressed. To date, evidence of the mitochondrial role as an initiator of AD is emerging. Multiple heteroplasmic mitochondrial DNA (mtDNA) mutations have been reported in AD patients [85,86,87,88,89]. Previous studies have shown that transferring platelet mtDNA from AD patients into cells with depletion of endogenous mtDNA promotes AD-like changes [90, 91], these findings as well as the association of mtDNA haplogroups with AD risk [92] add credit to the hypothesis of mitochondria-related mode of transmission in the development of this neurodegenerative disorder. In addition to mtDNA mutations, our findings of nuclear DNA (nDNA)-encoded SLC25-related susceptibility genes of AD yield new insight into the interaction between genetic variability and mitochondria in the development of dementia. Because the vast majority of mitochondria-associated proteins are encoded by nuclear genes [93], increased attention to the connection between mutations in mitochondrial protein-encoding nuclear genes and mitochondrial deficits will broaden our view of inherited mitochondrial dysfunction in the development of AD and further strengthen the mitochondrial cascade hypothesis of this neurodegenerative disorder [10, 94, 95]. Furthermore, increasing evidence implicates somatic genetic [96, 97] and epigenetic [98, 99] factors in dementia. To this end, in addition to hereditary mutations, the contribution of somatic mutations and epigenetic regulation of mitochondrial genes to AD risk also merits further investigation.
Several limitations of the current study should also be noted. Although we established the association of transcriptome-wide significant SLC25 family members, including SLC25A10, SLC25A17, and SLC25A22 with AD, further phenotypic association of SLC25 family genes with hippocampal volumetric loss was conducted using a cohort of a relatively smaller size. Moreover, due to the limitations of the reference transcriptomic data, the hippocampal expression of only forty-one out of the total of fifty-three SLC25 family genes was predicted and used in this study. Furthermore, genetic data was collected primarily from subjects with European ancestry. These caveats warrant the need for future validation of our findings through the analysis of neuroimaging and genetic datasets with larger sample size and broader diversity. Of note, in the current study, we primarily focused on the association of SLC25A22 regulation with the risk of late onset AD. A related question is whether SLC25A22 polymorphism may also play a role in the development of early-onset AD. It is well-documented that the etiopathogenesis of early-onset AD is closely related to autosomal dominant mutations in PSEN1, PSEN2, and APP [100]. These genetic alterations lead to an earlier onset and accelerated progression of the disease. We cannot fully exclude the contribution of SLC25A22 polymorphism to early onset AD, at least, in a subset of patients although its contribution, if exists, might be relatively weak given the genetic penetrance of the aforementioned early onset-AD-related genetic risks. This outstanding question warrants further investigation of the association of hippocampal SLC25A22 regulation with phenotype development in patients with early-onset AD and, to a further extent, in patients with other types of AD-related dementia (ADRD) such as Lewy body dementia and Down syndrome, which also demonstrate hippocampal lesions and AD-like cognitive deficits [101, 102].
In summary, our findings identified the association of hippocampal SLC25 genes, including SLC25A10, SLC25A17, and SLC25A22 with AD risk through TWAS-predicted hippocampal gene expression. Further gene-to-disease trait assessment supports the influence of hippocampal SLC25A22 downregulation on hippocampal lesions in AD and the transition from non-demented to AD. In contrast, hippocampal SLC25A22 upregulation is protective against AD. Therefore, we conclude that genetic variability of the identified SLC25 family genes, especially SLC25A22 contribute to AD. Further investigation on SLC25A22 in experimental and clinical settings holds promise to deepen our understanding of the mitochondrial pathway of AD and advance the development of strategies targeting this AD-susceptibility gene for the diagnosis and early prevention of this neurodegenerative disorder.
Data availability
The data of this study is available from the corresponding authors upon request with additional approval.
Code availability
The analyses were conducted using standard code that are available in the software instruction page and can be provided by the corresponding author upon request. All software used in this study is freely available online.
References
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44.
Henneman WJ, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC, et al. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology. 2009;72:999–1007.
Rao YL, Ganaraja B, Murlimanju BV, Joy T, Krishnamurthy A, Agrawal A. Hippocampus and its involvement in Alzheimer’s disease: a review. 3 Biotech. 2022;12:55.
Xiao Y, Hu Y, Huang K, Alzheimer’s Disease Neuroimaging I. Atrophy of hippocampal subfields relates to memory decline during the pathological progression of Alzheimer’s disease. Front Aging Neurosci. 2023;15:1287122.
Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp. 2010;31:1339–47.
Miao D, Zhou X, Wu X, Chen C, Tian L. Hippocampal morphological atrophy and distinct patterns of structural covariance network in Alzheimer’s disease and mild cognitive impairment. Front Psychol. 2022;13:980954.
Jia K, Tian J, Wang T, Guo L, Xuan Z, Swerdlow RH, et al. Mitochondria-sequestered Abeta renders synaptic mitochondria vulnerable in the elderly with a risk of Alzheimer disease. JCI Insight. 2023;8:e174290.
Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–36.
Swerdlow RH. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis. 2018;62:1403–16.
Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30.
Pradeepkiran JA, Baig J, Selman A, and Reddy PH. Mitochondria in Aging and Alzheimer’s Disease: Focus on Mitophagy. Neuroscientist. 2023:10738584221139761.
Guo L, Tian J, Du H. Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer’s Disease. J Alzheimers Dis. 2017;57:1071–86.
Watts A, Chalise P, Hu J, Hui D, Pa J, Andrews SJ, et al. A Mitochondrial DNA Haplogroup Defines Patterns of Five-Year Cognitive Change. J Alzheimers Dis. 2022;89:913–22.
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
Belloy ME, Andrews SJ, Le Guen Y, Cuccaro M, Farrer LA, Napolioni V, et al. APOE Genotype and Alzheimer Disease Risk Across Age, Sex, and Population Ancestry. JAMA Neurol. 2023;80:1284–94.
Chen HH, Petty LE, Sha J, Zhao Y, Kuzma A, Valladares O, et al. Genetically regulated expression in late-onset Alzheimer’s disease implicates risk genes within known and novel loci. Transl Psychiatry. 2021;11:618.
Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34:465–84.
Ruprecht JJ, Kunji ERS. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem Sci. 2020;45:244–58.
Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722–30.
Goubert E, Mircheva Y, Lasorsa FM, Melon C, Profilo E, Sutera J, et al. Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front Cell Neurosci. 2017;11:149.
Gutierrez-Aguilar M, Baines CP. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem J. 2013;454:371–86.
Saraste M, Walker JE. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982;144:250–4.
Henderson PJ, Lardy HA. Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem. 1970;245:1319–26.
Lauquin GJ, Duplaa AM, Klein G, Rousseau A, Vignais PV. Isobongkrekic acid, a new inhibitor of mitochondrial ADP-ATP transport: radioactive labeling and chemical and biological properties. Biochemistry. 1976;15:2323–7.
Edvardson S, Porcelli V, Jalas C, Soiferman D, Kellner Y, Shaag A, et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J Med Genet. 2013;50:240–5.
Mayr JA, Merkel O, Kohlwein SD, Gebhardt BR, Bohles H, Fotschl U, et al. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet. 2007;80:478–84.
Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, et al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–24.
Molinari F, Kaminska A, Fiermonte G, Boddaert N, Raas-Rothschild A, Plouin P, et al. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin Genet. 2009;76:188–94.
Palmieri F, Scarcia P, Monne M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules. 2020;10:655.
Ayka A, Sehirli AO. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin Psychopharmacol Neurosci. 2020;18:174–87.
Mai J, Lu M, Gao Q, Zeng J, Xiao J. Transcriptome-wide association studies: recent advances in methods, applications and available databases. Commun Biol. 2023;6:899.
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–9.
de Leeuw C, Werme J, Savage JE, Peyrot WJ, Posthuma D. On the interpretation of transcriptome-wide association studies. PLoS Genet. 2023;19:e1010921.
Consortium GT. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30.
Parrish RL, Gibson GC, Epstein MP, Yang J. TIGAR-V2: Efficient TWAS tool with nonparametric Bayesian eQTL weights of 49 tissue types from GTEx V8. HGG Adv. 2022;3:100068.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13.
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–8.
Gunter JL, Bernstein MA, Borowski BJ, Ward CP, Britson PJ, Felmlee JP, et al. Measurement of MRI scanner performance with the ADNI phantom. Med Phys. 2009;36:2193–205.
Hua X, Ching CRK, Mezher A, Gutman BA, Hibar DP, Bhatt P, et al. MRI-based brain atrophy rates in ADNI phase 2: acceleration and enrichment considerations for clinical trials. Neurobiol Aging. 2016;37:26–37.
Hua X, Hibar DP, Lee S, Toga AW, Jack CR Jr., Weiner MW, et al. Sex and age differences in atrophic rates: an ADNI study with n=1368 MRI scans. Neurobiol Aging. 2010;31:1463–80.
Jack CR Jr., Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91.
Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010;31:1401–18.
Westman E, Simmons A, Muehlboeck JS, Mecocci P, Vellas B, Tsolaki M, et al. AddNeuroMed and ADNI: similar patterns of Alzheimer’s atrophy and automated MRI classification accuracy in Europe and North America. Neuroimage. 2011;58:818–28.
Wyman BT, Harvey DJ, Crawford K, Bernstein MA, Carmichael O, Cole PE, et al. Standardization of analysis sets for reporting results from ADNI MRI data. Alzheimers Dement. 2013;9:332–7.
Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47:569–76.
Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–8.
Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–8.
Yoon S, Baik B, Park T, Nam D. Powerful p-value combination methods to detect incomplete association. Sci Rep. 2021;11:6980.
Krishnan A, Williams LJ, McIntosh AR, Abdi H. Partial Least Squares (PLS) methods for neuroimaging: a tutorial and review. Neuroimage. 2011;56:455–75.
Grady CL, Rieck JR, Nichol D, Rodrigue KM, Kennedy KM. Influence of sample size and analytic approach on stability and interpretation of brain-behavior correlations in task-related fMRI data. Hum Brain Mapp. 2021;42:204–19.
Johnson RN, Chappell JB. The inhibition of mitochondrial dicarboxylate transport by inorganic phosphate, some phosphate esters and some phosphonate compounds. Biochem J. 1974;138:171–5.
Agrimi G, Russo A, Scarcia P, Palmieri F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem J. 2012;443:241–7.
Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277:19289–94.
Wang X, Michaelis ML, Michaelis EK. Functional genomics of brain aging and Alzheimer’s disease: focus on selective neuronal vulnerability. Curr Genomics. 2010;11:618–33.
Evans SJ, Choudary PV, Vawter MP, Li J, Meador-Woodruff JH, Lopez JF, et al. DNA microarray analysis of functionally discrete human brain regions reveals divergent transcriptional profiles. Neurobiol Dis. 2003;14:240–50.
Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci USA. 2008;105:15605–10.
Franko E, Joly O, Alzheimer’s Disease Neuroimaging I. Evaluating Alzheimer’s disease progression using rate of regional hippocampal atrophy. PLoS One. 2013;8:e71354.
Leung KK, Shen KK, Barnes J, Ridgway GR, Clarkson MJ, Fripp J, et al. Increasing power to predict mild cognitive impairment conversion to Alzheimer’s disease using hippocampal atrophy rate and statistical shape models. Med Image Comput Comput Assist Interv. 2010;13:125–32.
Honea RA, Vidoni E, Harsha A, Burns JM. Impact of APOE on the healthy aging brain: a voxel-based MRI and DTI study. J Alzheimers Dis. 2009;18:553–64.
Markov NT, Lindbergh CA, Staffaroni AM, Perez K, Stevens M, Nguyen K, et al. Age-related brain atrophy is not a homogenous process: Different functional brain networks associate differentially with aging and blood factors. Proc Natl Acad Sci USA. 2022;119:e2207181119.
Filon JR, Intorcia AJ, Sue LI, Vazquez Arreola E, Wilson J, Davis KJ, et al. Gender Differences in Alzheimer Disease: Brain Atrophy, Histopathology Burden, and Cognition. J Neuropathol Exp Neurol. 2016;75:748–54.
Berkich DA, Ola MS, Cole J, Sweatt AJ, Hutson SM, LaNoue KF. Mitochondrial transport proteins of the brain. J Neurosci Res. 2007;85:3367–77.
Russo GL, Sonsalla G, Natarajan P, Breunig CT, Bulli G, Merl-Pham J, et al. CRISPR-Mediated Induction of Neuron-Enriched Mitochondrial Proteins Boosts Direct Glia-to-Neuron Conversion. Cell Stem Cell. 2021;28:524–34.e7.
Wynne ME, Lane AR, Singleton KS, Zlatic SA, Gokhale A, Werner E, et al. Heterogeneous Expression of Nuclear Encoded Mitochondrial Genes Distinguishes Inhibitory and Excitatory Neurons. eNeuro. 2021;8:ENEURO.0232-21.2021.
Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, Vargas-Rodriguez I, Cadena-Suarez AR, Sanchez-Garibay C, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int J Mol Sci. 2023;24:3754.
Li B, Ritchie MD. From GWAS to Gene: Transcriptome-Wide Association Studies and Other Methods to Functionally Understand GWAS Discoveries. Front Genet. 2021;12:713230.
Korologou-Linden R, Bhatta L, Brumpton BM, Howe LD, Millard LAC, Kolaric K, et al. The causes and consequences of Alzheimer’s disease: phenome-wide evidence from Mendelian randomization. Nat Commun. 2022;13:4726.
Kunji ERS, King MS, Ruprecht JJ, Thangaratnarajah C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology (Bethesda). 2020;35:302–27.
Liu AR, Liu YN, Shen SX, Yan LR, Lv Z, Ding HX, et al. Comprehensive Analysis and Validation of Solute Carrier Family 25 (SLC25) and Its Correlation with Immune Infiltration in Pan-Cancer. Biomed Res Int. 2022;2022:4009354.
Chen YJ, Hong WF, Liu ML, Guo X, Yu YY, Cui YH, et al. An integrated bioinformatic investigation of mitochondrial solute carrier family 25 (SLC25) in colon cancer followed by preliminary validation of member 5 (SLC25A5) in tumorigenesis. Cell Death Dis. 2022;13:237.
Ardekani BA, Izadi NO, Hadid SA, Meftah AM, Bachman AH, Alzheimer’s Disease Neuroimaging I. Effects of sex, age, and apolipoprotein E genotype on hippocampal parenchymal fraction in cognitively normal older adults. Psychiatry Res Neuroimaging. 2020;301:111107.
Ardekani BA, Hadid SA, Blessing E, Bachman AH. Sexual Dimorphism and Hemispheric Asymmetry of Hippocampal Volumetric Integrity in Normal Aging and Alzheimer Disease. AJNR Am J Neuroradiol. 2019;40:276–82.
Ledig C, Schuh A, Guerrero R, Heckemann RA, Rueckert D. Structural brain imaging in Alzheimer’s disease and mild cognitive impairment: biomarker analysis and shared morphometry database. Sci Rep. 2018;8:11258.
Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells. 2019;8:184.
Zhou Y, Hassel B, Eid T, Danbolt NC. Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochem Int. 2019;123:101–13.
Brymer KJ, Barnes JR, Parsons MP. Entering a new era of quantifying glutamate clearance in health and disease. J Neurosci Res. 2021;99:1598–617.
Perez-Liebana I, Juaristi I, Gonzalez-Sanchez P, Gonzalez-Moreno L, Rial E, Podunavac M, et al. A Ca(2+)-Dependent Mechanism Boosting Glycolysis and OXPHOS by Activating Aralar-Malate-Aspartate Shuttle, upon Neuronal Stimulation. J Neurosci. 2022;42:3879–95.
Dienel GA. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol Rev. 2019;99:949–1045.
McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. 2006;71:399–407.
Robinson MB, Lee ML, DaSilva S. Glutamate Transporters and Mitochondria: Signaling, Co-compartmentalization, Functional Coupling, and Future Directions. Neurochem Res. 2020;45:526–40.
Rigau M, Juan D, Valencia A, Rico D. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019;15:e1007902.
Heyn P, Kalinka AT, Tomancak P, Neugebauer KM. Introns and gene expression: cellular constraints, transcriptional regulation, and evolutionary consequences. Bioessays. 2015;37:148–54.
Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun. 2016;7:11483.
Miller B, Kim SJ, Mehta HH, Cao K, Kumagai H, Thumaty N, et al. Mitochondrial DNA variation in Alzheimer’s disease reveals a unique microprotein called SHMOOSE. Mol Psychiatry. 2023;28:1813–26.
Antonyova V, Kejik Z, Brogyanyi T, Kaplanek R, Pajkova M, Talianova V, et al. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair (Amst). 2020;91-92:102871.
Phillips NR, Simpkins JW, Roby RK. Mitochondrial DNA deletions in Alzheimer’s brains: a review. Alzheimers Dement. 2014;10:393–400.
Blanchard BJ, Park T, Fripp WJ, Lerman LS, Ingram VM. A mitochondrial DNA deletion in normally aging and in Alzheimer brain tissue. Neuroreport. 1993;4:799–802.
Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, et al. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–6.
Wilkins HM, Carl SM, Swerdlow RH. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014;2:619–31.
Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD Jr., Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann N Y Acad Sci. 1999;893:176–91.
Swerdlow RH, Hui D, Chalise P, Sharma P, Wang X, Andrews SJ, et al. Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimers Dement. 2020;16:1164–72.
Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019;20:267–84.
Swerdlow RH. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: A Current Overview. J Alzheimers Dis. 2023;92:751–68.
Dewanjee S, Chakraborty P, Bhattacharya H, Chacko L, Singh B, Chaudhary A, et al. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free Radic Biol Med. 2022;193:134–57.
Miller MB, Reed HC, Walsh CA. Brain Somatic Mutation in Aging and Alzheimer’s Disease. Annu Rev Genomics Hum Genet. 2021;22:239–56.
Miller MB, Huang AY, Kim J, Zhou Z, Kirkham SL, Maury EA, et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature. 2022;604:714–22.
Xiao X, Liu X, Jiao B. Epigenetics: Recent Advances and Its Role in the Treatment of Alzheimer’s Disease. Front Neurol. 2020;11:538301.
Migliore L, Coppede F. Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat Rev Neurol. 2022;18:643–60.
Mendez MF. Early-onset Alzheimer Disease and Its Variants. Continuum (Minneap Minn). 2019;25:34–51.
Izzo A, Mollo N, Nitti M, Paladino S, Cali G, Genesio R, et al. Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Mol Med. 2018;24:2.
Spano M, Signorelli M, Vitaliani R, Aguglia E, Giometto B. The possible involvement of mitochondrial dysfunctions in Lewy body dementia: a systematic review. Funct Neurol. 2015;30:151–8.
Acknowledgements
This work was supported by research fundings from NIH (R01AG053588, R01AG059753 and R01AG075108 to HD; P30 AG035982 to RS), Higuchi Biosciences Center research grant to HD and RS, Brightfocus Foundation research grant A20201159S to HD, Brightfocus Foundation research grant A2022036S to LG, KU career development grant 2302009 to LG, NIH P30 AG072973 to KU ADC, KU School of Medicine, and the Landon Center on Aging, NIH P30 AG072973 to the University of Kansas Alzheimer’s Disease Research Center’s Research Education Component, and REC fellowship to JT. Data collection and sharing for the Alzheimer’s Disease Neuroimaging Initiative (ADNI) is funded by the National Institute on Aging (National Institutes of Health Grant U19 AG024904). The grantee organization is the Northern California Institute for Research and Education. In the past, ADNI has also received funding from the National Institute of Biomedical Imaging and Bioengineering, the Canadian Institutes of Health Research, and private sector contributions through the Foundation for the National Institutes of Health (FNIH) including generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research &Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics.
Author information
Authors and Affiliations
Consortia
Contributions
The following authors carried out data collection and analysis: JT, KJ, TW, LG, and HD. ZX, EKM, RHS, and HD contributed to the design of experiments and helped with a critical reading of the manuscript. JT and HD wrote the manuscript. HD supervised the experiments and conceived the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Whole genomic sequencing data, neuroimaging data, and patient information from the ADNI program were collected in compliance with the Declaration of Helsinki and approved by the Ethics and Institutional Review Committees of the participating institutions, as detailed in the consortium section. Each site followed a protocol approved by its institutional review board (IRB), and informed consent was obtained from all participants at each site.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
*Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, J., Jia, K., Wang, T. et al. Hippocampal transcriptome-wide association study and pathway analysis of mitochondrial solute carriers in Alzheimer’s disease. Transl Psychiatry 14, 250 (2024). https://doi.org/10.1038/s41398-024-02958-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-02958-0
- Springer Nature Limited