
Abstract
There is a lack of knowledge regarding the relationship between proneness to dimensional psychopathological syndromes and the underlying pathogenesis across major psychiatric disorders, i.e., Major Depressive Disorder (MDD), Bipolar Disorder (BD), Schizoaffective Disorder (SZA), and Schizophrenia (SZ). Lifetime psychopathology was assessed using the OPerational CRITeria (OPCRIT) system in 1,038 patients meeting DSM-IV-TR criteria for MDD, BD, SZ, or SZA. The cohort was split into two samples for exploratory and confirmatory factor analyses. All patients were scanned with 3-T MRI, and data was analyzed with the CAT-12 toolbox in SPM12. Psychopathological factor scores were correlated with gray matter volume (GMV) and cortical thickness (CT). Finally, factor scores were used for exploratory genetic analyses including genome-wide association studies (GWAS) and polygenic risk score (PRS) association analyses. Three factors (paranoid-hallucinatory syndrome, PHS; mania, MA; depression, DEP) were identified and cross-validated. PHS was negatively correlated with four GMV clusters comprising parts of the hippocampus, amygdala, angular, middle occipital, and middle frontal gyri. PHS was also negatively associated with the bilateral superior temporal, left parietal operculum, and right angular gyrus CT. No significant brain correlates were observed for the two other psychopathological factors. We identified genome-wide significant associations for MA and DEP. PRS for MDD and SZ showed a positive effect on PHS, while PRS for BD showed a positive effect on all three factors. This study investigated the relationship of lifetime psychopathological factors and brain morphometric and genetic markers. Results highlight the need for dimensional approaches, overcoming the limitations of the current psychiatric nosology.
Similar content being viewed by others


Introduction
There is a long tradition of investigating the relationship between psychopathological syndromes and brain structure and function in patients suffering from schizophrenia (SZ) and schizoaffective disorder – henceforth referred to as schizophrenia spectrum disorders (SSD), as well as bipolar disorder (BD), and major depressive disorder (MDD). Several studies have linked specific symptoms such as verbal hallucinations to local brain structures, particularly the bilateral superior temporal gyri [1,2,3]. However, these have been either low in statistical power or variance [4], or limited to a specific diagnosis, such as SZ [5, 6]. This raises the question of generalizability across diagnostic categories: Since almost all symptoms can be present in different diagnoses (e.g., formal thought disorders are found in SZ, as well as in BD, and in MDD) [7,8,9], it is of major interest to study these syndromes transdiagnostically using dimensional approaches. Moreover, the phenotypic overlap between psychiatric disorders is also reflected at a brain structural [10,11,12,13] as well as genetic level [14].
Factor analyses of lifetime psychopathology have mostly been performed within one categorical disorder. Only a few studies are available, investigating transdiagnostic symptom dimensions of lifetime psychopathology across diagnoses: Investigating patients with DSM-IV diagnosed SZ, BD and delusional disorder, Serretti and Olgiati found that a five-factor model best described lifetime symptom dimensions [15]. In a sample consisting of patients with SZ, BD, MDD, delusional, and psychotic disorder not otherwise specified, a four-factor solution was obtained, consisting of excitement, psychotic features (hallucinations and delusions), depression and disorganization [16]. Studying the factor structure of the OPerational CRITeria (OPCRIT) system in the SZ spectrum and BD, Reininghaus and colleagues obtained a bifactor model with one transdiagnostic psychosis dimension and five specific factors comprising positive, negative, manic, disorganized and depressive symptoms [17].
Previously, most structural and functional magnetic resonance neuroimaging studies focused on categorical comparisons of one patient group (MDD, BD, or SSD) compared to a healthy control (HC) group. However, these studies failed to identify structural and functional brain correlates that separate disorders [18]. In contrast, studies and meta-analyses indicated common alterations across diagnoses [11,12,13, 19, 20]. Transdiagnostic studies of dimensional psychopathology might thus be more promising regarding identification of common risk factors and might especially lead to a more precise treatment of these syndromes on a transdiagnostic rather than diagnosis-based level. In addition, they should be able to take into account the heterogeneity of psychiatric disorders as well as potential comorbidities. This should also help to identify specific neurobiological markers which in turn can inform personalized treatment interventions.
Twin and family studies demonstrate that genetic factors contribute substantially to the development of MDD, BD and SZ, with heritability estimates of around 60% to 85% for SZ and BD [21,22,23] and around 40% for MDD [24]. Recent genome-wide association studies (GWAS) have identified numerous genome-wide significant loci for all three psychiatric disorders (e.g., refs. [25, 26]). Furthermore, transdiagnostic GWAS meta-analyses have demonstrated an extensive genetic overlap between MDD, BD and SZ [14]. Byrne et al. provided evidence that only a small subset of the genome-wide significant variants for SZ and MDD have disorder-specific effects [27]. One plausible hypothesis, therefore, is that pleiotropic genetic variants mediate their disease risk via effects on transdiagnostic symptom dimensions. In addition, an analysis of polygenic risk scores (PRS), which summarize the effects of multiple common genetic variants into an individual genetic risk profile [28], by Ruderfer et al. showed that the PRS for SZ was significantly increased in BD patients with psychotic features and SZ patients with prominent negative symptoms [29]. These results suggest that there are genetic factors underlying specific symptom dimensions within both disorders [29].
As symptom presentations can fluctuate within an individual patient over the course of life and even within a single episode, the aim of the present study was to i) assess lifetime symptoms in a transdiagnostic sample to identify underlying symptom factors; and ii) investigate the relationship of detected factors with local GMV and CT. Considering that brain structure is less variable within a short period of time, we hypothesize that this approach would yield more conclusive results than correlating GMV with psychopathology present at any given point in time. In addition, applying both GMV as well as CT measures should render a fuller picture of underlying mechanisms as we would not assume that all potential associations would be based on one measure alone. Finally, iii) it was explored if the detected factor structure can be linked to common genetic variation. Based on previous brain-morphometric and genetic studies, we hypothesized findings from specific DSM-IV diagnostic categories to be present across diagnoses, too.
Material and methods
Participants
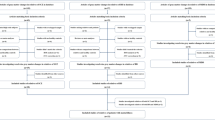
Patients were recruited as part of the FOR2107 cohort [30] (www.for2107.de). Patient recruitment took place via the in-patient facilities of the University hospitals in Marburg and Münster, Germany, through participating hospitals, and via postings in local newspapers. Written informed consent was obtained from all patients before participation. According to the Declaration of Helsinki, all procedures were approved by the local ethics committees. After study participation, all patients received financial compensation. After excluding patients with incomplete data, serious medical illnesses, neurological illnesses, and an IQ < 80, we analyzed a total of N = 1038 patients (see Table 1a, b, required sample size is based on [31]) suffering from MDD, BD, and SSD (aged 18–65).
Psychopathological assessment and factor score calculation
The German version of the structured interview SCID-I (DSM-IV-TR [32]) and the OPCRIT (version 4 [33]) were administered in all patients. Lifetime psychopathology was assessed as any occurrence of symptoms during the life span until data acquisition. Trained personnel assessed lifetime symptoms based on patients’ reports and additional hospital records, when available. Numerous interview trainings assured data quality. Interrater-reliability was assessed with the interclass coefficient, achieving good reliability of r > 0.86. For the present study, only symptomatic items were included (items 17–77). Following the procedures described in Stein et al. [34], we separated the total cohort (N = 1038) into two samples using the “mindiff” [35] package in R [36] accounting for age, sex, and diagnostic category (i.e., same distribution of categorical diagnoses across both samples). In the first sample of n = 520, we performed varimax rotated principal axis factor analyses with bootstrapping (5000 permutations) using the psych [37] and EFAutilities [38] packages in R (v4.0.5.) for models with 2–5 factors. Hereof, z-transformed values were used since the data was differently scaled. For interpretation purposes, items with factor loadings <0.5 were not considered in the analysis [34]. Cronbach’s alpha coefficients [39] were used to test the internal consistency of the explorative factors. Using the second sample of n = 518, we cross-validated the explorative models using confirmatory factor analysis in Mplus (version 8.4 [40]). Confirmatory model estimation was performed using the maximum-likelihood-method (MLM) since this estimator is robust to standard errors and is one of the most common estimators [41]. The following fit indices were used: chi-square significance test, comparative fit index (CFI [42]) and Root Mean Square Error of Approximation (RMSEA [43]). Based on these fit indices, we evaluated the different models and selected the one with best fit. After cross-validating the explorative model in the second sample, we tested the model for the whole sample (N = 1038).
As the DSM-IV-TR diagnostic groups were unequally distributed, we wanted to rule out potential confounding effects of formal diagnosis. Therefore, we tested the selected factorial model in an age- and sex-matched sample with an equal diagnosis distribution (each n = 108 of MDD, BD, SSD, total n = 324) (see supplement eTable1). Matching was performed using the “MatchIt” package [44] in R [36]. Furthermore, the factorial model was also tested within each of the three diagnostic categories and factor loadings were compared between DSM diagnosis using non-parametric Kruskal–Wallis tests (see supplement).
MRI assessment and preprocessing
Subjects were scanned with a 3-T MRI at the Department of Psychiatry and Psychotherapy in Marburg (Tim Trio, Siemens, Erlangen, Germany; 12-channel head coil) and the Institute for Translational Psychiatry in Münster (Prisma, Siemens, Erlangen, Germany; 20-channel head coil). MRI data were acquired according to an extensive quality assurance protocol [45]. A fast gradient echo MP-RAGE sequence with a slice thickness of 1.0 mm consisting of 176 sagittal orientated slices in Marburg and 192 in Münster and a FOV of 256 mm was used to acquire T1 weighted images. Parameters differed across sites: Marburg: TR = 1.9 s, TE = 2.26 ms, TI = 900 ms, flip angle = 9°; Münster: TR = 2.13 s, TE = 2.28 ms, TI = 900 ms, flip angle = 8°.
For a detailed description of the preprocessing of MRI data please see refs. [31, 46]. In short, both voxel-based-morphometry GMV and cortical thickness (CT) data were preprocessed using the default parameters as implemented in the CAT12-Toolbox (Computation Anatomy Toolbox for SPM, build 1184, Structural Brain Mapping group, Jena University Hospital, Germany) building on SPM12 (Statistical Parametric Mapping, Institute of Neurology, London, UK). We opted for GMV and CT over other MRI-derived metrics for two primary reasons. Firstly, volume and thickness measures, commonly employed in large-scale analyses such as those by the ENIGMA consortium, were selected to facilitate result comparisons. Second, recent neuroimaging research has underscored the complementary nature of GMV and CT measurements. GMV provides insight into overall gray matter volume, which can reflect global brain atrophy or neurodevelopmental factors. In contrast, CT offers information about the thickness of the cortical mantle, allowing for the detection of localized changes. By analyzing both GMV and CT, we aimed to capture both global and local structural alterations in the context of these psychiatric disorders [47, 48]. Images were spatially registered, segmented [49] and normalized [50]. CT preprocessing included fully-automated methods projecting local maxima to other GM voxels using a neighbor relationship described by the white matter distance [51]. Quality control of processed data was performed as implemented in CAT12. For GMV data, a Gaussian kernel of 8 mm FWHM was used for smoothing. For CT data, a Gaussian kernel of 20 mm FWHM was used.
Statistical analyses: gray matter volume and cortical thickness
For both GMV and CT analyses, we used separate linear regression models for each factor using CAT12 and SPM12. The following nuisance variables were included in brain structural analyses: age, sex, and two dummy-coded variables accounting for the different MRI scanners and a body coil exchange in Marburg (Marburg pre body coil: yes/no, Marburg post body coil: yes/no, Münster as reference category [30, 45]). To control for potential medication effects, we used three dummy coded (yes/no) covariates accounting for the current medication with antidepressants, mood stabilizers and antipsychotics. For GMV analyses total intracranial volume was additionally accounted as covariate of no interest and absolute threshold masking with a threshold value of 0.1 was used.
To further test confounding effects of unequally distributed diagnostic categories, we performed multiple regression analyses in the age and sex matched sample (n = 324) with same n per diagnosis, again. Besides this whole brain analysis, we additionally performed ROI analyses of the detected clusters from the total sample in the matched sub-sample (see supplement).
In addition to multiple regression analyses, we performed full factorial ANCOVA whole brain interaction analyses (factor x diagnosis) for each of the three factors to test whether transdiagnostic brain correlates were driven by single DSM-IV-TR diagnostic categories for both the total and the matched sample with same n per diagnosis. Moreover, post hoc interaction analyses (factor x diagnosis) were performed specifically within each detected cluster of the total sample using the “lm-function” in R.
Cluster labeling was applied using the dartel space Neuromorphometrics atlas (http://www.neuromorphometrics.com/) for GMV analyses and for CT analyses using the Desikan–Killiany atlas [52]. Results were suggested significant at p < 0.05 peak-level, family wise error (FWE) corrected, cluster extend k = 35 voxels in the total and matched sample.
GWAS and PRS association analysis
DNA extraction, genome-wide genotyping, quality control and imputation were carried out as previously described [53] in the full FOR2107 cohort. Briefly, genotyping was performed using genomic DNA from blood samples and the Infinium PsychArray BeadChip (Illumina, San Diego, CA, USA). Pre-imputation quality control (QC) was performed in GenomeStudio, PLINK v1.9 [54], and R [36], with removal of genetic variants and individuals according to standard filter criteria. Genotype data were imputed to the 1000 Genomes phase 3 reference panel [55] using SHAPEIT [56] and IMPUTE2 [57]. In post-imputation QC, variants with a minor allele frequency <1%, Hardy-Weinberg equilibrium p < 1e−6, and an INFO metric <0.8 were removed. From the total sample of the present study (N = 1038), high-quality genotype data were available for 951 individuals. From these, 13 individuals were excluded due to intra-sample relatedness (π ̂ > 0.125), resulting in a sample of n = 938 individuals used for genetic analyses.
For each of the three factors, GWAS, which should be considered exploratory at the given sample size, were conducted via linear regression in PLINK with rank-based inverse normal transformed values [58] as quantitative phenotypes due to the non-normal distribution of factor scores. Sex, age and the first four multidimensional scaling (MDS) components were included as covariates. All variables were z-scaled via the ‘standard-beta’ modifier for better comparability between factor dimensions. We performed clumping of genetic markers in the GWAS results using a maximum p value of 1e−4 for index variants (‘--clump-p1 1e−4’), an LD threshold of 0.1 (‘--clump-r2 0.1’), and a window size of 1000 kb (‘--clump-kb 500’). We considered genetic associations with p < 5e−8 to be genome-wide significant and with p < 1e−6 to be suggestive. We performed gene-based and gene-set analyses with MAGMA [59] as implemented in FUMA [60]. The resulting p values were corrected for multiple testing using the Bonferroni method taking into account the number of tested genes (n = 18,846) or gene sets (n = 10,678). We used LocusZoom [61] to generate regional plots.
PRS for MDD, BD and SZ were calculated based on publicly available summary statistics from three studies [25, 26, 62]. Variant weights for PRS calculation were estimated with PRS-CS [63] using default parameters and a set of pre-defined values for the global shrinkage parameter φ (1e−4, 1e−3, 1e−2). PRS were subsequently calculated in R [36] as described previously [64]. Linear additive models with rank-based inverse normal transformed factor scores as outcome, one of the z-scaled disorder-specific PRS as predictor and sex, age and the first four MDS components as covariates were fitted in R. The PRS association analysis was conducted for both the complete set of n = 938 individuals as well as for each diagnostic subgroup separately. Adjustment of p values for multiple testing was performed with the Benjamini–Hochberg approach [65] within each set of 27 tests (3 factor dimensions * 3 PRS models * 3 values for φ). Model coefficients were considered to be statistically significant at p < 0.05. We calculated the variance explained (R2) by each PRS as the difference between R2 of the full model and R2 of the null model containing only the covariates.
Results
Exploratory and confirmatory factor analyses
We tested explorative models ranging from 2-5 factors. Results of these models can be found in Supplementary eTables 2a–d. In a next step, we evaluated the four explorative models using confirmatory analyses in the second sample. Model fits were as follows: a) 2 factors: χ2 = 393.645, df = 224, p < 0.001, CFI = 0.903, RMSEA = 0.038; b) 3 factors: χ2 = 543.005, df = 316, p < 0.001, CFI = 0.904, RMSEA = 0.037; c) 4 factors: χ2 = 588.773, df = 314, p < 0.001, CFI = 0.875, RMSEA = 0.042; d) 5 factors: χ2 = 748.705, df = 391, p < 0.001, CFI = 0.884, RMSEA = 0.041. Based on the fit indices, we decided to use model b) with 3 factors (Table 2) for further analyses as this model outperformed the other ones. Moreover, a 3-factor model is also in line with the Scree Plot (Supplementary eFigure 1). The model included the factors paranoid-hallucinatory syndrome (PHS) (explaining 14% of total variance), mania (MA) (explaining 11% of total variance), and depression (DEP) (explaining 5% of total variance). Furthermore, we performed a confirmatory factor analysis in the whole sample (N = 1038) showing a good fit χ2 = 605.667, df = 316, p < 0.0001, CFI = 0.932, RMSEA = 0.03. Results of the confirmatory analyses of the matched sample and within each diagnostic category are presented in the supplement (Supplementary eResults1 and 2). We investigated differences of the factor loadings between diagnostic categories using a non-parametric ANOVA (Kruskal–Wallis). Diagnostic groups differed significantly in all three factors identified (Supplementary eResults3 and Supplementary eFigure 2).
Brain morphometric correlates of life-time psychopathology
Results of the multiple regression analyses of the total sample are displayed in Table 3 (GMV) and 4 (CT). For the paranoid-hallucinatory syndrome (PHS), negative GMV correlations were observed in the bilateral hippocampus, amygdala, and right angular gyrus (see Fig. 1). CT was negatively correlated with the paranoid-hallucinatory syndrome (PHS) comprising left supramariginal, bilateral superior temporal, and right lateral occipital clusters (see Fig. 2). Whole-brain interaction analyses revealed no significant interaction of psychopathological factor and DSM-IV-TR diagnosis for both GMV and CT. Post hoc interaction analyses on the significant clusters in Tables 3 and 4 revealed no significant interactions of factor x diagnosis (all ps > 0.05, see Supplementary eResults4 and Supplementary eFig. 3–10 for details). Results of the GMV and CT analyses in the matched sample are presented in the supplement (Supplementary eResults 5, Supplementary eTables 3 and 4). Here, results from the total sample could be replicated. We did not find any associations with the DEP and MA factors for both GMV and CT.

Negative association of factor 1 paranoid-hallucinatory syndrome (PHS) and gray matter volume (GMV) comprising parts of the bilateral hippocampus, amygdala, and right angular gyrus across patients with major depressive disorder, bipolar disorder, and schizophrenia spectrum disorders. Clusters are shown at p < 0.05 peak-level, family-wise error-corrected.

Negative association of factor 1 paranoid-hallucinatory syndrome (PHS) and cortical thickness (CT) comprising parts of left supramariginal, bilateral superior temporal, and right lateral occipital clusters across patients with major depressive disorder, bipolar disorder, and schizophrenia spectrum disorders. Clusters are shown at p < 0.05 peak-level, family-wise error-corrected.
Genetic correlates of life-time psychopathology
Our exploratory GWAS revealed genome-wide significant associations for MA and DEP (Fig. 3, Supplementary eFigs. 11–13, Supplementary eTable 5), with intronic lead variants rs10062519 (p = 1.10e−8) located in ADAMTS19 for MA and rs11131155 (p = 4.12e−8) located in RAD18 for DEP. In the MAGMA gene analysis, a genome-wide significant association was identified for SYTL1 (DEP, p = 1.79e−6). The MAGMA gene-set analysis yielded no statistically significant results for any of the three factors after correction for multiple testing (data not shown).

Regional plots with a window size of 500 kb are shown for the genome-wide significant associations with MA “mania” (A), and DEP “depression” (B). The respective lead variants rs10062519 and rs11131155 are depicted as linkage disequilibrium reference variants (purple diamonds). cM centimorgan, LD Ref Var, linkage disequilibrium reference variant, Mb megabase.
In the PRS association analysis of the complete sample (Fig. 4), we detected a positive effect of PRS for BD on all three factors (PHS: maximum β = 0.13 at φ = 1e−3 with R2 = 0.021 and adjusted p = 5.48e−5; MA: maximum β = 0.18 at φ = 1e−3 with R2 = 0.031 and adjusted p = 1.17e−6; DEP: maximum β = 0.08 at φ = 1e−4 with R2 = 0.006 and adjusted p = 0.038). Further, a positive effect on PHS was observed for the PRS for MDD (maximum β = 0.07 at φ = 1e−2 with R2 = 0.006 and adjusted p = 0.038) and SZ (maximum β = 0.13 at φ = 1e−3 with R2 = 0.020 and adjusted p = 7.06e−5). In the subset analysis of each diagnostic group, none of the effects observed in the complete transdiagnostic sample reached statistical significance (Supplementary eFig. 14).

Regression of the three factors on the PRS for MDD, BD, and SZ shows significant effects of PRS for MDD, BD, and SZ on PHS “paranoid-hallucinatory syndrome” and of PRS for BD on MA “mania” and DEP “depression” in the full transdiagnostic sample. BD bipolar disorder, BH Benjamini–Hochberg, MDD major depressive disorder, SZ schizophrenia.
Discussion
In the present study, exploratory and confirmatory factor analyses of lifetime psychopathology revealed a three-factor model with superior fit properties compared to models with less or more factors. Factors were the paranoid-hallucinatory syndrome (PHS), mania (MA) and depression (DEP). In addition, several associations with both brain morphometry and genetics were reported. This study represents a successful advancement of previous research by Stein et al. [34] and David et al. [66] all of them part of the FOR2107 cohort, wherein five factors of acute psychopathology were described and genetically investigated.
Compared to previous factor analytical research in the three diagnoses included in our study, utilizing the OPCRIT, the present factor solution features three factors, while other studies showed an additional negative factor, which was not present in our study. Nevertheless, an overlap exists as previous models also comprised a depression factor and a mania factor (e.g., refs. [15, 67]). The often-reported factors positive and negative symptoms are split into all three factors in the present results while disorganization best fits the present second factor MA.
The derived lifetime psychopathological factors were used to investigate underlying GMV and CT correlates. We were able to detect numerous associations between the PHS and both GMV and CT in both temporal and frontal regions. We did not detect any interactions for both factor x diagnosis on a whole-brain level, nor in post hoc analyses of the significant clusters. These findings do not exclude that the severity of both brain structural alterations and psychopathological syndromes may vary by diagnosis. Our study aligns with previous studies proposing overlaps in acute psychopathology, brain structure as well as genetics across MDD, BD and SSD [11,12,13, 68]. Combining a data-driven approach to psychopathology with studying neuroanatomical and genetic correlates may help elucidate the biological underpinnings of complex syndromes in psychiatric disorders. Approaches such as those applied in the present study can reveal intra- and inter-disorder heterogeneity and thus could support the establishment of treatments specific to symptom or syndrome in the next step.
When comparing our results to previous dimensional studies, a recent study also identified subcortical volume reductions associated with hallucinations as well as delusions [69], but reductions of superior temporal areas have also been well established in SSD [1, 3, 70, 71]. The present findings are also in line with a recent investigation where psychotic symptoms were negatively correlated with CT in a large sample of SSD patients, relatives and healthy controls [72]. Consistent with previous studies in SSD, we found cortical thinning in the bilateral STG to be correlated with the PHS factor [73], indicating this brain structure to be a core feature of positive symptomatology.
Exploratory GWAS and PRS analyses suggest a contribution of common genetic variants to all three factor dimensions, supporting the hypothesis that symptoms observed in different diagnostic groups may be influenced by the same genetic variants across diagnostic boundaries [14, 29]. Interestingly, the genome-wide significant loci of our GWAS implicated protein-coding genes that both might be linked to psychiatric disorders. ADAMTS19 is a member of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motif) family [74], which might be involved in neuroplasticity [75]. The RAD18 gene encodes for a DNA damage repair protein [74, 76]. Notably, a study by Alsulami and colleagues provided evidence that RAD18 interacts with SETD1A [76], which has previously been associated with SSD at the rare variant level [77, 78]. As it is known that genome-wide significant lead variants do not necessarily exert their effects through the nearest genes (e.g., ref. [79]), the above discussed functional interpretations should be viewed with caution, as further bioinformatic and functional analyses are needed to identify the gene(s) relevant at the identified loci.
Finally, despite the associations at the genetic level, we did not detect an association between the MA or DEP factor and brain morphology. This suggests that even though aspects of lifetime psychopathology might at least be partially influenced by genetic factors, this might not necessarily be detectable on a neural level. It could thus be argued that a dimensional approach is even more important than a narrow nosology as these associations might be subtle and implications for translation into treatment options are not as clear, yet.
Limitations
There are several limitations to be considered: First, as lifetime psychopathology was assessed only at one point in this study, a bias may arise in favor of symptoms that have occurred recently or are currently present, as they could be more salient in the individual’s memory. This bias could lead to an overemphasis on these symptoms during the assessment process [80]. As a result, symptoms that occurred in the past may be underreported or forgotten entirely. We tried to circumvent these biases by carefully examining every hospital record available for each patient, but these were not available for all patients included here. In addition, the used psychopathological scale did not include the full symptomatic spectrum, which restricted the identification of psychopathological factors.
Second, sample sizes of each diagnostic category were unequal. The aim of the present study was to investigate syndrome-brain structure and syndrome-genetic associations dimensionally rather than within categorical diagnoses. The presence of psychotic and manic symptoms in the MDD group might be limited, which may result in restricted variance found for the PHS factor. While results can be considered as diagnosis-shared, severity may be differing across diagnoses.
Third, MRI techniques in general might not be able to detect subtle differences in locations of effects if these occurs in close proximity. In addition, true effects between groups might be mapped onto the same neural circuit while in fact there are differences on the underlying cellular level [81].
Fourth, pharmacological treatment was considered as three dummy coded variables to account for the current intake of antidepressants, antipsychotics, and mood stabilizers. This approach does not take into account both the dosage and lifetime cumulative intake of psychotropic medication, which might have influenced our results.
Finally, the available sample size represents a limitation for the genetic analyses, as the robust detection of genetic associations with small effect sizes usually requires meta-analytical efforts involving multiple cohorts [82]. Thus, the exploratory nature of the presented GWAS should be considered in the interpretation of our findings.
Conclusion
This study comprehensively investigated the association of lifetime psychopathological dimensions and brain morphometric markers as well as underlying genetic factors. At the level of brain imaging, GMV and CT reductions in temporal, occipital, and limbic structures were found to be correlated with paranoid-hallucinatory symptoms in a transdiagnostic sample. On the genetic level, we identified genome-wide significant loci for MA and DEP factors, as well as positive effects of specific PRS on different factors. These findings suggest that genetic factors contribute to the identified factor dimensions. The results presented in this study highlight the importance of i) dimensional modeling and ii) transdiagnostic research gaining a better understanding of pathophysiological mechanisms underlying MDD, BD and SSD.
Data availability
The data and code supporting the findings of this study can be accessed by contacting the corresponding author (FS).
References
Modinos G, Costafreda SG, van Tol M-J, McGuire PK, Aleman A, Allen P. Neuroanatomy of auditory verbal hallucinations in schizophrenia: A quantitative meta-analysis of voxel-based morphometry studies. Cortex. 2013;49:1046–55.
Wong TY, Radua J, Pomarol-Clotet E, Salvador R, Albajes-Eizagirre A, Solanes A et al. An overlapping pattern of cerebral cortical thinning is associated with both positive symptoms and aggression in schizophrenia via the ENIGMA consortium. Psychol Med. 2020;50:2034–45.
Palaniyappan L, Balain V, Radua J, Liddle PF. Structural correlates of auditory hallucinations in schizophrenia: A meta-analysis. Schizophr Res. 2012;137:169–73.
Kim GW, Kim YH, Jeong GW. Whole brain volume changes and its correlation with clinical symptom severity in patients with schizophrenia: A DARTEL-based VBM study. PLoS One. 2017;12. https://doi.org/10.1371/journal.pone.0177251.
Kircher T, Markov V, Krug A, Eggermann T, Zerres K, Nöthen MM, et al. Association of the DTNBP1 genotype with cognition and personality traits in healthy subjects. Psychol Med. 2009;39:1657–65.
Nenadic I, Sauer H, Gaser C. Distinct pattern of brain structural deficits in subsyndromes of schizophrenia delineated by psychopathology. Neuroimage. 2010;49:1153–60.
Kircher T, Krug A, Stratmann M, Ghazi S, Schales C, Frauenheim M, et al. A rating scale for the assessment of objective and subjective formal thought and language disorder (TALD). Schizophr Res. 2014;160:216–21.
Andreasen NC, Grove WM. Thought, language, and communication in schizophrenia: diagnosis and prognosis. Schizophr Bull. 1986;12:348–59.
Stein F, Buckenmayer E, Brosch K, Meller T, Schmitt S, Ringwald KG et al. Dimensions of Formal Thought Disorder and Their Relation to Gray- and White Matter Brain Structure in Affective and Psychotic Disorders. Schizophr Bull. 2022. https://doi.org/10.1093/SCHBUL/SBAC002.
Patel Y, Parker N, Shin J, Howard D, French L, Thomopoulos SI et al. Virtual Histology of Cortical Thickness and Shared Neurobiology in 6 Psychiatric Disorders. JAMA Psychiatry. 2020. https://doi.org/10.1001/jamapsychiatry.2020.2694.
Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, et al. Identification of a Common Neurobiological Substrate for Mental Illness. JAMA Psychiatry. 2015;72:305.
Brosch K, Stein F, Schmitt S, Pfarr JK, Ringwald KG, Thomas-Odenthal F et al. Reduced hippocampal gray matter volume is a common feature of patients with major depression, bipolar disorder, and schizophrenia spectrum disorders. Mol Psychiatry. 2022;27. https://doi.org/10.1038/S41380-022-01687-4.
Repple J, Gruber M, Mauritz M, de Lange SC, Winter NR, Opel N et al. Shared and specific patterns of structural brain connectivity across affective and psychotic disorders. Biol Psychiatry. 2022. https://doi.org/10.1016/J.BIOPSYCH.2022.05.031.
Lee PH, Anttila V, Won H, Feng YCA, Rosenthal J, Zhu Z, et al. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell. 2019;179:1469–82.e11.
Serretti A, Olgiati P. Dimensions of major psychoses: A confirmatory factor analysis of six competing models. Psychiatry Res. 2004;127:101–9.
Serretti A, Rietschel M, Lattuada E, Krauss H, Schulze TG, Müller DJ, et al. Major psychoses symptomatology: Factor analysis of 2241 psychotic subjects. Eur Arch Psychiatry Clin Neurosci. 2001;251:193–8.
Reininghaus U, Böhnke JR, Hosang G, Farmer A, Burns T, McGuffin P, et al. Evaluation of the validity and utility of a transdiagnostic psychosis dimension encompassing schizophrenia and bipolar disorder. Br J Psychiatry. 2016;209:107–13.
Lalousis PA, Wood SJ, Schmaal L, Chisholm K, Griffiths SL, Reniers RLEP et al. Heterogeneity and Classification of Recent Onset Psychosis and Depression: A Multimodal Machine Learning Approach. Schizophr Bull. 2021. https://doi.org/10.1093/schbul/sbaa185.
Chang M, Womer FY, Edmiston EK, Bai C, Zhou Q, Jiang X, et al. Neurobiological Commonalities and Distinctions Among Three Major Psychiatric Diagnostic Categories: A Structural MRI Study. Schizophr Bull. 2018;44:65–74.
Koutsouleris N, Meisenzahl EM, Borgwardt S, Riecher-Rössler A, Frodl T, Kambeitz J, et al. Individualized differential diagnosis of schizophrenia and mood disorders using neuroanatomical biomarkers. Brain. 2015;138:2059–73.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a Complex Trait: Evidence from a Meta-analysis of Twin Studies. Arch Gen Psychiatry. 2003;60:1187–92.
Lichtenstein P, Yip BH, Björk C, Pawitan Y. Common genetic influences for schizophrenia and bipolar disorder: A population-based study of 2 million nuclear families. Lancet; 2009;373. https://doi.org/10.1016/S0140-6736(09)60072-6.
Bienvenu OJ, Davydow DS, Kendler KS. Psychiatric diseases versus behavioral disorders and degree of genetic influence. Psychol Med. 2011;41:33–40.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: Review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Byrne EM, Zhu Z, Qi T, Skene NG, Bryois J, Pardinas AF et al. Conditional GWAS analysis to identify disorder-specific SNPs for psychiatric disorders. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0705-9.
Lewis CM, Vassos E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020;12. https://doi.org/10.1186/s13073-020-00742-5.
Ruderfer DM, Ripke S, McQuillin A, Boocock J, Stahl EA, Pavlides JMW, et al. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell. 2018;173:1705–1715.e16.
Kircher T, Wöhr M, Nenadic I, Schwarting R, Schratt G, Alferink J, et al. Neurobiology of the major psychoses: a translational perspective on brain structure and function-the FOR2107 consortium. Eur Arch Psychiatry Clin Neurosci. 2018;1:3.
Stein F, Meller T, Brosch K, Schmitt S, Ringwald K, Pfarr JK et al. Psychopathological Syndromes Across Affective and Psychotic Disorders Correlate With Gray Matter Volumes. Schizophr Bull. 2021;47:1740–0.
Wittchen HU, Wunderlich U, Gruschwitz S, Zaudig M. SKID I. Strukturiertes Klinisches Interview für DSM-IV. Achse I: Psychische Störungen. Interviewheft und Beurteilungsheft. Eine deutschsprachige, erweiterte Bearb. d. amerikanischen Originalversion des SKID I. 1997. Göttingen: Hogrefe.
McGuffin P, Farmer A, Harvey I. A polydiagnostic application of operational criteria in studies of psychotic illness: Development and reliability of the OPCRIT system. Arch Gen Psychiatry. 1991;48:764–70.
Stein F, Lemmer G, Schmitt S, Brosch K, Meller T, Fischer E, et al. Factor analyses of multidimensional symptoms in a large group of patients with major depressive disorder, bipolar disorder, schizoaffective disorder and schizophrenia. Schizophr Res. 2020;218:38–47.
Papenberg M. minDiff: Minimize Differences Between Groups (R package version 0.01-3) 2019. [Computer software]. Available online at: https://github.com/m-Py/minDiff.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2021. Vienna, Austria. https://www.R-project.org/.
Revelle W. psych: Procedures for Psychological, Psychometric, and Personality Research. 2022. https://cran.r-project.org/web/packages/psych/citation.html. Accessed 16 Mar 2023.
Luo L, Arizmendi C, Gates KM. Exploratory Factor Analysis (EFA) Programs in R. Multidiscipl J. 2019;26:819–26.
Cronbach LJ. Coefficient alpha and the internal structure of tests. Psychometrika. 1951;16:234–97.
Muthén LK, Muthén BO. Mplus User’s Guide. Eighth Edition. (1998-2017) Los Angeles, CA: Muthén & Muthén.
Maydeu-Olivares A. Maximum Likelihood Estimation of Structural Equation Models for Continuous Data: Standard Errors and Goodness of Fit. Struct Equ Model. 2017;24:383–94.
Bentler PM. Comparative Fit Indexes in Structural Models. Psychol Bull. 1990;107:238–46.
Steiger JH. Structural Model Evaluation and Modification: An Interval Estimation Approach. Multivar Behav Res. 1990;25:173–80.
Ho D, Kosuke I, King G, Stuart E. Matchit: Nonparametric Preprocessing for Parametric Causal Inference. J Stat Softw. 2011;42:1–28.
Vogelbacher C, Möbius TWD, Sommer J, Schuster V, Dannlowski U, Kircher T, et al. The Marburg-Münster Affective Disorders Cohort Study (MACS): A quality assurance protocol for MR neuroimaging data. Neuroimage. 2018;172:450–60.
Brosch K, Stein F, Meller T, Schmitt S, Yuksel D, Ringwald KG et al. DLPFC volume is a neural correlate of resilience in healthy high-risk individuals with both childhood maltreatment and familial risk for depression. Psychol Med. 2022;52:4139–45.
Rimol LM, Hartberg CB, Nesvåg R, Fennema-Notestine C, Hagler DJ, Pung CJ, et al. Cortical Thickness and Subcortical Volumes in Schizophrenia and Bipolar Disorder. Biol Psychiatry. 2010;68:41–50.
Hibar DP, Westlye LT, Van Erp TGM, Rasmussen J, Leonardo CD, Faskowitz J, et al. Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry. 2016;21:1710–6.
Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51.
Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95–113.
Dahnke R, Yotter RA, Gaser C. Cortical thickness and central surface estimation. Neuroimage. 2013;65:336–48.
Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80.
Meller T, Schmitt S, Stein F, Brosch K, Mosebach J, Yüksel D et al. Associations of schizophrenia risk genes ZNF804A and CACNA1C with schizotypy and modulation of attention in healthy subjects. Schizophr Res. 2019;208. https://doi.org/10.1016/j.schres.2019.04.018.
Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience. 2015;4. https://doi.org/10.1186/s13742-015-0047-8.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5. https://doi.org/10.1371/journal.pgen.1000529.
McCaw ZR, Lane JM, Saxena R, Redline S, Lin X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics. 2020;76:1262–72.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput Biol. 2015;11:1–19.
Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826 https://doi.org/10.1038/s41467-017-01261-5.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics Appl Note. 2010;26:2336–7.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Ge T, Chen CY, Ni Y, Feng YCA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1–10.
Andlauer TFM, Guzman-Parra J, Streit F, Strohmaier J, González MJ, Gil Flores S, et al. Bipolar multiplex families have an increased burden of common risk variants for psychiatric disorders. Mol Psychiatry. 2021;26:1286–98.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57:289-300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x1995.
David FS, Stein F, Andlauer TFM, Streit F, Witt SH, Herms S, et al. Genetic contributions to transdiagnostic symptom dimensions in patients with major depressive disorder, bipolar disorder, and schizophrenia spectrum disorders. Schizophr Res. 2023;252:161–71.
Reininghaus U, Böhnke JR, Chavez-Baldini U, Gibbons R, Ivleva E, Clementz BA, et al. Transdiagnostic dimensions of psychosis in the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP). World Psychiatry. 2019;18:67–76.
Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:eaap8757.
Schoorl J, Barbu MC, Shen X, Harris MR, Adams MJ, Whalley HC et al. Grey and white matter associations of psychotic-like experiences in a general population sample (UK Biobank). Transl Psychiatry. 2021;11. https://doi.org/10.1038/s41398-020-01131-7.
Van Tol MJ, Van Der Meer L, Bruggeman R, Modinos G, Knegtering H, Aleman A. Voxel-based gray and white matter morphometry correlates of hallucinations in schizophrenia: The superior temporal gyrus does not stand alone. Neuroimage Clin. 2014;4:249–57.
Nickl-Jockschat T, Schneider F, Pagel AD, Laird AR, Fox PT, Eickhoff SB. Progressive pathology is functionally linked to the domains of language and emotion: Meta-analysis of brain structure changes in schizophrenia patients. Eur Arch Psychiatry Clin Neurosci. 2011;261. https://doi.org/10.1007/s00406-011-0249-8.
Stan AD, Tamminga CA, Han K, Kim JB, Padmanabhan J, Tandon N, et al. Associating Psychotic Symptoms with Altered Brain Anatomy in Psychotic Disorders Using Multidimensional Item Response Theory Models. Cereb Cortex. 2020;30:2939–47.
van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, et al. Cortical Brain Abnormalities in 4474 Individuals With Schizophrenia and 5098 Control Subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol Psychiatry. 2018;84:644–54.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745.
Gottschall PE, Howell MD. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 2015;44–46:70–76.
Alsulami M, Munawar N, DIllon E, Oliviero G, Wynne K, Alsolami M, et al. SETD1A methyltransferase is physically and functionally linked to the DNA damage repair protein RAD18. Mol Cell Proteom. 2019;18:1428–36.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–7.
Singh T, Neale BM, Daly MJ. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia on behalf of the Schizophrenia Exome Meta-Analysis (SCHEMA) Consortium. medRxiv. 2020. https://www.medrxiv.org/content/10.1101/2020.09.18.20192815v1.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
Wenze SJ, Gunthert KC, German RE. Biases in Affective Forecasting and Recall in Individuals With Depression and Anxiety Symptoms. Pers Soc Psychol Bull. 2012;38:895–906.
Zeighami Y, Bakken TE, Nickl-Jockschat T, Peterson Z, Jegga AG, Miller JA et al. A comparison of anatomic and cellular transcriptome structures across 40 human brain diseases. PLoS Biol. 2023;21. https://doi.org/10.1371/JOURNAL.PBIO.3002058.
Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, et al. Psychiatric Genomics: An Update and an Agenda. Am J Psychiatry. 2018;175:15–27.
Acknowledgements
We thank the patients participating in this study and the staff for their help with recruitment and data collection. A detailed list of acknowledgements can be found here: www.for2107.de/acknowledgements.
Funding
This work is part of the German multicentre consortium “Neurobiology of Affective Disorders. A translational perspective on brain structure and function“, funded by the German Research Foundation (Deutsche Forschungsgemeinschaft DFG; Forschungsgruppe/Research Unit FOR2107). Principal investigators (PIs) with respective areas of responsibility in the FOR2107 consortium are: Work Package WP1, FOR2107/MACS cohort and brainimaging: Tilo Kircher (speaker FOR2107; DFG grant numbers KI588/14-1, and KI588/14-2, and KI588/20-1, KI588/22-1), Udo Dannlowski (co-speaker FOR2107; DA 1151/5-1, DA 1151/5-2), Axel Krug (KR 3822/5-1, KR 3822/7-2), Igor Nenadić (NE2254/1-2, NE2254/2-1, NE2254/3-1, NE2254/4-1), Carsten Konrad (KO 4291/3-1). WP5, genetics: Marcella Rietschel (RI 908/11-1, RI 908/11-2), Markus Nöthen (NO 246/10-1, NO 246/10-2), Stephanie Witt (WI 3439/3-1, WI 3439/3-2). The study was in part supported by grants from UKGM and Forschungscampus Mittelhessen to Igor Nenadić. Biomedical financial interests or potential conflicts of interest: Tilo Kircher received unrestricted educational grants from Servier, Janssen, Recordati, Aristo, Otsuka, neuraxpharm. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AK* & FS*: data collection, study design, analysis, data interpretation, literature search, writing, figures, *contributed equally; FSD: data collection, study design, genetic analysis, data interpretation, figure, literature search, writing; TFMA, SS, KB, JKP, KGR, TM, FTO, SM, KT, AW, LW, HL, DG, NO, JR, FSTR, SW: data collection and curation, review; AP: data interpretation, review; TH, MR, MMN, IN, UD, TK: funding acquisition, data collection, data curation, review; AJF: data collection, study design, genetic analysis, data interpretation, writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Patients gave written informed consent to the study protocol. This study was approved by the local Ethics Committee Marburg (AZ:07/14) and Münster (AZ:2014-422-b-S) according to the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krug, A., Stein, F., David, F.S. et al. Factor analysis of lifetime psychopathology and its brain morphometric and genetic correlates in a transdiagnostic sample. Transl Psychiatry 14, 235 (2024). https://doi.org/10.1038/s41398-024-02936-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-02936-6
- Springer Nature Limited