
Abstract
Around 15–65% of women globally experience depression during pregnancy, prevalence being particularly high in low- and middle-income countries. Prenatal depression has been associated with adverse birth and child development outcomes. DNA methylation (DNAm) may aid in understanding this association. In this project, we analyzed associations between prenatal depression and DNAm from cord blood from participants of the South African Drakenstein Child Health Study. We examined DNAm in an epigenome-wide association study (EWAS) of 248 mother-child pairs. DNAm was measured using the Infinium MethylationEPIC (N = 145) and the Infinium HumanMethylation450 (N = 103) arrays. Prenatal depression scores, obtained with the Edinburgh Postnatal Depression Scale (EPDS) and the Beck Depression Inventory-II (BDI-II), were analyzed as continuous and dichotomized variables. We used linear robust models to estimate associations between depression and newborn DNAm, adjusted for measured (smoking status, household income, sex, preterm birth, cell type proportions, and genetic principal components) and unmeasured confounding using Cate and Bacon algorithms. Bonferroni correction was used to adjust for multiple testing. DMRcate and dmrff were used to test for differentially methylated regions (DMRs). Differential DNAm was significantly associated with BDI-II variables, in cg16473797 (Δ beta = −1.10E-02, p = 6.87E-08), cg23262030 (Δ beta per BDI-II total IQR = 1.47E-03, p = 1.18E-07), and cg04859497 (Δ beta = −6.42E-02, p = 1.06E-09). Five DMRs were associated with at least two depression variables. Further studies are needed to replicate these findings and investigate their biological impact.
Similar content being viewed by others


Introduction
Prenatal depression affects about 15–65% of women around the world with a higher percentage in low to middle-income countries (LMICs) than high-income countries (HICs) [1]. Adverse birth and child development outcomes, such as low birth weight, pre-term birth, and developmental delay, have been observed in children whose mothers experienced prenatal depression [1, 2]. Epigenetics has been hypothesized to play a role in this association. Prenatal development is a crucial and vulnerable period for the epigenome due to epigenetic reprogramming that occurs for both DNA methylation (DNAm) and histone modifications during this time [3]. With the exception of a few regions such as imprinted genes, the epigenome is reprogrammed by the global decrease in DNAm pre-implantation and then increase in DNAm following implantation for processes such as organogenesis [4]. Prenatal exposures such as tobacco smoke [5], maternal stress [6], or toxins [7] can affect the child’s epigenome during prenatal development. Changes in the infant’s epigenetic mechanisms, such as DNAm, as a result of prenatal depression, may provide insight into this association either as a biomarker or as a possible mediating factor in biological pathways.
Previous studies investigating the association between prenatal depression and differential DNAm have focused on candidate genes such as NR3C1 and SLC6A4 [8, 9]. Children exposed to prenatal depression have been shown to have increased DNAm in NR3C1 and decreased DNAm in SLC6A4 [8, 9]. Epigenome-wide association studies (EWAS) have also investigated associations between prenatal depression and differential DNAm [10, 11]. Two of these studies found a combined total of five CpG sites (cg08667740, cg22868225, cg06808585, cg05245515, and cg15264806) and 39 differentially methylated regions (DMRs) associated with prenatal depression [10, 11]. These previous studies included mother-child pairs only from high-income countries including Norway, the Netherlands, the United Kingdom, and the United States [10,11,12].
As women from LMICs are particularly vulnerable to prenatal depression [1], this study aimed to investigate this association using the Drakenstein child health study (DCHS), a population-based birth cohort in South Africa [13, 14]. This cohort is representative of several aspects of the LMIC context and allows for the study of potential associations between prenatal depression and DNAm in this setting [13].
Materials and methods
Study population
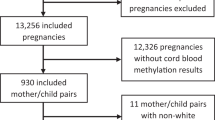
The study population consisted of 248 mother-child pairs from the DCHS with data available for the Edinburgh Postnatal Depression Scale (EPDS) and the Beck Depression Inventory-II (BDI-II) scores, cord blood DNAm, and covariates. Participants were recruited between March 2012 and March 2015 from two primary care clinics, TC Newman or Mbekweni [13, 15]. Mothers were enrolled during their second trimester and followed until the child was at least five years old [13,14,15,16].
Ethical approval was given from the Human Research Ethics Committee of the Faculty of Health Sciences of the University of Cape Town for human subjects’ research and written consent was obtained from the mothers [13, 15].
DNA methylation measurements
DNA methylation was measured from cord blood collected at delivery by either the MethylationEPIC BeadChips (EPIC; n = 145) or the Illumina Infinium HumanMethylation450 BeadChips (450K; n = 103) [13, 15]. The subgroup that was selected for the second set of DNAm analyses (EPIC, n = 145) was enriched for maternal trauma exposure/post-traumatic stress disorder (PTSD).
Pre-processing and statistics were done using R 3.5.1. Raw iDat files were imported to RStudio where intensity values were converted into beta values. The 450K array had 426,378 probes while the EPIC array contained 781,536 probes. Pre-processing was performed in each array separately but with identical pre-processing steps. Background subtraction, colour correction and normalization were performed using the preprocessFunnorm function. After sample and probe filtering, 120 samples and 426,378 probes remained for the 450K dataset with 153 samples and 781,536 probes with the EPIC dataset. Batch effects were removed using ComBat from the R package sva. Cord blood cell type composition was predicted using the most recent cord blood reference data set and the IDOL algorithm and probe selection.
Depression measurements
Prenatal depression was assessed with both the EPDS and BDI-II administered at 28–32 weeks gestation [13, 15]. The EPDS scale has 10 questions, scores ranging from 0 to 30, and was designed to screen for postnatal depression [17]. This scale has been verified for prenatal depression in an African setting [18]. The BDI-II scale has 21 questions, scores ranging from 0 to 63, and is used to screen for depression [19]. The BDI-II scale has been validated for prenatal depression [20] and used for prenatal depression in countries such as Ethiopia and Kenya [21, 22]. For EPDS, thresholds of 10 and 13 are commonly used to screen for depression with 10 having a higher sensitivity and 13 having a higher specificity [23]. For BDI-II, the lower threshold of 14 is the threshold for mild depression, and a higher threshold of 20 is the threshold for moderate depression [19].
Statistical analysis
The association between prenatal maternal depression and newborn differential methylation at individual CpG sites and DMRs were assessed in epigenome-wide association studies (EWAS). We conducted EWAS for the 450K and EPIC data separately, followed by a meta-analysis to combine the results of the CpG sites that were measured with both arrays. For each of the EWAS analyses, a multivariable robust linear regression model with empirical Bayes using the limma R package was fitted [24]. The dependent variable was cord blood DNAm, with depression variables as the independent variable while adjusting for the following covariates: mother’s smoking status, household income, sex of the child, gestational age at birth, first three cell type principal components (PCs) which explained 90% of heterogeneity due to cell type [25], and first five genotype PCs for population stratification. The meta-analysis was performed imputing the individual 450K and EPIC EWAS results into METAL and running a fixed-effect model with inverse variance weighting [26]. A sensitivity analysis was performed to determine the effect of including HIV exposure as a potential confounder in the model. P-values were additionally adjusted for bias and unmeasured confounding using the Bacon and Cate R packages respectively [27] (Table S1). The continuous depression scale was used as primary outcomes, followed by analyses of the dichotomized variables (screening for depression) as secondary outcomes. To account for multiple testing, the Bonferroni threshold was used for statistical significance (EPIC: 0.05 / 781536 CpGs = 6.40 × 10−8, 450K: 0.05/426378 CpGs = 1.17 × 10−7, meta-analysis: 0.05/386685 CpGs = 1.29 × 10−7). Fine-mapping of our epigenome-wide associations was done with the R package comet, that displays the region surrounding any significant CpG sites [28]. DMRs were assessed from the meta-analysis of the overlapping CpG sites from EPIC and 450K using the R package DMRcate (version 1.20.0) [29]. The input files for these analyses included the regression coefficients, standard deviations, and p-values from the meta-analysis of single-CpG analyses. The R package dmrff [30] was also used to evaluate DMRs by meta-analysis of the EPIC and 450K arrays. DMRs for both the DMRcate and dmrff analysis methods were defined by requiring at least two CpG sites within 1,000 bps apart and the region having a Bonferroni corrected p-value < 0.05. Furthermore, we used the robustness of DMRs across different depression scales as an additional validation criterion.
For any significant CpG sites or DMRs, we looked up the correlations between blood and brain DNA methylation using the public data source IMAGE-CpG, that is based on blood, saliva, buccal, and live brain tissue samples from 27 patients with medically intractable epilepsy undergoing brain resection [31].
Results
Study population characteristics
The analysis sample included 248 mother-child pairs with complete information for depression scores, cord blood DNAm, and relevant covariates (Table 1). DNAm was measured in the cord blood of 145 infants (58%) using the EPIC array and in 103 infants (42%) using the 450K array. Overall, 44% of the infants were female and the mean gestational age at birth was 38.75 weeks. About 21% of the mothers were smokers during pregnancy with a higher proportion of smokers in 450K data than in the EPIC data. The average EPDS score was 10.52 (sd = 5.09) with 56% defined as depressed according to the threshold of 10 and 31% according to the threshold of 13. The average BDI-II score among mothers was 13.19 (sd = 11.17) with 44% and 25% defined as depressed according to the thresholds of 14 or 20, respectively. The women in the 450K array group (n = 103) tended to have higher depression scores (EPDS and BDI-II) than in the EPIC data.
Primary outcomes: Maternal depression scores and newborn DNAm
After accounting for bias and measured and unmeasured confounding, the EWAS of the individual 450K and EPIC data did not produce significant CpG sites for either of the continuous depression variables (EPDS and BDI-II) (Figure S1–S4). After combining the overlapping CpG sites from both arrays in a meta-analysis, we found a significant association between prenatal depression and differential DNAm in cg23262030 for the BDI-II continuous variable (Δ beta per BDI-II total IQR = 1.47E-03, p-value = 1.18E-07) (Fig. 1). This CpG site also had suggestive p-values for the BDI-II 14 threshold (Δ beta = 4.10E-04, p-value = 3.76E-06) and nominally significant p-values for the BDI-II 20 threshold, EPDS continuous, and EPDS 13 threshold variables (Table 2, Fig. 1, Figures S5–S8). Associations with DNAm in cg23262030 were similar for data from both arrays, however, the association for the 450K array was not significant (Fig. 2). For CpG sites that reached p-values less than 5 × 10−4 for at least one of the depression scales in the meta-analysis, beta estimates for the BDI-II and EPDS continuous variables were correlated (Figure S9).

Manhattan and QQ plots for the meta-analysis of EWAS of maternal depression (primary outcomes, A. EPDS and B. BDI-II). Adjusted for covariates: mother’s smoking status, average household income, child’s sex, gestational age at birth, first three cell type PCs, and first five genotype PCs. Unmeasured confounding and bias were adjusted with Cate and Bacon R packages. Bonferroni threshold: meta-analysis = 1.29 × 10−7.

Adjusted for covariates: mother’s smoking status, average household income, child’s sex, gestational age at birth, first three cell type PCs, and first five genotype PCs. Unmeasured confounding and bias were adjusted with Cate and Bacon R packages. A For cg16473797 using the BDI-II 14 threshold. B For cg23262030 using the BDI-II continuous variable.
The DMRcate meta-analysis resulted in seven DMRs for the EPDS continuous score and seven DMRs for the BDI-II continuous score (Tables S2, S3). The DMR, chr18: 67069959–67070461, was significant for both depression variables (EPDS: max Δ beta estimate = −2.60E-03, p-value = 8.29E-10, BDI-II: max Δ beta estimate = −1.09E-03, p-value = 6.84E-05) (Table 3). The meta-analysis using dmrff resulted in one DMR for the EPDS continuous score, however, this DMR was not found with any other depression variable (Table S4).
Secondary outcomes: Screening for maternal depression and newborn DNAm
The analyses of our secondary outcomes identified additional significant CpG sites that were also at least suggestive for our primary outcomes (Figure S10–S17). Two CpG sites, cg03489382 and cg19350511, were significant for the 450K array for the BDI-II 14 threshold variable but not for the EPIC array (Figure S10, S11). The EWAS of the EPIC data resulted in cg04859497 being statistically significant for the BDI-II threshold 20 (Δ beta = −6.42E-02, p-value = 1.06E-09) (Table 2, Figure S12). This CpG site is unique to the EPIC array and not available on the 450K array. The p-values for the CpG site were suggestive for the primary outcome BDI-II but not for the primary outcome EPDS (Table 2). The meta-analysis for both the arrays resulted in a significant association between differential DNAm in cg16473797 (BDI-II 14 threshold: Δ beta = −1.10E-02, p-value = 6.87E-08) and prenatal depression (Figure S6). This CpG site had suggestive p-values for the continuous BDI-II variable (Δ beta per BDI-II total IQR = −5.64E-03, p-value = 5.72E-05) and the BDI-II 20 threshold (Δ beta = −8.61E-03, p-value = 1.72E-04) and nominally significant p-values for the EPDS continuous and EPDS 13 threshold variables (Table 2). Associations with DNAm in cg16473797 were similar for data from both arrays (Fig. 2).
The DMRcate meta-analysis resulted in 24 DMRs for the binary variables (EPDS threshold-10: four DMRs, EPDS threshold-13: four DMRs, BDI-II threshold-14: eight DMRS, BDI-II threshold-20: eight DMRs) (Tables S5–S8). Five DMRs were significant for more than one variable, continuous or dichotomized, and two of the five DMRs, chr18: 67069959–67070461, chr7: 155174726–155175340, were significant for more than one dichotomized variable (Table 3). The meta-analysis using dmrff did not produce significant DMRs for the dichotomized depression variables.
Discussion
In this study of infants from a peri-urban region in a low-resourced community in South Africa, we found prenatal depression to be associated with differential methylation in three CpG sites and within multiple DMRs measured in cord blood of newborns from the Drakenstein Child Health Study.
Comparison with previous studies
The association between maternal prenatal depression and differences in infant DNAm is not completely understood. Previous studies measuring DNAm from cord blood have shown mixed results. A study by Viuff et al. (2018), which used the EPDS threshold-12 variable to screen for prenatal depression, found differential methylation in two CpG sites to be associated with prenatal depression while a study by Cardenas, A. et al. (2019), which used the Brief Symptom Inventory threshold-0.80 variable to screen for prenatal depression, found differential methylation in three different CpGs to be associated with prenatal depression [10, 11]. However, the significant sites for both of these studies were unable to be replicated using the Generation R study [10, 11]. The five CpG sites identified in previous studies were not significantly associated with prenatal depression in our cohort, which is in line with the results from the Generation R study (Tables S9, S10). As for DMRs, Cardenas et al. (2019) did not find any DMRs significantly associated with prenatal depression [11]. However, Viuff, et al. (2018) found 39 DMRs to be associated with prenatal depression [10]. Of these 39 DMRs, a DMR containing eight CpGs, chr8:70378380–70378995 [10], overlapped with a seven CpG DMR, chr8: 70378380–70378994 found to be significantly associated with the BDI-II continuous and BDI-II threshold-20 in our study. This replicated DMR in chr8 was previously found to be significant for mid-pregnancy maternal depression, which is defined as the depression between 18 to 32 weeks gestation [10]. This overlaps with the time maternal depression was assessed in our study, which was between 28 and 32 weeks gestation [32]. The DMR in chr8: 70378380–70378994 overlaps with the promoter region for SULF1 which codes for the extracellular sulfatase Sulf-1 and is involved in regulating heparin sulfate (HS)-dependent signalling pathways [31]. In mice, deficiencies in SULF1 were associated with impaired neurite outgrowth, providing evidence for the role of SULF1 in nervous system development [33, 34]. One site within this DMR, cg07051728, was found to have a significant correlation between DNAm in brain tissue and DNAm in the blood (Table S11).
Primary outcomes: Maternal depression scores and newborn DNAm
In our meta-analysis of overlapping CpG sites from the 450K and EPIC arrays, one CpG site, cg23262030, was significant for the BDI-II continuous depression variable (Figure S18). This CpG site is located within a promoter region near the SCRN2. The full role of the SCRN2 gene does not appear to be well understood so it is unknown whether this gene or the overlapping promoter region have an important role in child development.
The meta-analysis resulted in seven DMRs for the continuous EPDS score and seven DMRs for the continuous BDI-II score with a DMR in chr18: 67069959–67070461 being significantly associated with all depression variables except for the BDI-II threshold-14 variable. This DMR overlaps with the promoter region for docking protein 6 (DOK6), specifically the protein-coding transcript DOK6–001, previously shown to perform a role in Ret-mediated neurite growth [35] and nervous system development through NT-3 mediation in mice [36]. Up to now, there has not been human research for this protein and neurodevelopment. However, in human tissues, DOK6 has been shown to have high expression in the fetal brain [35]. This DMR contained one site with a significant correlation for DNAm between brain tissue and blood (Table S10). This DMR may be important for studying adverse developmental outcomes in children born to mothers who experienced prenatal depression. The DMR, chr15: 98195808–98196247, was significantly associated with a continuous and threshold variable in the meta-analysis however no clear link between this location and adverse birth or developmental outcomes or prenatal depression has been reported in the literature.
Secondary outcomes: Screening for maternal depression and newborn DNAm
Our secondary outcomes include dichotomized versions of our primary outcomes using different thresholds commonly used for screening for depression. For the secondary outcomes, we found two additional significant CpG sites that were also at least suggestive for our primary outcome BDI-II. The CpG site, cg16473797, was found to be significant in the meta-analysis for the BDI-II 14 threshold (Figure S19). This site does not appear to be located within a known gene or regulatory region but does share the same position as SNP rs1358065399. The major allele for this SNP is C and the minor allele is T with a minor allele frequency less than 0.01 among the global population.
A single CpG site found from the EPIC array specific analysis was significantly associated with the BDI-II threshold-20 variable and also suggestive for the other BDI-II variables (Figure S20). This site also occupies the same position as SNP, rs140401989, which has C as the major allele and T as the minor allele (minor allele frequency = 0.01–0.04 among the global population). This site is located in the second intron within CTNNA2 which codes for the catenin alpha-2 protein which plays an important role in neurodevelopment by acting as a regulator for actin branching, with mutations in this gene associated with a neuronal migration disorder [37]. CTNNA2 has been shown to have higher expression in the brain than most other tissues [37]. However, the CpG site, cg04859497, was not found to have a significant correlation for DNAm across brain tissue and blood (Table S11). As a result, it is unknown whether this site would also have differential methylation in cells within prenatal brain tissue due to prenatal depression.
The meta-analysis resulted in 24 DMRs for the dichotomized variables (EPDS threshold-10: four DMRs, EPDS threshold-13: four DMRs, BDI-II threshold-14: eight DMRS, BDI-II threshold-20: eight DMRs) and five of these were significant for more than one depression variable (including the replicated DMR in chr8: 70378380–70378994 discussed above). The DMR, chr7: 155174726–155175340, and chr19: 18698825–18699631 were significant for more than one depression variable, however, the connection between these regions and adverse outcomes due to prenatal depression is not clear. Overall, these sites and DMRs may be useful for investigating the biological pathway for the association between maternal prenatal depression and adverse birth and child development outcomes.
Strengths and limitations
Our study has several strengths. Previous studies have only focused on one dichotomized depression scale [10, 11]. In our study, we used more than one scale with continuous variables as primary outcomes and dichotomized variables as secondary outcomes to reflect the complexity of depression and to validate the robustness of our findings across different depression scales. Another strength lies in the study population being of African and mixed ancestry and from a low to a middle-income country, underrepresented populations among genetic and epigenetic studies [38, 39] (Figure S21). A major contributor to the variation in DNA methylation is genetic variation. This study includes genome-wide genotype data which was used to correct for population stratification. However, genetic variation could also contribute to differential DNA methylation through methylation quantitative trait loci (meQTLs) [40]. None of the significant individual CpG sites was found to be associated meQTLs, but CpG sites within the significant DMRs were found to be associated with meQTLs (Table S12). The meQTLs could indicate a genetic contribution to the association between the DMRs and prenatal depression. Another issue that plagues EWASs is unknown confounding. This study used Cate and Bacon to control for bias and unmeasured confounding, which are state-of-the-art confounder adjustment methods based on the calculation of surrogate variables and the empirical null distribution, respectively [28].
This study does include limitations such as a relatively small study size which reduces the power to detect differences in methylation [41]. The depression scores were collected at a single time point resulting in the inability to know the complete timeframe for the onset and duration of the prenatal depression [32]. Also, this analysis is based on depression scales instead of clinical diagnosis of prenatal depression, though the depression scales are used to screen for probable depression but are not equivalent to clinical diagnosis [21]. While using more than one scale with continuous variables as primary outcomes and dichotomized variables as secondary outcomes helps to reflect the complexity of depression and to validate the robustness of our findings across different depression scales, we acknowledge that this approach also increases the chances of false-positive findings due to the burden of multiple testing. Furthermore, having two datasets measured on different array platforms resulted in EPIC specific sites not being assessed in the meta-analysis excluding these results from benefiting from larger samples sizes. Another limitation in this study is the use of heterogeneous tissue. Although the variance in the proportions of cell types was controlled for using methylation predicted estimated cell counts [42], the specific cell type origin for a given change in methylation was impossible to determine. This is an issue that is alleviated using techniques such as single-cell methylation measurements. Another limitation is the use of IMAGE-CpG to determine if there is a significant correlation between differential methylation in significant CpG sites in blood and brain tissue. The blood samples for this study were taken at birth while the samples for IMAGE-CpG have an age range of 5–61 years [31]. DNA methylation changes with age [43] which may result in incorrect associations or lack of associations for significant CpG sites in blood and brain tissue. A large limitation in most EWASs is in interpreting what changes in methylation actually mean. Even though there have been studies showing the impact of DNAm on gene expression, interpretations must be taken with caution. Additionally, many of the genes which were differentially methylated in association with maternal depression in our study have been linked to various neurological outcomes. We know that DNAm varies most greatly between tissues, as establishing cellular identity is one of the main functions of DNAm. As such, the methylation status observed in the blood cannot be extrapolated to the methylation status in the brain. While there are datasets with matched brain and blood methylation available to help support the link between blood methylation and neurological outcomes, interpretations of differential methylation need to be taken with caution.
Conclusion
Maternal depression was associated with differential DNAm in three CpG sites and within multiple DMRs. The DMR chr8: 70378380–70378994 has been associated with maternal depression in a previous study [10]. The remaining sites and DMRs, to our knowledge, have not been previously associated with maternal depression. Further research is needed to replicate this finding and to investigate its impact on birth outcomes and child development.
Web resources
Ensembl, NCBI’s Gene resources, Human Protein Atlas, mQTLdb:
Accessed on 4/9/2021
-
Overlapping genes for DMR chr8: 70378380-70378995
-
Overlapping genes for DMR chr18: 67069959-67070461
-
Overlapping genes for DMR chr15: 98195808-98196247: http://grch37.ensembl.org/Homo_sapiens/Location/View?r=15:98196029-98196118;db=core
-
CTNNA2 position of cg04859497: https://grch37.ensembl.org/Homo_sapiens/Location/View?db=core;g=ENSG00000066032;r=2:79900309-79939089;t=ENST00000496558
Accessed on 8/28/2021
-
Position of cg23262030 in the genome. https://grch37.ensembl.org/Homo_sapiens/Location/View?r=17:45918302-45919105;db=core
-
Position of cg16473797 in the genome. https://grch37.ensembl.org/Homo_sapiens/Location/View?r=20%3A61754945-61755145
-
Function of the SCRN2 gene: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SCRN2
-
Overlapping genes for DMR chr7: 55174991-155175340
-
Overlapping genes for DMR chr19: 18698825-18699423
-
rs1358065399 genomic position and allele frequency: https://grch37.ensembl.org/Homo_sapiens/Variation/Explore?db=core;r=20:61754945-61755145;v=rs1358065399;vdb=variation;vf=440868348
-
rs140401989 genomic position and allele frequency: https://grch37.ensembl.org/Homo_sapiens/Variation/Explore?db=core;r=2:79923800-79923830;v=rs140401989;vdb=variation;vf=160926159
Accessed on 8/31/2021
-
Presence or absence of meQTLs at birth for CpG sites and DMRs: http://www.mqtldb.org/cgi-bin/search.cgi
References
Dadi AF, Miller ER, Bisetegn TA, Mwanri L. Global burden of antenatal depression and its association with adverse birth outcomes: An umbrella review. BMC Public Health. https://doi.org/10.1186/s12889-020-8293-9 (2020).
Deave T, Heron J, Evans J, Emond A. The impact of maternal depression in pregnancy on early child development. BJOG. 2008;115:1043–51.
Kundakovic M, Jaric I. The epigenetic link between prenatal adverse environments and neurodevelopmental disorders. Genes (Basel). 2017;18:104.
Thorsell A, Nätt D. Maternal stress and diet may influence affective behavior and stress-response in offspring via epigenetic regulation of central peptidergic function. Environ. Epigenetics 2016;2:dvw012.
Suter MA, Aagaard K. What changes in DNA methylation take place in individuals exposed to maternal smoking in utero? Epigenomics 2012;4:115–8.
Kertes DA, Kamin HS, Hughes DA, Rodney NC, Bhatt S, Mulligan CJ. Prenatal maternal stress predicts methylation of genes regulating the hypothalamic-pituitary-adrenocortical system in mothers and newborns in the democratic Republic of Congo. Child Dev. 2016;87:61–72.
Appleton AA, Jackson BP, Karagas M, Marsit CJ. Prenatal exposure to neurotoxic metals is associated with increased placental glucocorticoid receptor DNA methylation. Epigenetics 2017;12:607–15.
Devlin AM, Brain U, Austin J, Oberlander TF. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS One. 2010;5:e12201.
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106.
Viuff AC, Sharp GC, Rai D, Henriksen TB, Pedersen LH, Kyng KJ, et al. Maternal depression during pregnancy and cord blood DNA methylation: Findings from the Avon Longitudinal Study of Parents and Children. Transl Psychiatry. 2018;8:244.
Cardenas A, Faleschini S, Cortes Hidalgo A, Rifas-Shiman SL, Baccarelli AA, DeMeo DL, et al. Prenatal maternal antidepressants, anxiety, and depression and offspring DNA methylation: Epigenome-wide associations at birth and persistence into early childhood. Clin Epigenetics. 2019;11:56.
Wikenius E, Myhre AM, Page CM, Moe V, Smith L, Heiervang ER, et al. Prenatal maternal depressive symptoms and infant DNA methylation: a longitudinal epigenome-wide study. Nord J Psychiatry. 2019;73:257–63.
Zar HJ, Barnett W, Myer L, Stein DJ, Nicol MP. Investigating the early-life determinants of illness in Africa: The Drakenstein child health study. Thorax 2015;70:592–4.
Brittain K, Myer L, Koen N, Koopowitz S, Donald KA, Barnett W, et al. Risk factors for antenatal depression and associations with infant birth outcomes: Results from a South African birth cohort study. Paediatr Perinat Epidemiol. 2015;29:505–14.
Stein DJ, Koen N, Donald KA, Adnams CM, Koopowitz S, Lund C, et al. Investigating the psychosocial determinants of child health in Africa: The Drakenstein child health study. J Neurosci Methods. 2015;252:27–35.
Donald KA, Hoogenhout M, du Plooy CP, Wedderburn CJ, Nhapi RT, Barnett W, et al. Drakenstein Child Health Study (DCHS): Investigating determinants of early child development and cognition. BMJ Paediatr Open. 2018;2:e000282.
Gibson J, McKenzie-Mcharg K, Shakespeare J, Price J, Gray R. A systematic review of studies validating the Edinburgh Postnatal Depression Scale in antepartum and postpartum women. Acta Psychiatr Scand. 2009;119:350–64.
Heyningen T, Honikman S, Tomlinson M, Field S, Myer L. Comparison of mental health screening tools for detecting antenatal depression and anxiety disorders in South African women. PLoS One. 2018;13:e0193697.
Graham RM, Jiang L, McCorkle G, Bellando BJ, Sorensen ST, Glasier CM, et al. Maternal anxiety and depression during late pregnancy and newborn brain white matter development. AJNR Am J Neuroradiol. 2020;41:1908–15.
Naja S, Al-Kubaisi N, Chehab M, Al-Dahshan A, Abuhashem N, Bougmiza I. Psychometric properties of the Arabic version of EPDS and BDI-II as a screening tool for antenatal depression: Evidence from Qatar. BMJ Open. 2019;9:e030365.
Alenko A, Dejene S, Girma S. Sociodemographic and obstetric determinants of antenatal depression in Jimma Medical Center, Southwest Ethiopia: Facility based case-control study. Int J Women’s Health. 2020;12:557–65.
Opiyo RO, Nyasulu PS, Koigi RK, Obondo A, Ogoyi D, Kogi-Makau W. Effect of fish oil omega-3 fatty acids on reduction of depressive symptoms among HIV-seropositive pregnant women: A randomized, double-blind controlled trial. Ann Gen Psychiatry. 2018;17:49.
Khanlari S, Barnett AmB, Ogbo FA, Eastwood J. Re-examination of perinatal mental health policy frameworks for women signalling distress on the Edinburgh Postnatal Depression Scale (EPDS) completed during their antenatal booking-in consultation: A call for population health intervention. BMC Pregnancy Childbirth. 2019;19:221.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;34:e47.
Pawlowsky-Glahn V, Egozcue JJ. Compositional data and their analysis: An introduction. Geol Soc Spec Publ. 2006;246:1–10.
Willer C, Li Y, Abecasis G. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–1.
Van Iterson M, Van Zwet E. BIOS Consortium, Heijmans B. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18:19.
Martin TC, Yet I, Tsai PC, Bell JT. coMET: Visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinforma. 2015;16:131.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Suderman M. dmrff: identifying differentially methylated regions efficiently with power and control. Preprint at https://www.biorxiv.org/content/biorxiv/early/2018/12/31/508556.full.pdf:508556. (2018).
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47.
Koen N, Brittain K, Donald KA, Barnett W, Koopowitz S, Maré K, et al. Psychological trauma and posttraumatic stress disorder: Risk factors and associatiosn with birth outcomes in the Drakenstein Child Health Study. Eur J Psychotraumatol. 2016;7:28720.
Kalus I, Salmen B, Viebahn C, von Figura K, Schmitz D, D'Hooge R, et al. Differential involvement of the extracellular 6-O-endosulfatases Sulf1 and Sulf2 in brain development and neuronal and behavioural plasticity. J Cell Mol Med. 2009;13:4505–21.
Kalus I, Rohn S, Puvirajesinghe TM, Guimond SE, Eyckerman-Kölln PJ, Ten Dam G, et al. Sulf1 and Sulf2 differentially modulate heparan sulfate proteoglycan sulfation during postnatal cerebellum development: Evidence for neuroprotective and neurite outgrowth promoting functions. PLOS One. 2015;10:e0139853.
Crowder R, Enomoto H, Yang M, Johnson E, Milbrandt J. Dok-6, a Novel p62 Dok family member, promotes Ret-mediated neurite outgrowth. J Biol Chem. 2004;279:42072–81.
Li WQ, Shi L, You YG, Gong YH, Yin B, Yuan JG, et al. Downstream of tyrosine kinase/docking protein 6, as a novel substrate of tropomyosin-related kinase C receptor, is involved in neurotrophin 3-mediated neurite outgrowth in mouse cortex neurons. BMC Biol. 2010;8:86.
Schaffer AE, Breuss MW, Caglayan AO, Al-Sanaa N, Al-Abdulwahed HY, Kaymakçalan H, et al. Biallelic loss of human CTNNA2, encoding αN-catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nat Genet. 2018;50:1093–101.
Popejoy A Genomics is failing on diversity. Nature. https://doi.org/10.1038/538161a (2016).
Cronjé H, Elliott H, Nienaber-Rousseau C, Pieters M. Replication and expansion of epigenome-wide association literature in a black South African population. Clin Epigenetics. 2020;12:6.
Lemire M, Zaidi SHE, Ban M, Ge B, Aïssi D, Germain M, et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat Commun. 2015;6:6326.
Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20:366.
Kaushal A. Comparison of different cell type correction methods for genome-scale epigenetics studies. BMC Bioinformatics. https://doi.org/10.1186/s12859-017-1611-2 (2017).
Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15:483–94.
Acknowledgements
The authors thank the study and clinical staff at Paarl Hospital, Mbekweni and TC Newman clinics, as well as the CEO of Paarl Hospital, and the Western Cape Health Department for their support of the study. The authors thank the families and children who participated in this study.
Funding
The Drakenstein child health study was funded by the Bill & Melinda Gates Foundation (OPP 1017641), Discovery Foundation, Medical Research Council South Africa, National Research Foundation South Africa, CIDRI Clinical Fellowship and Wellcome Trust (204755/2/16/z). Additional support for the DNA methylation work was by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (NICHD) under Award Number R21HD085849, and the Fogarty International Center (FIC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. AH is supported by the HERCULES Center (NIEHS P30ES019776). MPE was supported by NIH grant R01 GM117946. DJS and HJZ are supported by the South African Medical Research Council (SAMRC). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Drzymalla, E., Gladish, N., Koen, N. et al. Association between maternal depression during pregnancy and newborn DNA methylation. Transl Psychiatry 11, 572 (2021). https://doi.org/10.1038/s41398-021-01697-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01697-w
- Springer Nature Limited