
Abstract
Oligogenic inheritance of autism spectrum disorder (ASD) has been supported by several studies. However, little is known about how the risk variants interact and converge on causative neurobiological pathways. We identified in an ASD proband deleterious compound heterozygous missense variants in the Reelin (RELN) gene, and a de novo splicing variant in the Cav3.2 calcium channel (CACNA1H) gene. Here, by using iPSC-derived neural progenitor cells (NPCs) and a heterologous expression system, we show that the variant in Cav3.2 leads to increased calcium influx into cells, which overactivates mTORC1 pathway and, consequently, further exacerbates the impairment of Reelin signaling. Also, we show that Cav3.2/mTORC1 overactivation induces proliferation of NPCs and that both mutant Cav3.2 and Reelin cause abnormal migration of these cells. Finally, analysis of the sequencing data from two ASD cohorts—a Brazilian cohort of 861 samples, 291 with ASD; the MSSNG cohort of 11,181 samples, 5,102 with ASD—revealed that the co-occurrence of risk variants in both alleles of Reelin pathway genes and in one allele of calcium channel genes confer significant liability for ASD. Our results support the notion that genes with co-occurring deleterious variants tend to have interconnected pathways underlying oligogenic forms of ASD.
Similar content being viewed by others


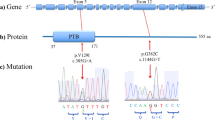
Introduction
Large-scale genomic analyses have revealed that the genetic susceptibility to autism spectrum disorder (ASD) is shaped by rare and common variants and varies from one individual to another. While in a few cases a single rare genetic variant with large effect appears sufficient to cause ASD [1,2,3,4], in most cases the genetic landscape of the disease is much more complex and involves thousands of common risk alleles, individually of small effect but collectively conferring susceptibility above a threshold of liability [5,6,7,8]. In addition, in several individuals with ASD the clinical outcome seem to be the result of the joint combination of more than one rare variant with reduced penetrance in an oligogenic (≥2 hits) mode of inheritance [9,10,11,12,13]. Therefore, a key step in understanding the neurobiological basis of ASD is to decipher the genetic interactions between ASD-risk variants. However, the additive or epistatic interactions of these risk alleles are still largely unexplored.
Recently, we have identified in one Brazilian individual with autism and macrocephaly rare compound heterozygous missense variants in the RELN gene and a de novo splice site variant in the CACNA1H gene [14,15,16]. The RELN gene encodes Reelin, a large secreted glycoprotein that controls neuronal migration and plasticity of synapses [17, 18]. The CACNA1H gene encodes the α1-subunit of the T-type low voltage-dependent calcium (Ca2+) channel Cav3.2, known to control neuronal excitability [19, 20]. Although rare variants have been identified in RELN and CACNA1H genes in ASD individuals, several of them did not segregate with the disease in the affected families and seem to require additional genetic, epigenetic or environmental factors to cause the clinical phenotype [1, 21,22,23].
We have previously shown that the variants in RELN identified in the Brazilian proband are deleterious and lead to diminished Reelin secretion and impaired Reelin signal transduction in induced pluripotent stem cells (iPSC)-derived neural progenitor cells (NPCs). Also, our results suggested that mTORC1 signaling is overactivated in RELN mutant NPCs and contributes to the downregulation of the Reelin cascade [14]. However, the underlying molecular mechanism of mTORC1 pathway hyperfunction remains unknown. Because mTORC1 pathway can be regulated by intracellular Ca2+ levels [24, 25], in this study we investigated whether the variant identified in CACNA1H in the Brazilian proband functions as a ‘second hit’, leading to changes in Ca2+ influx and contributing to overactivation of the mTORC1 pathway and to downregulation of the Reelin signaling in NPCs, a cell population that endogenously express low-voltage-activated Ca2+ channels [16, 26, 27], and that can reveal important clues about both early stages of brain development and ASD pathophysiology [14, 16, 28,29,30]. We also investigated whether the CACNA1H and RELN variants cause abnormal NPC phenotypes. Finally, we examined whether the co-occurrence of damaging variants in genes for the Reelin cascade and Ca2+ channels may also be observed in other ASD individuals using sequencing data from different ASD cohorts.
Methods
Subjects and genetic analysis
This study was approved by the Ethics Committees of the Instituto de Biociências da Universidade de São Paulo and of the Hospital Israelita Albert Einstein, as well as by the Western Institutional Review Board, Montreal Children’s Hospital–McGill University Health Center Research Ethics Board, McMaster University–Hamilton Integrated Research Ethics Board, Eastern Health Research Ethics Board, Holland Bloorview Research Ethics Board and The Hospital for Sick Children Research Ethics Board. Informed consent was obtained from all participants. All individuals whose cells were evaluated in this study were described previously [14,15,16]. Whole-exome sequencing (WES) was performed using genomic DNA from peripheral blood of 861 Brazilian individuals from 279 trio and 6 quartet families of children diagnosed with ASD (n = 291 individuals with ASD, 570 unaffected parents) and analyzed as described previously [4, 14,15,16]. By using a custom script, WES data was subsequently re-analyzed for the presence of co-occurring rare (Global MAF ≤ 0.01 in gnomAD, 1000G, ESP6500, and AbraOM) and predicted impactful (CADD score ≥20) variants in both alleles of Reelin pathway genes and in at least one allele of Ca2+ channel genes (Supplementary Table S1). The findings were validated using whole-genome sequencing (WGS) data from the MSSNG cohort [3] (n = 4258 families of children with ASD; 11,181 samples, 5102 with ASD) and the same selection criteria. For details in WES and WGS and data processing see Supplementary information. The selected variants in Reelin pathway and Ca2+ channel genes (Table 1) were subsequently reanalyzed with 13 additional pathogenic prediction tools (Supplementary Table S2) and were validated by Sanger sequencing. Clinical characteristics of the 10 individuals with ASD from both cohorts who harbor these variants are described in Supplementary Table S3.
Three-dimensional modeling
The 3D models of wild-type and mutant Cav3.2 channels were built using the online I-Tasser server [31]. Three sets of models for each protein were built using the amino acids corresponding to domains I–II, II–III, and III–IV. The motifs were then superimposed onto the crystal structure coordinates of the complex of voltage-gated sodium channel NavPaS from the American cockroach Periplaneta americana (PDB code 6A91) using the secondary-structure matching tool [32] and Coot v.0.8.2 [33]. Pymol [34] was used to create the structural figures. Protein hydrophobicity plots were produced using Expasy Protscale [35].
Neural progenitor cells culture
All NPC samples used in this study have been previously differentiated from iPSCs and were cultured as described previously [14, 16]. NPCs derived from 2 iPSC clones of the Brazilian patient harboring the rare variants in the RELN and CACNA1H genes (referred to as F2688-1), and either 1 or 2 iPSC clones of the other subjects (n = 4 control individuals) were used in all experiments described herein.
Cloning of the mutated CACNA1H and HEK293T transfection
cDNA samples from patient F2688-1 were used to amplify by conventional PCR the region of the CACNA1H gene containing the 156 bp insertion (intron 13). The purified PCR fragment was then introduced into the wild-type human Cav3.2 cDNA (plasmid a1Ha-pcDNA3, #45809 Addgene) between exons 13 and 14 using standards molecular cloning techniques. The presence of the mutation and integrity of the construct were verified by Sanger sequencing. HEK293T cells were transfected with either wild-type or mutated a1Ha-pcDNA3, or empty pcDNA3 vector (negative control) using the Lipofectamine™ 3000 Transfection Reagent (ThermoFisher). RNA and protein were extracted from cells 48 h after transfection. The overexpression of WT- and MUT-a1Ha was confirmed by quantitative reverse transcription-PCR (RT-qPCR), which showed very similar levels of expression (data not shown).
Conventional and quantitative reverse transcription-PCR analyses
Extraction of total RNA from each cell sample and cDNA synthesis was performed as described previously [14]. Conventional PCR reactions were performed with a forward primer designed to anneal in exon 12 and a reverse primer designed to anneal in exon 15 of CACNA1H. Sanger sequencing was carried out to confirm intron 13 retention in the mature CACNA1H transcript. Quantitative PCR (qPCR) reactions were performed with predesigned TaqMan gene expression assays (ThermoFisher), and HMBS as a housekeeping gene. All qPCR samples were run in triplicate. The comparative CT method (ΔΔCT Method) was used to analyze the expression levels of CACNA1H. The experiments were repeated twice with similar results.
Cell treatments
The cells were cultured in the presence of vehicle (DMSO) or 10 μM of the T-type Ca2+ channels blocker NNC 55-0396 dihydrochloride (Tocrisis Bioscience) for 30 min prior to protein extraction for western blot analysis or measurements of extracellular Ca2+ influx. For the proliferation and migration assays, NPCs were cultured in the presence of vehicle (DMSO), 1 uM of NNC 55-0396 dihydrochloride, 100 nM of rapamycin (Sigma Aldrich), wild-type Reelin- or mock-conditioned medium (prepared as previously described [14]) during the entire course of the experiments.
Measurement of extracellular Ca2+ influx
The fluorescence-based FLIPR® Calcium 4 Assay Kit (Molecular Devices) was used to detect changes in intracellular Ca2+. Briefly, the cells (3–5 × 104 cells/well) were seeded in black 96-well plates, and cultured in growth medium for 48 h. Before initiating the assay, the medium was removed and cells were incubated with the Ca2+ dye containing 2.5 mM probenecid for 60 min at 37 °C. The cells were then treated with vehicle or NNC 55-0396 dihydrochloride, and the dye was excited at 480 nm and its fluorescence signals detected at 525 nM. The basal fluorescence intensity was monitored for 15 s at 1.65 s intervals, and then cells were challenged with 100 mM KCl and the cellular fluorescence continued to be recorded over 200 s. KCl-evoked increases in intracellular Ca2+ were determined as ΔF/F0 (F0 is basal fluorescence) using the SoftMax2Pro software (Molecular Devices Corp.). The ΔF/F0 values obtained from the same group did not differ significantly at either time points after KCl stimulation. Each experimental condition was performed in three replicate wells for NPCs and five replicates wells for HEK293T cells. The experiments were repeated at least twice with similar results.
Protein extraction and immunoblotting
Extraction of total proteins from cell samples, standard western blotting, and quantification of band intensity were carried out as described previously [14]. The anti-DAB1, anti-pRPS6, anti-pSRC, anti-SRC, and anti-ßactin antibodies used are described in [14], and results shown are from two independent experiments. In addition, the following primary antibodies were used in order to detect endogenous Cav3.2 protein in NPCs and other cell types (including Hela and HEK293T cells transfected with WT-a1Ha) but, despite several attempts, neither of them worked properly: ab135974 and ab128251 (Abcam), sc-377510 (Santa Cruz Biotechnology).
Analysis of cell morphology, size, and proliferation
NPC samples were seeded into 12-well plates at density of 1.0 × 105 cells per well and cultured in complete NPC medium for 24 h. Phase-contrast images were obtained at 20× magnification using the IncuCyte® system (Essen Bioscience). Analysis of cell morphology and measurements of cell body sizes (including soma size and all cell surface projections) were performed using the IncuCyte Neurotrack software. For the proliferation assay, 24 h after initial plating (Day 0), cells were counted at 48 h and 72 h using trypan blue exclusion and automated cell counting. Each experimental condition was performed in 2 replicate wells. The experiments were repeated at least twice with similar results.
Analysis of cell migration (scratch-wound healing assay)
NPC samples were seeded into 96-well plates at density of 3.5 × 104 cells per well and cultured in complete NPC medium until 90% confluence. The cells were then grown in DMEM/F12 without FGF and EGF for 48 h to inhibit cell proliferation, and the 96-well WoundMaker (Essen Bioscience) was used to generate a wound area in the confluent monolayer of cells. Wound closure was monitored and quantified with the IncuCyte® system (Essen Bioscience). Relative wound density was defined as cell density in the wound area expressed relative to the cell density outside of the wound area over time. Each experimental condition was performed in 6 replicate wells. The experiments were repeated at least twice with similar results.
Statistical analysis
Statistical analyses were conducted using SPSS statistical software (IBM SPSS Statistics 20). The generalized linear mixed-effect models were used to account for dependency between biological replicates (when NPCs derived from different iPSC clones of the same individual were used) and independent technical replicates per individual, and to investigate association between the response and the explanatory variables (time period and treatments). Fisher’s Exact Test was used to determine if there is an enrichment of co-occurring rare variants in Reelin pathway and Ca2+ channel genes in ASD individuals. The PASS (Power Analysis and Sample Size) software was used to calculate statistical power, and the results showed that the sample size ensured a statistical power ≥0.80 for most analyses in this study. Differences were considered significant when Bonferroni-corrected p < 0.05. Data are presented as medians with interquartile ranges.
Results
The CACNA1H splice site variant identified in patient F2688-1 causes intron retention in the mature transcript
Through WES, we have identified in one individual with ASD and macrocephaly, referred to as F2688-1, the previously characterized compound heterozygous missense variants in the RELN gene [14], and a de novo heterozygous splice donor site variant in intron 13 of the CACNA1H gene (NM_001005407:c.2907+1 G>A) (Table 1; Supplementary Table S2; Supplementary Fig. S1A). This variant was absent from the population databases, and is predicted to inactivate the 5′ splice site of intron 13, which is only 156 bp long, causing the retention of this intron in the mature CACNA1H mRNA. Because the retained intron does not cause a frameshift in the CACNA1H coding sequence, it is expected to introduce an in-frame insertion of 52 novel amino acid residues in the pore-forming region of the Cav3.2 channel.
We, therefore, sought to investigate whether the removal of intron 13 from the pre-mRNA of CACNA1H was abnormal in neural cells derived from patient F2688-1 using cDNA samples from iPSC-derived NPCs from this patient and from control individuals. We observed, as expected, that the variant causes abnormal splicing and intron 13 retention in the mature CACNA1H transcript (Fig. 1A–C). Despite the defective RNA splicing, we observed no significant change in the transcript levels of CACNA1H in F2688-1 NPCs compared to control NPCs (Fig. 1D).

A Schematic representation of intron 13 (In13) retention in the CACNA1H transcript showing the locations of primers for conventional RT-PCR. The PCR band corresponding to the wild-type allele is 370 bp long, whereas the band corresponding to the mutant allele is 526 pb long due to retention of intron 13 in the mature transcript. B Representative image of agarose gel electrophoresis showing conventional RT-PCR products. The 526 bp PCR product of the mutant allele is only observed in F2688-1-derived NPCs (n = 2 iPSC clones-derived NPCs). C Sanger sequencing of the cloned 526 bp PCR band confirmed intron 13 retention in the mature CACNA1H transcript. The location of the variant NM_001005407:c.2907+1 G>A is shown. D Quantitative RT-PCR demonstrated that the relative transcription levels of CACNA1H are not changed in F2688-1-derived NPCs (n = 2 iPSC clones-derived NPCs) compared with control NPCs (Controls, n = 4). The bar graph shows the median value and interquartile range for each group.
Structural modeling of the mutant Cav3.2 channel suggests a larger pore diameter than the wild-type channel
Cav3.2 channel consists of four homologous domains (I–IV), each one composed of six α-helical transmembrane segments (S1–S6). The voltage-sensing module of the channel is formed by the positively charged S4 segments, while the ion conductivity and selectivity lie on the negatively charged extracellular membrane-reentrant pore loop (P-loop) between S5 and S6 [20, 36] (Fig. 2A). We observed that the 52-amino acid insertion, located between segments S5 and S6 of domain II, alters the hydropathy profile in the pore region and causes significant conformational changes in the next domain (Fig. 2A, yellow). To evaluate the impact of the insertion on the Cav3.2 channel structure, we built 3D models of the wild-type and mutant proteins. We observed, as pointed by the domain and hydropathy profile analyses, that the insertion (Fig. 2Bi, yellow) alters the secondary structure of domain II (Fig. 2Bi, magenta) and also impacts the folding of domain III (Fig. 2Bi, blue), changing the position of transmembrane helices that compose the pore-forming domain and increasing the pore diameter in the mutant protein (Fig. 2Bii). Together, these analyses suggest that the insertion induces significant structural changes in the mutant channel, resulting in larger open pores.

A Predicted topologies of the wild type (WT) and mutant (MUT) Cav3.2 channels showing the four homologous domains (I–IV) in different colors, each composed of six transmembrane segments (bars S1–S6). The 52-amino acid insertion (yellow) is located between segments S5 and S6 of domain II (between residues Q969 and I970). The hydropathy profiles of the amino acid sequences are shown below the channel topologies. B Structural features of the WT and MUT Cav3.2 channels. Bi Simulations of the proteins viewed from the cytosolic side of the plasma membrane are shown in cartoon representation and colored by domain (DI–DIV). Domain II is colored in magenta and the 52-amino acid insertion is detached in yellow. Bii Structural view of the pore center showing the differences in the pore diameter between WT and MUT Cav3.2 channels.
Calcium influx through the mutant Cav3.2 channel is increased and induces exaggerated mTORC1 signaling in F2688-1 neural progenitor cells
To assess the functional impact of the 52-amino acid insertion on Cav3.2 channel activity, we measured the influx of extracellular Ca2+ in F2688-1 and control-derived NPCs. We found that F2688-1 NPCs show significantly increased Ca2+ influx after depolarization compared with control NPCs, which was abolished by treatment with the T-type Ca2+ channels blocker NNC 55-0396 (Fig. 3A). These results suggest that the identified variant in the pore region of Cav3.2 allows greater Ca2+ influx into NPCs derived from patient F2688-1, which is consistent with the enlarged pore size predicted by the 3D-model structure of the mutant channel.
Next, we sought to verify whether the mTORC1 signaling overactivation previously observed in F2688-1 NPCs [14] is due to greater influx of Ca2+ into cells via the mutant Cav3.2 channel. We treated F2688-1 and control NPCs with NNC 55-0396 and analyzed the phosphorylation status of RPS6, a downstream target of the mTORC1 pathway. We observed that whereas vehicle-treated F2688-1 NPCs showed, as expected, elevated levels of pRPS6, the blockade of T-type Ca2+ channels significantly reduced pRPS6 expression in F2688-1 NPCs to levels similar to the untreated control NPCs (Fig. 3A). These results suggest that the enhanced Ca2+ entry into F2688-1 NPCs through the mutant Cav3.2 channel causes excessive mTORC1 signaling activity in these cells.
Mutant Cav3.2-mediated increase in mTORC1 signaling contributes to impaired Reelin signaling in F2688-1 neural progenitor cells
To test the hypothesis that the exaggerated mTORC1 signaling activation due to increased influx of Ca2+ through the mutant Cav3.2 channel in F2688-1 NPCs contributes to the impaired Reelin signaling in these cells, we treated F2688-1 and control-derived NPCs with NNC 55-0396 and analyzed the phosphorylation levels of SRC and DAB1, downstream targets of Reelin [37, 38]. We found that whereas vehicle-treated F2688-1 NPCs showed, as expected, decreased expression of pSRC and pDAB1 [14], the T-type Ca2+ channels blocker significantly enhanced pSRC expression to levels similar to the untreated control NPCs (Fig. 3B). On the other hand, while the T-type Ca2+ channels blocker significantly improved pDab1 levels in F2688-1 NPCs, there is still a clear trend towards diminished expression of pDAB1 in these cells compared to untreated control cells (Fig. 3B), corroborating our previous findings indicating that the variants in the RELN gene contribute to defective levels of this key regulator of Reelin signaling [14]. These results support our hypothesis of an abnormal interaction between mutated CACNA1H and RELN genes, showing that the mutant Cav3.2 further exacerbates impaired Reelin signaling in F2688-1 NPCs through the overactivation of mTORC1 pathway.

A Intracellular Ca2+ measurements in control-derived NPCs (n = 4) and in F2688-1-derived NPCs (NPCs derived from 2 iPSC clones) depolarized with 100 mM KCl and cultured in the absence (vehicle) or presence of 10 μM of the T-type Ca2+ channels blocker NNC 55-0396 (NNC). The bar graph shows the median value and interquartile range for each group. F2688-1 NPCs show significantly increased Ca2+ influx compared with control NPCs, which was rescued by treatment with NNC. B Representative immunoblots showing the expression levels of pRPS6, pSRC, SRC, pDAB1, DAB1, and β-actin (used as loading control) in control-derived NPCs (n = 4) and in F2688-1-derived NPCs (NPCs derived from 2 iPSC clones) cultured in the absence (−) or presence (+) of 10uM of NNC. The bar graph shows the median levels and interquartile ranges of normalized pRPS6, pSRC, and pDAB1 for each group. While F2688-1-derived NPCs cultured in the absence of NNC show elevated levels of pRPS6 and diminished levels of pSRC and pDAB1, treatment with NNC significantly rescued pRPS6 and pSRC expression to levels similar to the untreated control NPCs. On the other hand, while treatment with NNC significantly improved pDab1 levels in F2688-1 NPCs, there is still a clear trend towards diminished expression of pDAB1 in these cells compared to untreated control cells. *p < 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
Exaggerated Cav3.2/mTORC1 signaling causes enhanced proliferation of F2688-1 neural progenitor cells
Hyperactivation of mTORC1 signaling has been associated with abnormal morphology, size, and number of neural cells [39,40,41,42,43]. Therefore, we sought to determine whether the enhanced Cav3.2/mTORC1 signaling in F2688-1 NPCs, which in turn further aggravates Reelin signaling impairment, leads to changes in the morphology, size, and proliferation rates of these cells. While no gross alterations in cell morphology and no significant differences in cell body size (including soma size and all cell surface projections) were observed between control- and F2688-1-derived NPCs (Fig. 4A), F2688-1 NPCs show significantly higher proliferation rates compared with control NPCs, which were completely rescued by treatment with either the mTOR inhibitor rapamycin or NNC 55-0396. On the other hand, treatment with wild-type Reelin had no effect on the proliferation of F2688-1 NPCs (Fig. 4B). Since we have previously demonstrated that treatment of F2688-1 NPCs with wild-type Reelin significantly improved the defective Reelin pathway [14], these results suggest that mutant Cav3.2-mediated hyperfunction of mTORC1 signaling acts independently of the defective Reelin signaling to cause enhanced proliferation of F2688-1 NPCs.
Abnormal Cav3.2 and Reelin induce aberrant migration of F2688-1 neural progenitor cells
Impaired Reelin signaling due to constitutive activation of the mTORC1 pathway plays an important role in abnormal neuronal migration in tuberous sclerosis complex (TSC) pathology [44]. Therefore, we next sought to investigate whether the mutant Cav3.2-mediated overactivation of mTORC1 signaling and reduction of Reelin signaling could affect the migration pattern of F2688-1 NPCs. Using the wound healing assay, we observed that the NPCs polarize toward the wound, extend filopodia protrusions, migrate, and closure the wound (Fig. S2). Also, we found that F2688-1 NPCs show significantly higher migration rates compared with control NPCs, which were significantly attenuated by treatment with rapamycin and completely rescued to levels equal to those of the untreated control NPCs in the exponential phase of the healing curve by treatment with either NNC 55-0396 or wild-type Reelin (Fig. 4C). Since we have previously shown that treatment of F2688-1 NPCs with rapamycin or wild-type Reelin led to a significant improvement in Reelin signaling deficits [14], these findings suggest that altered Cav3.2/mTORC1/Reelin pathways are all involved in the abnormal migration of F2688-1 NPCs.

A Representative phase-contrast microscopy photographs of control- and F2688-1-derived NPCs. The bar graph shows the median cell body size (including soma size and all cell surface projections) of control-derived NPCs (n = 4) and F2688-1-derived NPCs (NPCs derived from 2 iPSC clones). There were no significant differences in median body size between the groups. B Line graph showing cell proliferation curves (live cell numbers at Day 0, 48 h, 72 h) of control-derived NPCs (n = 4) and F2688-1-derived (NPCs derived from 2 iPSC clones) cultured in the presence of either vehicle, 1 μM of NNC 55-0396 (NNC), or 100 nM of rapamycin; and in the presence of either mock-conditioned medium (mock) or Reelin-conditioned medium (Reelin). The graph shows the median value and interquartile range for each group. F2688-1 NPCs show significantly higher proliferation rates compared with control NPCs, which were completely rescued by treatment with either rapamycin or NNC. On the other hand, treatment with Reelin had no effect on the proliferation of F2688-1 NPCs. C Line graph showing the percentage relative wound density (RWD) over time in control-derived NPCs (n = 4) and in F2688-1-derived NPCs (NPCs derived from 2 iPSC clones) cultured in the presence of either vehicle, 1 μM of NNC, or 100 nM of rapamycin; and in the presence of either mock or Reelin. The graph shows the median value and interquartile range for each group. F2688-1 NPCs exhibit significantly higher migration rates compared with control NPCs, which was significantly attenuated by treatment with rapamycin, and completely rescued to levels equal to those of the untreated control NPCs in the exponential phase of the healing curve by treatment with either NNC or Reelin (analyzed time interval: 15 h–25 h). **p ≤ 0.01, ****p ≤ 0.0001.
Heterologous expression of mutant Cav3.2 channel confirms its functional relevance
To provide additional support for our results, we next examined whether mutant Cav3.2 channel when heterologously expressed in HEK293T cells would alter Ca2+ influx and affect endogenous mTORC1 and Reelin pathway components. To this end, HEK293T cells were transfected with an empty pcDNA3 vector or with plasmids expressing either wild-type (WT-a1Ha) or mutant (MUT-a1Ha) Cav3.2 channels, and the influx of extracellular Ca2+ and the phosphorylation status of RPS6 and SRC were measured (Dab1 is not endogenously expressed in HEK293T cells). We observed that cells overexpressing both wild-type and mutant Cav3.2 channels show significantly increased Ca2+ influx after depolarization (Fig. 5A) and significantly augmented pRPS6 levels (Fig. 5B) compared with cells overexpressing the empty vector, suggesting that both the amount of Ca2+ ions entering the cells and the activity of the mTORC1 pathway are dependent on the number of Cav3.2 channels expressed in the plasma membrane. Also, in accordance with the results using F2688-1 NPCs, we observed that Ca2+ entry and pRPS6 levels were significantly greater in cells overexpressing mutant Cav3.2, and that treatment with NNC 55-0396 rescued these phenotypes (Fig. 5B). Finally, we observed that cells overexpressing mutant Cav3.2 show significantly decreased pSRC levels compared with cells overexpressing both the wild-type Cav3.2 and the empty vector, and that treatment with NNC 55-0396 also rescued this phenotype (Fig. 5B).

HEK293T cells were transfected with an empty vector (EMPTY-vector; n = 5) or with plasmids expressing either wild-type (WT-a1Ha; n = 5) or mutant (MUT-a1Ha; n = 5) Cav3.2 channels. A Intracellular Ca2+ measurements in transfected cells depolarized with 100 mM KCl and cultured in the absence (vehicle) or presence of 10 μM of the T-type Ca2+ channels blocker NNC 55-0396 (NNC). The bar graph shows the median value and interquartile range for each group. Cells overexpressing mutant Cav3.2 show significantly increased Ca2+ influx, which was rescued by treatment with NNC. Only the p-values obtained from the analyses using MUT-a1Ha are shown. B Representative immunoblots showing the expression levels of pRPS6, pSRC, SRC, and β-actin (used as loading control) in transfected cells cultured in the absence (−) or presence (+) of 10 μM of NNC. The bar graph shows the median levels and interquartile ranges of normalized pRPS6 and pSRC for each group; only the p-values obtained from the analyses using MUT-a1Ha are shown. Cells overexpressing mutant Cav3.2 show significantly increased pRPS6 levels and significantly decreased pSRC levels, which was rescued by treatment with NNC. *p < 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
Taken together, these results strongly support that the variant identified in CACNA1H in patient F2688-1 exerts a gain-of-function effect by enhancing Ca2+ influx through the mutant Cav3.2 channel, which overactivates mTORC1 signaling and, in turn, contributes to impaired activation of Reelin signaling components in these cells.
Increased burden of co-occurring risk variants in both alleles of Reelin pathway genes and in one allele of Ca2+ channel genes in ASD individuals
We hypothesized that the concomitant occurrence of rare deleterious variants (MAF ≤ 0.01; CADD score ≥20) in both alleles of genes for the Reelin cascade and in at least one allele of genes for Ca2+ channels may also be observed in other ASD individuals. To test this hypothesis, we initially analyzed the WES results from our Brazilian cohort of 279 trio and 6 quartet families with ASD (n = 291 individuals with ASD, 570 unaffected parents), and identified in two additional probands, referred to as F10832-1 and F11463-1, this specific combination of hits (Table 1; Supplementary Table S2; Supplementary Fig. S1B, C). Proband F10832-1 carries rare compound heterozygous variants in VLDLR, which encodes one of the receptors for Reelin [45,46,47], and a rare heterozygous variant in CACNA2D4, which encodes the voltage-gated L-type Ca2+ channel auxiliary subunit of the α2δ4 type [48,49,50]. Proband F11463-1 harbors rare compound heterozygous variants in RELN, and a rare heterozygous variant in RYR3, which encodes the intracellular Ca2+ release channel ryanodine receptor 3 [51,52,53]. Importantly, in silico analysis using several pathogenicity prediction tools strongly suggested that these variants are deleterious (Supplementary Table S2), and this particular combination of rare putatively damaging variants was not found in the parents, suggesting a significant enrichment of such combination in ASD individuals (p = 0.04). It is also noteworthy that it was not observed in the probands a significant excess of other combinations of risk variants in these genes, such as compound heterozygous variants in genes for the Reelin pathway alone (without co-occurring variants in Ca2+ channel genes), or variants in Ca2+ channel genes alone (without co-occurring variants in Reelin cascade genes), or two in cis variants in a Reelin pathway gene with co-occurring variants in Ca2+ channel genes, or heterozygous variants in a Reelin pathway gene with co-occurring variants in Ca2+ channel genes.
In order to further strengthen our findings, we next analyzed the WGS data from a large and independent cohort of ASD cases, the MSSNG resource [3], presently consisting of 11,181 samples from families with ASD, including 5102 ASD subjects. We found that 7 ASD individuals carry compound heterozygous risk variants in either the RELN or the VLDLR gene and also a risk variant in at least one Ca2+ channel gene, including RYR2 [54, 55]; RYR1 [56], ORAI1 [57], CACNA1B [58]; GRIN2C [59], CACNA1A [60], CACNB2 [61], CACNA1H [21], CACNA2D4 [48,49,50] (Table 1; Supplementary Table S2; Supplementary Fig. S1D–J). This specific combination of risk variants was not found in 6079 unaffected parents or siblings, and different combinations of risk variants in these genes were not found to be significantly enriched in the ASD subjects as observed in the Brazilian cohort, which provides corroborating evidence for a significantly increased burden of co-occurring impactful variants in both copies of either the RELN or the VLDLR gene and in one copy of Ca2+ channel genes in ASD (p = 0.004). Finally, it is also noteworthy that the ASD subjects from both the Brazilian and the MSSNG cohorts who harbor this specific combination of variants do not carry rare variants that cause a known deleterious loss of function (LoF) of a high-penetrant ASD gene. However, some of these individuals also carry risk variants in genes previously shown to contribute to ASD with reduced penetrance: the affected sisters 2-1259-003 and 2-1259-004 harbor a CNV disrupting the ELP4 gene [62] (Supplementary Fig. S1E), and the proband AU2168301 carries a de novo LoF variant in the KDM6B gene [63] (Supplementary Fig. S1H).
Discussion
Identifying the combinations of genetic variants required for determining ASD causality will aid in the understanding of the complex genetic architecture of ASD and of disease pathophysiology. In this study, we describe the functional analysis of a de novo splice site variant identified in the CACNA1H gene in an individual with ASD and macrocephaly who also carries deleterious compound heterozygous missense variants in the RELN gene [14]. We found that the variant in CACNA1H leads to the retention of an intronic sequence within the mature transcript, which is predicted to cause an in-frame insertion of 52 novel amino acid residues in the pore-forming domain of the Cav3.2 channel. The 3D model-structure of the mutated channel suggested that the pore may be enlarged in size, and we showed that Ca2+ influx through the mutant Cav3.2 is increased in patient-derived NPCs, which overactivates mTORC1 pathway and, consequently, further aggravates the impairment of Reelin signaling. Heterologous expression of the mutated Cav3.2 channel confirmed these results.
Within the CNS, Cav3.2 is predominantly expressed in the hippocampus, and Cav3.2 knockout mice show some behavioral abnormalities reminiscent of human autism [64, 65]. Rare missense variants in the human Cav3.2 have previously been reported in ASD individuals and appear to contribute to ASD susceptibility [21, 66,67,68]. Electrophysiological analysis of some of these variants using a non-neuronal exogenous expression system revealed that mutated Cav3.2 channels conducted substantially less current than wild-type channels. However, two of these variants located within the voltage-sensing region of Cav3.2 were shown to decrease the inactivation rates of the mutated channels and, thus, are expected to allow larger Ca2+ influx once activated [21], an effect similar to the variant here described. Therefore, a detailed biophysical analysis investigating the effects of ASD-associated Cav3.2 variants in more native conditions are still necessary to understand their characteristics and functional impact on neuronal excitability and function.
It should also be noted that common and rare variants in other Ca2+ channel genes (CACNA1A, CACNA1C, CACNA1G, CACNA1I, CACNA1D, CACNA2D3, and CACNB2) have been found to be associated with ASD [59, 60, 67,68,69,70,71,72,73], and functional analysis of some of these variants showed either gain- or loss-of-function of channel activity [71, 73]. Altogether, these findings suggest that perturbed intracellular Ca2+ homeostasis due to voltage-gated Ca2+ channel dysfunctions may affect neurodevelopmental processes contributing to ASD. In this sense, it is possible that the enhanced proliferation of F2688-1 NPCs caused by aberrant Cav3.2/Ca2+-mediated increase in mTORC1 signaling, which in turn further exacerbates the Reelin signal transduction defect and leads to abnormal migration of these cells might contribute to the macrocephaly and ASD symptoms observed in the patient. It has been shown that Ca2+ and mTORC1 pathways regulate neural cell proliferation [74, 75] and that hyperactive mTORC1 signaling is associated with enlarged head/brain size [24, 76], which is commonly observed in children with ASD [77]. It is also noteworthy that several neuropathological findings in the brains from autistic subjects support neuronal migration abnormalities [78], and that, in addition to the TSC mouse model that shows mTORC1/Reelin-mediated deficits in neuronal migration [44], aberrant neuronal migration have also been observed in many rodent genetic models of ASD that mimic disease-causing mutations in patients [79,80,81].
Analysis of WES data from our Brazilian cohort of 285 families with ASD and the analysis of WGS data from the MSSNG cohort of 4,258 families with ASD suggested that the co-occurrence of rare and potentially damaging variants in both alleles of either the RELN or the VLDLR gene and in one allele of Ca2+ channel genes confer significant liability for ASD. The existence of an oligogenic inheritance of ASD has been unrevealed by a series of studies [2, 10, 82,83,84,85], and recent analyses using large scale genomic data have suggested that genes involved in this multi-hit model show higher functional connectivity [12, 13, 86]. The data reported here corroborate these results, lending further support to the concept that genes with co-occurring deleterious variants tend to have interconnected signaling pathways.
References
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional, and chromatin genes disrupted in autism A. Nature. 2014;515:209–15.
Yuen C, Merico RK, Bookman D, M, L Howe J, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell.2020;180:568–84.e23.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–5.
Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21.
Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet. 2017;49:978–85.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–9.
Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521.
Chilian B, Abdollahpour H, Bierhals T, Haltrich I, Fekete G, Nagel I, et al. Dysfunction of SHANK2 and CHRNA7 in a patient with intellectual disability and language impairment supports genetic epistasis of the two loci. Clin Genet. 2013;84:560–5.
Du Y, Li Z, Liu Z, Zhang N, Wang R, Li F, et al. Nonrandom occurrence of multiple de novo coding variants in a proband indicates the existence of an oligogenic model in autism. Genet Med. 2019;22:170–80.
Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. 2019;21:816–25.
Sánchez-Sánchez SM, Magdalon J, Griesi-Oliveira K, Yamamoto GL, Santacruz-Perez C, Fogo M, et al. Rare RELN variants affect Reelin–DAB1 signal transduction in autism spectrum disorder. Hum Mutat. 2018;39:1372–83.
da Silva Montenegro EM, Costa CS, Campos G, Scliar M, de Almeida TF, Zachi EC, et al. Meta-analyses support previous and novel autism candidate genes: Outcomes of an unexplored Brazilian cohort. Autism Res. 2020;13:199–206.
Griesi-Oliveira K, Fogo MS, Pinto BGG, Alves AY, Suzuki AM, Morales AG, et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol Psychiatry. 2020;26:1589–605.
Jossin Y, Goffinet AM. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 2007;27:7113–24.
Lee GH, D’Arcangelo G. New insights into Reelin-mediated signaling pathways. Front Cell Neurosci. 2016;10:122.
Leresche N, Lambert RC. T-type calcium channels in synaptic plasticity. Channels. 2017;1:121–39.
Weiss N, Zamponi GW. Genetic T-type calcium channelopathies. J Med Genet. 2020;57:1–10.
Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. J Biol Chem. 2006;281:22085–91.
Lammert DB, Howell BW. RELN mutations in autism spectrum disorder. Front Cell Neurosci. 2016;10:84.
Lammert DB, Middleton FA, Pan J, Olson EC, Howell BW. The de novo autism spectrum disorder RELN R2290C mutation reduces Reelin secretion and increases protein disulfide isomerase expression. J Neurochem. 2017;142:89–102.
Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–65.
Li RJ, Xu J, Fu C, Zhang J, Zheng YG, Jia H, et al. Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife. 2016;5:1–16.
Louhivuori LM, Louhivuori V, Wigren HK, Hakala E, Jansson LC, Nordström T, et al. Role of low voltage activated calcium channels in neuritogenesis and active migration of embryonic neural progenitor cells. Stem Cells Dev. 2013;22:1206–19.
Kim JW, Oh HA, Lee SH, et al. T-Type calcium channels are required to maintain viability of neural progenitor cells. Biomol Ther. 2018;26:439–45.
Anderson DJ. Stem cells and pattern formation in the nervous system: The possible versus the actual. Neuron. 2001;30:19–35.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–90.
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2017;22:820–35.
Yang J, Zhang Y. Protein structure and function prediction using I-TASSER. Curr Protoc Bioinform. 2015;52:5.8.1–15.
Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Struct Biol. 2004;60:2256–68.
Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Struct Biol. 2004;60:2126–32.
DeLano WL. Unraveling hot spots in binding interfaces: Progress and challenges. Curr Opin Struct Biol. 2002;12:14–20.
Kyte J, Doolittle RF. simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. 1
Catterall WA. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett. 2020;486:107–16.
Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26.
Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13:643–8.
Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, et al. Pharmacological inhibition of mTORCl suppresses anatomical, cellular, and behavioral abnormalities in neural-specific PTEN knock-out mice. J Neurosci. 2009;29:1773–83.
Feliciano DM, Su T, Lopez J, Platel JC, Bordey A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J Clin Invest. 2011;121:1596–607.
Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011;9:447–62.
Iffland PH 2nd, Baybis M, Barnes AE, Leventer RJ, Lockhart PJ, Crino PB. DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol Dis. 2018;114:184–93.
Martin P, Wagh V, Reis SA, Erdin S, Beauchamp RL, Shaikh G, et al. TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol Autism. 2020;11:2.
Moon UY, Park JY, Park R, Cho JY, Hughes LJ, McKenna J 3rd, et al. Impaired Reelin-Dab1 signaling contributes to neuronal migration deficits of tuberous sclerosis complex. Cell Rep. 2015;12:965–78.
D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–9.
Fraley ER, Burkett ZD, Day NF, Schwartz BA, Phelps PE, White SA. Mice with Dab1 or Vldlr insufficiency exhibit abnormal neonatal vocalization patterns. Sci Rep. 2016;6:25807.
Iwata K, Izumo N, Matsuzaki H, Manabe T, Ishibashi Y, Ichitani Y, et al. Vldlr overexpression causes hyperactivity in rats. Mol Autism. 2012;3:11.
Van Den Bossche MJ, Strazisar M, De Bruyne S, Bervoets C, Lenaerts AS, et al. Identification of a CACNA2D4 deletion in late onset bipolar disorder patients and implications for the involvement of voltage-dependent calcium channels in psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:465–75.
Abdelmoity AT, Hall JJ, Bittel DC, Yu S. 1.39 Mb inherited interstitial deletion in 12p13.33 associated with developmental delay. Eur J Med Genet. 2011;54:198–203.
Smith M, Flodman PL, Gargus JJ, Simon MT, Verrell K, Haas R, et al. Mitochondrial and ion channel gene alterations in autism. Biochim Biophys Acta Bioenerg. 2012;1817:1796–802.
Frank DF, Miller GW, Connon RE, Geist J, Lein PJ. Transcriptomic profiling of mTOR and ryanodine receptor signaling molecules in developing zebrafish in the absence and presence of PCB 95. PeerJ. 2017;29:e4106.
Matsuo N, Tanda K, Nakanishi K, Yamasaki N, Toyama K, Takao K, et al. Comprehensive behavioral phenotyping of ryanodine receptor type 3 (RyR3) knockout mice: decreased social contact duration in two social interaction tests. Front Behav Neurosci. 2009;7:3.
Tedoldi A, Ludwig P, Fulgenzi G, Takeshima H, Pedarzani P, Stocker M. Calcium-induced calcium release and type 3 ryanodine receptors modulate the slow afterhyperpolarising current, sIAHP, and its potentiation in hippocampal pyramidal neurons. PLoS One. 2020;15:e0230465.
Soueid J, Kourtian S, Makhoul NJ, Makoukji J, Haddad S, et al. RYR2, PTDSS1, and AREG genes are implicated in a Lebanese population-based study of copy number variation in autism. Sci Rep. 2016;8:19088.
Bertan F, Wischhof L, Sosulina L, Mittag M, Dalügge D, et al. Loss of ryanodine receptor 2 impairs neuronal activity-dependent remodeling of dendritic spines and triggers compensatory neuronal hyperexcitability. Cell Death Differ. 2020;27:3354–73.
De Crescenzo V, Fogarty KE, Lefkowitz JJ, Bellve KD, Zvaritch E, MacLennan DH, et al. Type 1 ryanodine receptor knock-in mutation causing central core disease of skeletal muscle also displays a neuronal phenotype. Proc Natl Acad Sci USA. 2012;109:610–5.
Dou Y, Xia J, Gao R, Gao X, Munoz FM, Wei D, et al. Orai1 plays a crucial role in central sensitization by modulating neuronal excitability. J Neurosci. 2018;38:887–900.
Gorman KM, Meyer E, Grozeva D, Spinelli E, McTague A, Sanchis-Juan A, et al. Bi-allelic loss-of-function CACNA1B mutations in progressive epilepsy-dyskinesia. Am J Hum Genet. 2019;104:948–56.
Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55.
Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism, and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015;23:1505–12.
Breitenkamp AF, Matthes J, Nass RD, Sinzig J, Lehmkuhl G, Nürnberg P, et al. Rare mutations of CACNB2 found in autism spectrum disease-affected families alter calcium channel function. PLoS One. 2014;9:e95579.
Addis L, Ahn JW, Dobson R, Dixit A, Ogilvie CM, Pinto D, et al. Microdeletions of ELP4 are associated with language impairment, autism spectrum disorder, and mental retardation. Hum Mutat. 2015;36:842–50.
Stolerman ES, Francisco E, Stallworth JL, Jones JR, Monaghan KG, Keller-Ramey J, et al. Genetic variants in the KDM6B gene are associated with neurodevelopmental delays and dysmorphic features. Am J Med Genet. A 2019;179:1276–86.
Gangarossa G, Laffray S, Bourinet E, Valjent E. T-type calcium channel Cav3.2 deficient mice show elevated anxiety, impaired memory, and reduced sensitivity to psychostimulants. Front Behav Neurosci. 2014;8:92.
Chung NC, Huang YH, Chang CH, Liao JC, Yang CH, et al. Behavior training reverses asymmetry in hippocampal transcriptome of the Cav3.2 knockout mice. PLoS One. 2015;10:e0118832.
Devlin B, Boone BE, Levy SE, Lihm J, Buxbaum JD, Wu Y, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;85:242–6.
Roak O’, BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50.
Chen R, Davis LK, Guter S, Wei Q, Jacob S, Potter MH, et al. Leveraging blood serotonin as an endophenotype to identify de novo and rare variants involved in autism. Mol Autism. 2017;8:14.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31.
Strom SP, Stone JL, Ten Bosch JR, Merriman B, Cantor RM, Geschwind SF, et al. High-density SNP association study of the 17q21 chromosomal region linked to autism identifies CACNA1G as a novel candidate gene. Mol Psychiatry. 2010;15:996–1005.
Lu AT-H, Dai X, Martinez-Agosto JA, Cantor RM. Support for calcium channel gene defects in autism spectrum disorders. Mol Autism. 2012;3:18.
Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet. 2013;92:221–37.
Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, et al. CACNA1D de novo mutations in autism spectrum disorders activate cav1.3 l-type calcium channels. Biol Psychiatry. 2015;77:816–22.
Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444:707–12.
Ryskalin L, Lazzeri G, Flaibani M, Biagioni F, Gambardella S, Frati A, et al. mTOR-dependent cell proliferation in the brain. Biomed Res Int. 2017;2017:7082696.
Reijnders MRF, Kousi M, Van Woerden GM, Klein M, Bralten J, Mancini G, et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat Commun. 2017;8:1052.
Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–10.
Pan YH, Wu N, Yuan XB. Toward a better understanding of neuronal migration deficits in autism spectrum disorders. Front Cell Dev Biol. 2019;7:205.
Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–46.
Feliciano DM, Quon JL, Su T, Taylor MM, Bordey A. Postnatal neurogenesis generates heterotopias, olfactory micronodules, and cortical infiltration following single-cell TSC1 deletion. Hum Mol Genet. 2012;21:799–810.
Provenzano G, Chelini G, Bozzi Y. Genetic control of social behavior: Lessons from mutant mice. Behav Brain Res. 2017;325:237–50.
O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–9.
Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366–75.
Iakoucheva LM, Muotri AR, Sebat J. Getting to the cores of autism. Cell. 2019;178:1287–98.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–8.
Ruzzo EK, Pérez-Cano L, Jung JY, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–.e26.
Acknowledgements
We are grateful to all the subjects who participated in this work and their family members. We thank Elaine Cristina Zachi for performing formal psychological evaluation of patient F2688-1. We thank Danielle Moreira, Claudia Ismania Samogy Costa, Gerson Kobayashi, Luciano Abreu Brito, and Angela May Suzuki for technical assistance. We thank Carolina Pecaibes de Oliveira and Sandra Regina Malagutti for statistical assistance. The authors wish to acknowledge the resources of Sao Paulo Research Foundation (FAPESP) [grant number 2019/08655-2], National Council for Scientific and Technological Development (CNPq), MSSNG (https://research.mss.ng), Autism Speaks, and The Center for Applied Genomics at The Hospital for Sick Children, Toronto, Canada.
Author information
Authors and Affiliations
Contributions
ALTS, TG, JC, and AB conducted the experiments and contributed to the acquisition, analysis, and interpretation of the data. JYTW, GSC, MZ, and EJH conducted the sequencing data analysis. ALTS and ALS wrote the manuscript and prepared all figures with input from all authors. KGO, MRPB, HU, and SWS contributed to the research project's design and discussion of the data. ALS conceived the project, handled the funding, supervised the results, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. SWS is on the Scientific Advisory Committees of Deep Genomics, Population Bio, and an Academic Consultant for the King Abdulaziz University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teles e Silva, A.L., Glaser, T., Griesi-Oliveira, K. et al. Rare CACNA1H and RELN variants interact through mTORC1 pathway in oligogenic autism spectrum disorder. Transl Psychiatry 12, 234 (2022). https://doi.org/10.1038/s41398-022-01997-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01997-9
- Springer Nature Limited