
Abstract
The identification of genetic variants underlying autism spectrum disorders (ASDs) may contribute to a better understanding of their underlying biology. To examine the possible role of a specific type of compound heterozygosity in ASD, namely, the occurrence of a deletion together with a functional nucleotide variant on the remaining allele, we sequenced 550 genes in 149 individuals with ASD and their deletion-transmitting parents. This approach allowed us to identify additional sequence variants occurring in the remaining allele of the deletion. Our main goal was to compare the rate of sequence variants in remaining alleles of deleted regions between probands and the deletion-transmitting parents. We also examined the predicted functional effect of the identified variants using Combined Annotation-Dependent Depletion (CADD) scores. The single nucleotide variant-deletion co-occurrence was observed in 13.4% of probands, compared with 8.1% of parents. The cumulative burden of sequence variants (n = 68) in pooled proband sequences was higher than the burden in pooled sequences from the deletion-transmitting parents (n = 41, X2 = 6.69, p = 0.0097). After filtering for those variants predicted to be most deleterious, we observed 21 of such variants in probands versus 8 in their deletion-transmitting parents (X2 = 5.82, p = 0.016). Finally, cumulative CADD scores conferred by these variants were significantly higher in probands than in deletion-transmitting parents (burden test, β = 0.13; p = 1.0 × 10−5). Our findings suggest that the compound heterozygosity described in the current study may be one of several mechanisms explaining variable penetrance of CNVs with known pathogenicity for ASD.
Similar content being viewed by others

Introduction
Autism spectrum disorders (ASDs) are a group of neurodevelopmental disorders characterized by social and communicative deficits, a marked insistence on sameness and/or repetitive behaviors1. The estimated population prevalence of ASDs is ~1%2. It is well established that genetic factors contribute to the risk of ASDs3. The identification of the genetic risk variants associated with ASDs constitutes an appealing strategy to elucidate their underlying biology4,5. Genetic variants identified so far include single nucleotide variants (SNVs), as well as structural abnormalities in copy number (CNVs), leading to a loss or gain of up to several millions of base pairs. These variants can be inherited or can occur de novo, i.e., a novel change in the genetic code emerges in the child while not part of the DNA sequence of either parent.
Common variants occur frequently in the population (minor allele frequency (MAF) of 5% or more) and are associated with small risk increases6,7. However, current estimates of the cumulative effect of such common variants account for 12% of the variance in autism (SNP heritability (h2 = 0.118)7,8. There is also evidence for the role of rare variants in ASD; these are alleles that occur infrequently in the population (e.g., MAF < 1%) but may be associated with larger risk effects in the individual carrier. It is estimated that causative rare genetic variants, both de novo and inherited, can be identified in 10–30% of patients with ASD9,10,11.
When a deletion affects a genomic region with optimally functioning genes on the remaining allele, the most likely effect of that deletion is a change in gene expression with potential to result in a phenotypic effect12. However, a pathogenic impact may be more likely if the performance of a gene on the remaining allele is also impacted by a functional variant (“compound heterozygosity”). The co-occurrence of impactful variation on both copies of a gene, a deletion on the one and a functional variant on the other allele, may thus be a relevant genetic mechanism in ASD (see Fig. 1). The psychiatric genetics literature provides precedents for this “double hit” mechanism, which can be considered as a specific type of compound heterozygosity: several case studies report the co-occurrence of an inherited deletion and a functional variant on the remaining allele in probands with autism13,14,15 and in schizophrenia16,17. Furthermore, the rate of a slightly different type of compound heterozygosity, i.e., two rare loss-of-function sequence variants co-occurring at the same locus, is found to be significantly increased in autism compared with controls18,19.

Scenario 1: a gene is included, partly or entirely, in a deletion. A sequence variant occurs at the remaining allele of the gene, within the boundaries of the deleted region. Scenario 2: a gene is partly included in a deletion. A sequence variant occurs at the remaining allele of the gene, but outside the boundaries of the deleted region.
Here, we hypothesize that compound heterozygosity of a deletion and a functional sequence variant at the remaining allele occurs more often in patients with ASDs compared with their parents transmitting the deletions. We speculate that this compound heterozygosity mechanism may provide an explanation for the penetrance of the inherited CNVs identified in individuals with ASD, compared with unaffected parents. The current study aims to provide empirical evidence for the proposed compound heterozygosity mechanism as a relevant causative factor in a proportion of ASD cases.
Material and methods
Project overview
We selected proband–parent pairs and trios from an existing dataset (Autism Genome Project, AGP) of 2191 families for which previous studies had already provided data from genome-wide CNV screening20. In brief, diagnosis of ASD was based on standardized assessments and/or clinical evaluation, as described previously20. DNA samples were available from six European sites and one American site from the AGP. Ethical approval was obtained from all participating sites’ IRBs and all participants provided written informed consent. We collected DNA aliquots that remained after the major genetic analyses of the AGP had been performed21,22,23,24. We abided by the principles laid out in the Declaration of Helsinki.
From the available AGP dataset we prioritized those probands who had inherited at least one deletion from a parent. We prioritized inherited deletions that involved one or more genes with probable relevance to the brain. We annotated genes as brain relevant on the basis of concordance between three different data categories: (1) sequence tags expressed in the brain (ESTs)25; (2) results from a large gene expression analysis26; and (3) biological functions inferred by matching a vocabulary of brain-related terms against gene ontologies from the AmiGO database27 (see Supplementary methods). After prioritization of subjects (see below), we investigated in our selected study population the rate of additional sequence variants in those genes affected by inherited deletions. We used targeted genomic enrichment followed by next-generation sequencing28 to identify the co-occurrences of inherited deletions with a functional sequence variant in the remaining allele in our entire sample of pedigrees. In essence, we examined the rate of these compound heterozygous events by comparing the sum of sequence variants in all deleted gene regions in probands to the sum of sequence variants identified in the same deleted gene regions in the parent who transmitted the deletion to each proband (Figs. 1 and 2). In addition, we investigated whether the cumulative predicted functional impact, as expressed by the Combined Annotation-Dependent Depletion v1.4 (CADD)29 scores (see below) of the genetic variants is different in probands compared with deletion-transmitting parents.

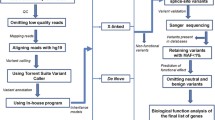
a Identification of inherited deletions in probands. In this example, the proband inherited a deletion from the father. The deletion involves one gene (red). We prioritized inherited deletions that involved one or more genes with probable relevance to the brain. b Targeted sequencing of deleted gene(s) in each proband and his/her parent(s) who transmitted the deletion. We analyzed 102 proband–parent pairs and 47 proband–parent trios. (in this figure, only proband–parent pairs are shown). c Comparing the rate of sequence variants (*) in the pooled set of sequenced genes between probands and their deletion-transmitting parents. For our analyses, for each of the 149 families we only queried the sequence of gene(s) affected by inherited deletion(s) in that specific family.
DNA sample collection and subject prioritization steps
We considered families from the seven sites that participate in the AGP, i.e., France, Germany, United Kingdom (International Molecular Genetic Study of Autism families) England, Ireland, Italy, Portugal, and the United States. There were N = 2191 families (mostly trios) for a total of 6986 samples. We prioritized CNV calls based on the following criteria: (1) called by two or more algorithms (QuantiSNP30, PennCNV31, and iPattern32); (2) <10% frequency in the AGP dataset to exclude common CNVs that are likely to be benign; and (3) length >5 kb to ensure adequate reliability of CNV detection algorithms33.
Furthermore, we attempted to enrich the sample for families with a theoretically higher likelihood of a compound heterozygous event. To that end, first, we excluded families with more than one affected proband, given that the likelihood of the same compound heterozygous event in more than one proband in a multiplex family is <0.25, assuming that in a proportion of cases the origin of a functional sequence variant in the remaining allele is de novo. Second, under the assumption that homozygous deletions affecting brain-expressed genes are likely pathogenic, we excluded probands with homozygous deletions. Third, we prioritized those probands with at least one deletion involving one or more genes relevant to the brain (defined hereafter). Finally, genetic variants, even those considered highly pathogenic, are often not completely penetrant34, suggesting that additional genetic variants in the genome may contribute to phenotypic expression. Therefore, rather than categorically excluding certain families based on a likely pathogenic variant, we chose a prioritization strategy. Hence, we prioritized probands with the smallest numbers of de novo CNVs (deletions and duplications) as de novo CNVs are more likely causative, thereby reducing the likelihood of a causative compound heterozygous event. Finally, we prioritized probands with the largest number of inherited CNVs, in particular those involving brain-relevant genes, while attributing a double weight to deletions compared with duplications:
Applying these criteria to the AGP families, we retrieved DNA samples from the participating sites of 254 families.
Targeted genomic enrichment and sequencing
We custom-designed a target sequence footprint, applying 60-mer tiling probes based on the selected genes for this study. Agilent SureSelect (Santa Clara) in solution capture assays were used for the enrichment procedure. The library preparation has been described in detail elsewhere35. Briefly, DNA samples were sheared into 100–120 nucleotide fragments, followed by ligation of double-stranded short adapters and, subsequently, ligation-mediated polymerase chain reaction (PCR) amplification. The pooled library fragments were then hybridized to the Agilent capture assays and underwent post enrichment PCR before sequencing.
We performed sequencing of enriched barcoded samples on a SOLiD 5500XL sequencer (Applied Biosystems) with V3 chemistry according to the manufacturer instructions to produce 50 bp sequencing reads. Reads were mapped onto the human genome (GRCh37), using BWA36 as default settings with the following parameters (-c -l 25 -k 2 -n 10).
Variant calling and quality control
A custom PERL pipeline (https://github.com/UMCUGenetics/SAP42) was developed to parse the BAM files and extract SNP genotypes with the following criteria: at least 10× coverage, sequencing quality Q >20, >15% non-reference alleles at variant sites (this is a cut-off criterion for individual sample positions), and support from >3 independent reads on both strands. A maximum number of five identical reads calling the same allele is set to suppress excessive co-linearity effects. The genetic variants calling was performed for each sample from BAM files and then merged.
The processed VCF file contained 357 individuals from 161 families, with a total of 50,729 SNVs (47 complete trios and 102 proband–parent pairs, as well as 12 singletons without sequence data from their transmitting parents; these 12 singletons were excluded from further analysis). Variants were annotated using SnpEff software, version 4.3 T37. All results of this study are reported in GRCh37/hg19 build. The CNV regions previously reported in this sample20 were reported in NCBI/hg18build. CNV coordinates were re-mapped to GRCh37/hg19 build using a publicly available LiftOver application (https://genome.ucsc.edu/cgi-bin/hgLiftOver).
The gene content of a CNV was defined as all genes located within the CNV region; an additional 500 kb fuzzy border was applied at both the 5′ and 3′ ends of the reported CNV. We extracted all SNVs located in the genes affected by inherited deletions; thus, in this study compound heterozygotes were defined as a second variant occurring in the gene and within the boundaries of the deletion region (Fig. 1, scenario 1). Alternatively, a genic sequence variant can be identified in a gene affected by a deletion, but outside breakpoints of the deletion (Fig. 1, scenario 2). In an attempt to maximize a conservative selection of potentially impactful compound heterozygous events, scenario 2 was not considered as an SNV-deletion event in the current study. Within these regions, we used the biomaRt package38 in R to identify genic regions for our downstream analyses; the output contained ~50.5% intronic sequence, and 16.5% sequence up and downstream from the outer exons, as well as the 3′ and 5′ UTRs. All genotyping results of variants within the deletion region were haploid, i.e., showing as homozygous calls. We excluded variants showing identical (“homozygous”) calls in both proband and deletion-transmitting parent (n = 276) under the assumption that parents were not affected with ASD. In order to identify homozygote reference alleles and missing genotypes, we used FixVcfMissingGenotypes39. We thus excluded variants that were not called (n = 76), based on the depth of coverage from the BAM files. Hence, after merging the VCFs files, we coded both homozygotes reference and genotypes not called as missing. After these quality control steps, we retained 109 SNVs identified in inherited deleted gene sequences.
Statistical analyses
We designed our study to detect an overall difference in rates of compound heterozygous events between probands and transmitting parents among 47 complete trios and 102 proband–parent pairs. Hence, we combined all deleted gene sequence in probands and tallied the number of SNVs in this pooled proband sequence. Similarly, we calculated the rate of variants in the pooled deleted gene sequence of their deletion-transmitting parents. By design, the combined proband sequence is equal in identity and length as the combined transmitting parent sequence (see Fig. 2). Therefore, to test the difference between the number of variants in the proband and the transmitting parent sequences, we have used the chi-square test.
Further, we annotated the identified sequence variants using CADD scores29, a publicly available online tool that integrates multiple variables to calculate an estimation of the predicted deleteriousness of sequence variants in the human genome. The output metric of CADD is a scaled “PHRED” score, which relies on the ranking of the predicted deleteriousness in the context of all ~8.6 billion sequence variants in the human genome29. In the group of individuals in whom SNV-deletion events were identified, we used a burden test40 to compare the cumulative scaled CADD scores between probands and parents. More specifically, all the SNVs’ CADD scores (in inherited CNV deletion regions, Supplementary Table 1) were aggregated for each individual. In other words, we calculated the sum score of CADD scores of the SNVs in the regions of interest for each individual. We then used logistic regression to compare the aggregated CADD scores between probands and parents.
Subsequently, we combined two filters to select for variants that are putatively most deleterious: (1) a CADD-10 score (defined as SNVs at the 10th% of CADD scores) to select only those sequence variants predicted to be most deleterious;29 and (2) variants predicted to change the properties of the encoded protein (in our data: missense variants and or splice-site altering variants)11,41,42. We retained variants that were identified by either one or both of these two filters.
Because of these three analyses conducted (1) the difference between the number of variants in the proband and the transmitting parent sequences; (2) burden test; and (3) analysis of most deleterious SNVs, we considered p values < 0.05/3 (Bonferroni correction for multiple testing) as statistically significant.
The data analyzed for the current study is derived from the AGP20, available through dbGap (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000267.v5.p2).
Results
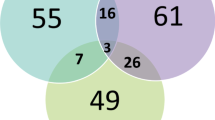
We obtained sequence data from 201 brain-relevant genes in 149 families (see Supplementary Table 2). For each family we restricted our analyses to the genes affected by the deletion transmitted in that family. We observed an average of 3.08 brain-relevant genes affected by a deletion per family. We identified a total of 109 SNVs in these deletions. There were 20 probands (13.4%) with at least one SNV-deletion compared with 16 deletion-transmitting parents (8.1%). There was a significant difference in distribution between probands and parents: 68 variants were identified in the pooled sequence of probands versus 41 variants in the pooled deletion-transmitting parent sequence (X2 = 6.69, p = 0.0097). Table 1 provides an overview of the identified SNVs in inherited deletion regions, along with their annotations. Supplementary Tables 1 and 3, and Supplementary Fig. 1 provide more detailed information, including distribution of variants and boundaries of the deletions involved in the observed SNV-deletion events. Of note, six probands in the subset of 47 complete trios carried a compound heterozygous event, which consisted of an inherited deletion and a de novo SNV (see Supplementary Table 4).
The burden test showed a significantly higher cumulative CADD score conferred by 68 SNVs observed in inherited deletions in 20 probands compared with 41 SNV-deletion events observed in 16 transmitting parents (β = 0.13, p = 1.0 × 10−5). However, the burden test applied to the entire sample, i.e., including the 129 probands and 180 parents without SNV-deletion events, was not significant (β = 0.019, p = 0.25).
Then we examined the SNVs yielded from the union of the two deleteriousness filters (Table 2). Of these 29 putatively most deleterious SNVs, 21 were detected in proband sequences versus 8 in parents (X2 = 5.82, p = 0.016; Supplementary Table 5). Post hoc we reiterated this analysis after omitting rs75355616 as this variant is located in a segmental duplication region overlapping with PRAMEF4, which implies highly homologous sequences elsewhere in the genome43, yielding unaltered results (20 SNVs in probands versus 8 in parents; X2 = 5.14, p = 0.023).
Discussion
This study provides tentative evidence for the role of a specific type of compound heterozygosity in the genetic architecture of ASD. Results indicate that in individuals with ASD, inherited deletions may co-occur more often with a predicted functional SNV affecting the remaining allele at the same locus than in their unaffected parents. Our burden analysis shows that, cumulatively, the burden of predicted deleteriousness inferred by variants on the remaining allele is significantly higher in probands than in their deletion-transmitting parents, providing further evidence for our “compound heterozygosity” hypothesis in ASD.
The pathogenic potential of some CNVs, in particular deletions, may sometimes be contingent on the presence of an additional genetic variant on the remaining allele. Vice versa, the phenotypic impact of the latter may in turn only be revealed when not compensated by a second wild-type allele, such as is the case in the presence of a deletion. A deletion, in such situation, can be said to “unmask the functional effect of a variant”44 which would otherwise have remained without phenotypic consequences. The compound nature implies a mutual rapport: a functional variant can equally be said to “uncover the pathogenicity of a deletion”. In the clinic, putatively pathogenic deletions identified in some patients often turn out to be inherited from seemingly unaffected parents45. This scenario strongly suggests the requirement of additional factors to mediate the pathogenic potential of the CNV. Although not currently applicable to clinical settings, we propose that the compound heterozygosity described in the current study is one of several mechanisms explaining variable penetrance of CNVs with known pathogenicity for ASD34.
Findings reported here are limited by the relatively small sample size. Given this, we restricted the statistical analysis in this work to only test the main hypothesis—that compound heterozygosity of a deletion and a functional sequence variant at the remaining allele occurs more often in patients with ASDs compared with the parents carrying the same deletion. In this study, we focused on deletions assuming a model of loss-of-function. This is a limitation by design, as duplications may also contribute to the etiology of ASD through dosage and gain-of-function. Arguably, compound heterozygous events may also occur under these scenarios. The annotation of SNVs included synonymous variants. In light of the overall small number of variants, we chose to retain this subset of SNVs in our analyses, even though they do not alter protein sequence and therefore have a lower probability of functional impact. In support of our approach, several recent studies suggest that synonymous variants can be pathogenic46. However, our main finding remained significant when comparing the burden of SNVs after excluding the synonymous variants (X2 = 7.67, p = 0.006). In addition, when we restricted the analyses to a subset of 29 variants predicted to be amongst the most deleterious variants in the genome (Supplementary Table 5), we observed a significantly higher burden of these in compound heterozygous events in probands compared with their unaffected parents. However, given our overall low event rate, we were not able to apply both filters (i.e., the intersection of CADD-10 and missense/splice-site altering variants) in a single analysis, which would have been a more stringent approach. The low overall event rate also prevents us from discriminating individual true versus false positive signals within the higher burden observed in probands. Given the limitations described above, we present our results as exploratory, to show the potential contribution of compound heterozygous events involving deletions. Hence, replication of our findings in independent studies is required: whole genome or exome sequencing would be the most appropriate method for such an endeavor47 within a sample with reliable matched CNV calls.
In conclusion, our results provide initial evidence for a role of compound heterozygosity in ASD. We propose that the compound heterozygosity described in the current study is one of several mechanisms explaining variable penetrance of CNVs, in particular deletions, with known pathogenicity for ASD. This mechanism can be taken into account in studies aiming to identify genetic variants contributing to ASD. Compound heterozygosity may be one factor that explains the frequently observed inconsistent phenotypic expression amongst carriers of the same putatively pathogenic deletion.
Data availability
The data analyzed for the current study are derived from the Autism Genome Project, available through dbGap (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs000267.v5.p2). The data generated during the current study are not publicly available due to individual privacy concerns but are available from the corresponding author on reasonable request.
References
Lai, M. C., Lombardo, M. V. & Baron-Cohen, S. Autism. Lancet 383, 896–910 (2014).
Lyall, K. et al. The changing epidemiology of autism spectrum disorders. Annu. Rev. Publ. Health 38, 81–102 (2017).
Vorstman, J. A. S. et al. Autism genetics: opportunities and challenges for clinical translation. Nat. Rev. Genet. 18, 362–376 (2017).
de la Torre-Ubieta, L., Won, H., Stein, J. L. & Geschwind, D. H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361 (2016).
D’Gama, A. M. et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917 (2015).
Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21 (2017).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Weiner, D. J. et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017).
Ronemus, M., Iossifov, I., Levy, D. & Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 15, 133–141 (2014).
Buxbaum, J. D. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues Clin. Neurosci. 11, 35–43 (2009).
Sanders, S. J. et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015).
Toro, R. et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 26, 363–372 (2010).
Vorstman, J. A. et al. A double hit implicates DIAPH3 as an autism risk gene. Mol. Psychiatry 16, 442–451 (2011).
Siu, W. K. et al. Unmasking a novel disease gene NEO1 associated with autism spectrum disorders by a hemizygous deletion on chromosome 15 and a functional polymorphism. Behav. Brain Res. 30, 135–142 (2015).
Bacchelli, E. et al. A CTNNA3 compound heterozygous deletion implicates a role for alphaT-catenin in susceptibility to autism spectrum disorder. J. Neurodev. Disord. 6, 17 (2014).
Knight, H. M. et al. A cytogenetic abnormality and rare coding variants identify ABCA13 as a candidate gene in schizophrenia, bipolar disorder, and depression. Am. J. Hum. Genet. 85, 833–846 (2009).
Vorstman, J. A. S., Olde Loohuis, L. M., Investigators, G., Kahn, R. S. & Ophoff, R. A. Double hits in schizophrenia. Hum. Mol. Genet. 15, 2755–2761 (2018).
Lim, E. T. et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 77, 235–242 (2013).
Doan, R. N. et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 51, 1092–1098 (2019).
Szatmari, P. et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328 (2007).
Hadley, D. et al. The impact of the metabotropic glutamate receptor and other gene family interaction networks on autism. Nat. Commun. 5, 4074 (2014).
Anney, R. et al. A genomewide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082 (2010).
Vieland, V. J. et al. Novel method for combined linkage and genome-wide association analysis finds evidence of distinct genetic architecture for two subtypes of autism. J. Neurodev. Disord. 3, 113–123 (2011).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694 (2014).
Wheeler, D. L. et al. Database resources of the national center for biotechnology. Nucleic Acids Res. 31, 28–33 (2003).
Fehrmann, R. S. et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 47, 115–125 (2015).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29 (2000).
Nijman, I. J. et al. Mutation discovery by targeted genomic enrichment of multiplexed barcoded samples. Nat. Methods 7, 913–915 (2010).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Colella, S. et al. QuantiSNP: an objective Bayes Hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 35, 2013–2025 (2007).
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674 (2007).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372 (2010).
Trost, B. et al. A Comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am. J. Hum. Genet. 102, 142–155 (2018).
Vorstman, J. A. & Ophoff, R. A. Genetic causes of developmental disorders. Curr. Opin. Neurol. 26, 128–136 (2013).
Harakalova, M. et al. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat. Protoc. 6, 1870–1886 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Lindenbaum, P. JVarkit: Java-based Utilities for Bioinformatics (Figshare, 2015).
Lee, S., Abecasis, G. R., Boehnke, M. & Lin, X. Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23 (2014).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–U136 (2014).
Yuen, R. K. C. et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 1, 1–10 (2016).
Bailey, J. A. et al. Recent segmental duplications in the human genome. Science 297, 1003–1007 (2002).
Hochstenbach, R. et al. Discovery of variants unmasked by hemizygous deletions. Eur. J. Hum. Genet. 20, 748–753 (2012).
Klopocki, E. et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 80, 232–240 (2007).
Hunt, R. C., Simhadri, V. L., Iandoli, M., Sauna, Z. E. & Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. 30, 308–321 (2014).
Yuen, R. K. et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 4, 602 (2017).
Moitra, K., et al. ABCC6 and pseudoxanthoma elasticum: the face of a rare disease from genetics to advocacy. Int. J. Mol. Sci. 18, (2017).
Huang, J., Snook, A. E., Uitto, J. & Li, Q. Adenovirus-mediated ABCC6 gene therapy for heritable ectopic mineralization disorders. J. Invest. Dermatol. 139, 1254–1263 (2019).
Chen, L. et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl Acad. Sci. USA 104, 20990–20995 (2007).
Priori, S. G. et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 15, 1389–1406 (2013).
Kury, S. et al. De Novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am. J. Hum. Genet. 101, 768–788 (2017).
Lehrmann, E. et al. Transcriptional changes common to human cocaine, cannabis and phencyclidine abuse. PLoS ONE 1, e114 (2006).
Perlman, E. J., Valentine, M. B., Griffin, C. A. & Look, A. T. Deletion of 1p36 in childhood endodermal sinus tumors by two-color fluorescence in situ hybridization: a pediatric oncology group study. Genes Chromosomes Cancer 16, 15–20 (1996).
Bottega, R. et al. Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica 103, 417–426 (2018).
Velmurugan, K. R. et al. repair pathway via defective FANCD2 gene engenders multifarious exomic and transcriptomic effects in Fanconi anemia. Mol. Genet. Genom. Med. 6, 1199–1208 (2018).
Pannu, H. et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 16, 2453–2462 (2007).
Khau Van Kien, P. et al. Familial thoracic aortic aneurysm/dissection with patent ductus arteriosus: genetic arguments for a particular pathophysiological entity. Eur. J. Hum. Genet. 12, 173–180 (2004).
Zhu, L. et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 38, 343–349 (2006).
Alkuraya, F. S. et al. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am. J. Hum. Genet. 88, 536–547 (2011).
Desikan, R. S. & Barkovich, A. J. Malformations of cortical development. Ann. Neurol. 80, 797–810 (2016).
Kridin, K. & Bergman, R. The usefulness of indirect immunofluorescence in pemphigus and the natural history of patients with initial false-positive results: a retrospective cohort study. Front. Med. 5, 266 (2018).
Witte, M., Zillikens, D. & Schmidt, E. Diagnosis of autoimmune blistering diseases. Front. Med. 5, 296 (2018).
Acknowledgements
The authors wish to express gratitude toward all the individuals and their families for their commitment to scientific research. This study has been funded by the Dutch Brain Foundation (Hersenstichting Nederland) to JV.
Author information
Authors and Affiliations
Contributions
F.C. performed all data preparation steps related to the selection of families within the AGP dataset. B.D.L. performed statistical analyses and wrote the first draft. J.J.L. and J.V. supervised the project and wrote the final version of the manuscript. I.J.N., J.M., and W.B. performed the wet lab analyses. K.v.E. provided bioinformatics support. All other authors were involved in recruitment and critically revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Ethics approval was obtained by multiple medical ethics committees (University Medical Center Utrecht, Henan University, Newcastle University, German Cancer Research Center, JW Goethe University Frankfurt, University of Bologna, University of Utah School of Medicine, Instituto Nacional de Saúde Doutor Ricardo Jorge, Centro Hospitalar de Coimbra, University of Oxford, Trinity College Dublin, University College Dublin, Cardiff University, Université de Paris, GGNet Mental Health, The Hospital for Sick Children, and University of Toronto).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, B.D., Colas, F., Nijman, I.J. et al. The role of rare compound heterozygous events in autism spectrum disorder. Transl Psychiatry 10, 204 (2020). https://doi.org/10.1038/s41398-020-00866-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-00866-7
- Springer Nature Limited