
Abstract
Inflammasomes are large protein complexes that play a major role in sensing inflammatory signals and triggering the innate immune response. Each inflammasome complex has three major components: an upstream sensor molecule that is connected to a downstream effector protein such as caspase-1 through the adapter protein ASC. Inflammasome formation typically occurs in response to infectious agents or cellular damage. The active inflammasome then triggers caspase-1 activation, followed by the secretion of pro-inflammatory cytokines and pyroptotic cell death. Aberrant inflammasome activation and activity contribute to the development of diabetes, cancer, and several cardiovascular and neurodegenerative disorders. As a result, recent research has increasingly focused on investigating the mechanisms that regulate inflammasome assembly and activation, as well as the potential of targeting inflammasomes to treat various diseases. Multiple clinical trials are currently underway to evaluate the therapeutic potential of several distinct inflammasome-targeting therapies. Therefore, understanding how different inflammasomes contribute to disease pathology may have significant implications for developing novel therapeutic strategies. In this article, we provide a summary of the biological and pathological roles of inflammasomes in health and disease. We also highlight key evidence that suggests targeting inflammasomes could be a novel strategy for developing new disease-modifying therapies that may be effective in several conditions.
Similar content being viewed by others
Introduction
The innate immune response enables humans to defend against new pathogens, environmental irritants, and tissue damage in part by triggering inflammation when immune cells recognize molecules that are commonly found in many pathogens or damaged cells but are otherwise absent in the body.1 This inflammatory response is mediated by large protein complexes called inflammasomes that have been increasingly shown to play a vital role in the immune system. The history of inflammasome-related research dates back to 1985, when Hanazawa and colleagues first showed that exposure to Lipopolysaccharide (LPS) induces interleukin-1 (IL-1) production in murine peritoneal macrophages (Fig. 1).2 Ultimately, this study was the first to suggest the existence of specific intracellular molecular platforms that could trigger the inflammatory response by inducing proinflammatory caspase activation and pro-IL-1β or pro-IL-18 processing.3 In the 1990s, caspase-1-mediated IL-1β processing and secretion was discovered and characterized, which provided the first tangible evidence that a molecular complex was responsible for this process.4 However, it was not until 2002 that the term “inflammasome” was coined to describe this multi-protein complex.3 The first inflammasome to be identified was NACHT, LRR, and PYD domains-containing protein 1 (NLRP1) in 2002, and NLRP3 quickly followed this in 2004.3,5 Henceforward, many inflammasomes have been identified, each with unique immune functions and roles.

Milestone events in inflammasome-related research and their applications. Blue box: discoveries of key components of different inflammasomes, purple box: representative clinical applications of inflammasome-related modulators, green box: the association between inflammasomes and various human diseases, pink box: the origin of the term “inflammasome”. The figure was created with the assistance of FIGDRAW
Over the past few decades, there has been a growing number of different types of inflammasomes. What has ultimately allowed for distinct inflammasomes to be characterized is that each type contains unique scaffolding proteins. Most of the scaffolding proteins belong to the nucleotide-binding domain, leucine-rich repeat-containing proteins (NLRs) family, or the absent in melanoma 2-like receptors (ALRs), also known as PYRIN-HIN-200 (PYHIN) proteins family (Fig. 2a).6,7,8,9,10 NLRs play an essential role in inflammation and belong to the pattern recognition receptors (PRRs) family that sense stress signals to generate immune responses to prevent further damage.11 Alternatively, the HIN-200 family’s function in the mammalian innate immune system is to detect cytoplasmic stimuli in order to regulate the immune response.10

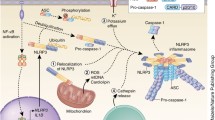
Representative structures of the inflammasome sensor proteins and the inflammasomes. a Representative structure of the inflammasome sensor proteins, including NLRPs (NLRP1 and NLRP3), NLRCs (NLRC4), and ALRs (AIM2). b Representative structure of the inflammasomes, including the NLRP1 inflammasome, the NLRP3 inflammasome, the NLRC4 inflammasome, and the AIM2 inflammasome. NLRs, nucleotide-binding domain, leucine-rich repeat-containing proteins; NLRPs, NLRs containing an N-terminal pyrin domain (PYD); NLRCs, NLRs containing a caspase activation and recruitment domain (CARD); ALRs, absent in melanoma 2 (AIM2)-like receptor; NAIP, NLR apoptosis inhibitory protein. The figure was created by FIGDRAW
NLRs consist of three main components: an N-terminal effector domain, a central nucleotide-binding (NACHT) domain, and a C-terminal leucine-rich repeat (LRR) domain.12 Differences in the N-terminal effector domain further divide them into two subgroups: NLRs containing a pyrin domain (PYD) are members of the NLRP subgroup, and NLRs with a caspase activation and recruitment domain (CARD) are members of the NLRC subgroup. Currently, known members of the NLR family that mediate the assembly of inflammasomes include NLRP-1,3,6,7,9 and NLRC4.3,13,14,15,16,17 Upon activation, NLRs typically form an inflammasome complex with the adaptor protein ASC (apoptosis-associated speck-like protein containing CARD) connected to a downstream effector or signaling protein, such as caspase-1 or 5 (Fig. 2b).18
NLRs act as the sensor components of inflammasomes that recognize foreign pathogen-associated molecular patterns (PAMPs) or endogenous damage-associated molecular patterns (DAMPs). Once activated, NLRs homo-oligomerize via NACHT domains, enabling them to bind to the ASC adapter protein.19 The ASC adaptor protein consists of two protein-protein interaction domains: an N-terminal PYD and a C-terminal CARD.20 Upon recruitment to the oligomerized NLRs, ASC releases its CARD domain from the auto-inhibited conformation. The assembled ASC subsequently recruits pro-caspase via CARD-CARD interactions, thereby inducing heterodimerization, auto-cleavage, and caspase-1 activation.21 Active caspase-1 cleaves the intracellular pro-inflammatory cytokines, such as IL-1β and IL-18, resulting in their maturation and activation. Once activated, IL-1β and IL-18 are then secreted out of the cell where they stimulate inflammation in other cells nearby.22 Additionally, active caspase-1 also cleaves gasdermin D (GSDMD), releasing the N-terminal fragment of GSDMD, which induces pyroptosis and promotes further IL-1β secretion.23 Notably, PYHIN proteins contain a DNA-binding HIN200 domain and one or several PYD domains. This conformation allows for the formation of macromolecular complexes with other PYD-containing proteins that ultimately play a vital role in recognizing the cytosolic DNA.9,24 Among the PYHIN proteins, absent in melanoma 2 (AIM2) and IFI16 are the members known to be capable of caspase-1 activation.25,26 The C-terminal HIN200 domain of AIM2, also known as the oligonucleotide/oligosaccharide-binding domain of AIM2, acts as the sensor that recognizes DNA. Alternatively, the PYD domain of AIM2 can interact with the adapter protein ASC to induce both NF-κB and caspase-1 activation.9
Since their initial discovery, a growing body of research has implicated that aberrant inflammasome activity contributes to the development of multiple disorders, including metabolic, neurodegenerative, and inflammatory conditions. In recent years, there has been significant progress in identifying the mechanisms that activate inflammasomes and their role in different diseases. These discoveries have led to an increasing interest in developing new inflammasome-targeting therapies, which are presently under evaluation in numerous clinical trials. Herein, we introduce the structural basis of different inflammasomes and the mechanisms that drive inflammasome activation. We also summarize our current understanding of the various roles each inflammasome plays in the development of different diseases.
Structure of inflammasome sensors
In the following section, we will present an overview of the well-described inflammasome structures, including the NLRP1, NLRP3, NLRC4, and AIM2 molecules.3,9,14,27 We will also report on the structure of molecules known to form inflammasome complexes under specific conditions, such as IFI16 (interferon-inducible protein 16), NLRC5, NLRP6, NLRP7, and NLRP9.16,28,29,30,31 Collectively, these studies provide insights into the molecular mechanisms of inflammasome formation and offer a basis on which to better understand the pathological consequences of various diseases on inflammasome function.
Structure of NLRP1 inflammasomes
Multiple alternatively spliced transcript variants encoding up to 5 distinct isoforms have been found for the Human NLRP1 gene, with the longest isoform (isoform 1) encoded by NLRP1 transcript variant 1 (Gene ID: 22861). Isoform 1 contains several conserved domains, including the PYD (Pyrin death domain), NACHT domain, NOD2_WH (NOD2 winged helix) domain, NLRC4_HD2 (NLRC4 helical domain), LRR_RI (LRRs, ribonuclease inhibitor RI-like subfamily) domain, LRR_AMN1 (LRR [structural motif]) domain, FIIND (function to find) domain, and CARD. Compared to isoform 1, the encoded isoform 2 lacks an internal segment in the FIIND domain, isoform 3 lacks an internal segment in the LRR_RI domain, isoform 4 lacks two internal segments in the FIIND and LRR_RI domain, and isoform 5 has a shorter and distinct C-terminus. However, the functional variance of distinct isoforms has yet to be determined.
The PYD and CARD domains belong to the death domain (DD) superfamily. NMR spectroscopy analysis has shown that the structure of NLRP1-PYD differs from the DD superfamily because a flexibly disordered loop replaces its third α-helix, and this difference may influence how NLRP1 functions in protein-protein interactions.32 Furthermore, NLRP1 activity depends on ASC, which interacts with its C-terminal CARD domain, and on autolytic cleavage at Ser1213 within the FIIND, both of which are essential for NLRP1 inflammasome activity.33 NLRP1-CARD contains prominently charged surface patches, which can interact with the procaspase-1-CARD via a complementary charge surface.18,34 NLRP1-CARD forms central helical filaments, which are sufficient to induce ASC speck formation.35,36 Structural analysis shows that NLRP1-CARD interacts with ASC-CARD to form the filament complex via an interaction between a conserved set of interaction surfaces (Type I, II, and III).37 Moreover, NLRP1-FIIND is a type of ZU5-UPA domain. It contains a conserved SF/S motif and conserved glutamic acid (Glu) and histidine (His) residues adjacent to the cleavage site, which execute post-translational autocleavage and regulate the auto-processing efficiency, respectively.38 NLRP1-FIIND functionally reduces the NLRP1-CARD oligomerization and filament formation threshold.36 Alternatively, NLRP1-ZU5 inhibits NLRP1 activation by downregulating NLRP1-UPA-CARD filament formation.38 Additionally, mutation of His residues caused the loss of NLRP1 autocleavage.38,39 Isoform 2 of NLRP1 lacks exon 14 and insights from research on a disease-associated single nucleotide polymorphism (SNP) near the highly conserved distal residue His1186 suggest this region is important for autolytic cleavage and NLRP1 activation.33,38 The SNP rs11651270 (M1184V) is a common NLRP1 variant, significantly associated with asthma, that can keep NLRP1-FIIND monomeric and subsequently promote the full-length NLRP1 assembly, but is independent of autoproteolysis.40,41
Structure of NLRP3 inflammasomes
Human NLRP3 has several alternatively spliced transcript variants (Gene ID: 114548). The most commonly referred to variant for full-length NLRP3 is isoform e, which is encoded by NLRP3 transcript variant 3 or 6. Isoform e has several conserved domains, including the PYD, FISNA (Fish-specific NATCH associated domain), NATCH domain, NOD2_WH domain, and LRR_RI domains. Compared to isoform e, isoform a contains a less conserved translational start codon and possesses two more amino acids at N-terminus; isoform b is encoded by NLRP3 transcript variant 2, isoform c is encoded by NLRP3 transcript variant 4, and isoform d is encoded by NLRP3 transcript variant 5. These isoforms have shorter but different internal segments in the LRR_RI domain than isoform e, as variant 2 lacks two in-frame exons and variant 4/5 lacks one in-frame exon.
NLRP3 integrates different inflammatory stimuli and relies on distinct structural features within the N-terminus, NATCH, and LRR domains.42 ATP shows high binding affinity with the NLRP3-NATCH domain that mediates NLRP3 self-oligomerization, and the Walker A, B, and extended Walker B motifs are the proposed key ATP binding regions in NACHT.43,44 NLRP3 mutations are predicted to disrupt the structure around these ATP binding regions, changing the dynamics of the hydrogen-bond and charge interactions and enhancing their ATP binding affinity.45 Notably, NLRP3 assembles via PYD-PYD interactions between NLRP3 and ASC. Structural and sequence analyses have indicated that NLRP3-PYD interacts with ASC-PYD using equivalent binding interfaces composed of hydrophobic residues and charged conserved surface residues.46,47,48 NLRP3-PYD, ASC- PYD, and ASC-CARD interactions form filaments, activate NLRP3 nucleate ASC-PYD filaments, and subsequently cluster the ASC-CARD, which in turn nucleates caspase-1-CARD filaments leading to NLRP3 inflammasome activation.21 Moreover, there is a disulfide bond between conserved Cys8 and Cys108 that appears to be important for NLRP3 activation by sterile insults (i.e., ischemia), but not infections.42,46
In a resting state, an NLRP3 PYD-PYD interaction exists that forms cylindrical filaments composed of 3 major asymmetric interfaces. The most dominant interface consists of highly polar residues that mediate homomeric interactions.49,50 The PYD-PYD oligomerization is facilitated by the flexible linker sequence and the NLRP3-FISNA domain, and the NLRP3 conformational change is activated by K+ efflux.51 Additionally, NLRP3 forms a decamer (or dodecamer) ring cage that is held together by LRR homomeric interactions. Inside the cage, PYD forms a dimer with the NACHT domain located at the top of the ring. The acidic loop, which extends from a transition segment of LRR, mediates molecular interactions between opposing concave sites of LRRs.50,52,53 The ring cage structure is an inactive form of NLRP3 which localizes to membranes and is essential for NLRP3 activation and inhibition. The NLRP3 isoform lacking in-frame exons in the LRR domain cannot be activated under certain conditions.54 One possibility is that the alternative splicing of the LRR domain could regulate the stochastic activity of NLRs. Nevertheless, variants that arise in NLRP3 have been implicated in several diseases.55,56,57,58,59 Therefore, further studies focused on the structure of NLRP3 are warranted to help guide further efforts in disease diagnosis and treatment.
Structure of NLRC4 inflammasomes
The human NLRC4 gene has 2 isoforms: a and b (Gene ID: 58484). NLRC4 transcript variants 1, 2, and 3 encode the same protein, with the longest transcript being isoform a. Isoform a contains conserved domains, including CARD, NATCH domain, LRR_AMN1 domain, and LRR_RI domains. Compared to isoform a, isoform b is encoded by NLRC4 transcript variant 4 and it lacks the NATCH domain and an AMN1 motif in the LRR domain; thus, isoform b has a shorter LRR domain than isoform a.
The NATCH domain of NLRC4 contains a central nucleotide-binding domain (NBD) and a winged helix domain (WHD). The ADP-mediated interaction between the NBD and WHD stabilizes NLRC4’s closed conformation and the NLRC4 helical domain inhibits conserved and functional α-helixes of the NBD. The NLRC4 protein is kept in a monomeric state due to the C-terminal LRR domain blocking the NBD domain.60 ICE-protease Activating Factor (IPAF), also called the NLRC4, was discovered along with the finding that caspase-1 cannot be activated by full-length NLRC4, but instead by the truncated protein lacking the C-terminal LRRs.61 Bacterial ligands recognized by the NLR apoptosis inhibitory proteins (NAIPs) are essential for NAIP-NLRC4 inflammasome formation. Evidence suggests that NAIPs are the upstream receptors that recognize bacterial ligands, while NLRC4 functions as the downstream adaptor that congregates NAIPs for inflammasome formation.62,63,64 The NBD-associated α-helical domains of NAIP, but not the LRR domain, are believed to mediate this ligand specificity.65 Evidence also shows that the BIR1, pre-BIR, and HD1 domains in NAIP2 and NAIP5 are implicated as enabling the specific recognition of their respective ligands.62,63 Moreover, the bacterial protein PrgJ directly binds to NAIP2, forming a single ligand-bound NAIP2 molecule and sequentially triggering the formation of the NAIP2-NLRC4 inflammasome complex.66,67,68 Specifically, in response to stimuli or pathogens, NLRC4 undergoes structural remodeling and forms a wheel-like structure with a single catalytic surface. Once active, NLRC4 uses this surface to catalyze NAIP2-NLRC4 inflammasome activation via a self-propagating mechanism. This self-activation happens because the NAIP2 proteins contain a catalytic surface that matches the complementary NLRC4 oligomerization surface (the receptor surface), and together, these surfaces form the wheel-like double-ring structure of the PrgJ-NAIP2-NLRC4 complex.68
NAIP5-NLRC4 complexes are also large constructions containing 11 or 12 subunits that have a crucial function in the immune response to the bacterial protein flagellin. The assembly of NAIP5-NLRC4 complexes occurs in response to flagellin binding to NAIP5, which induces the recruitment of NLRC4 and subsequent formation of a disk-shaped hetero-oligomeric complex.69 Unliganded mouse NAIP5 recruits inactive NLRC4 via a fully exposed nucleating surface.64 Upon flagellin binding, the WHD of NAIP5 undergoes a steric rotation that activates NLRC4, consequently enabling NAIP5 to integrate with the NLRC4 protein, and stabilizing the NAIP5-NLRC4 complex.64 Flagellin-induced NAIP5-NLRC4 multimers subsequently form left- and right-handed helixes with a pitch of ∼6.5 nm and a diameter of ∼28 nm.70 Furthermore, NLRC4-CARD can nucleate caspase-1 assembly and activate caspase-1.71 The NLRC4-CARD filament is a left-handed helix consisting of 3.6 subunits per helical turn, similar to the ASC-CARD and CASP1-CARD filament.71,72 The upstream NLRC4-CARD and downstream CASP1-CARD interact based on the consistent helical assemblies.73 Mutations that have been reported influencing the NATCH or LRR domains of NLRC4 reinforce the likely pathogenicity in autoinflammatory disorders.74,75,76,77,78,79,80 Mechanistically, the p.W655C NLRC4 mutation activates the NLRC4 inflammasome via engaging 2 LRR interfaces.78
Structure of AIM2 inflammasomes
The human AIM2 gene is expressed as two isoforms (Gene ID: 9447). The longer isoform 1 is encoded by the AIM2 variant 1, which contains two conserved domains, including the PYD and DNA-binding HIN (HIN-200/IF120x domain) domain. Alternatively, isoform 2 lacks a conserved PYD domain.
AIM2 is a member of the PYHIN family, which is characterized by an N-terminal pyrin domain that allows for the formation of multimolecular complexes via PYD-PYD interactions with other pyrin-containing proteins. Researchers found that the AIM2 PYD self-oligomerizes, and mutations on these residues could disrupt AIM2 PYD self-association (e.g., the F27G mutation).81 Structural analysis reveals that the AIM2 PYD domain is similar to a B-DNA cylinder, which could bind to the AIM2 HIN domain at the concave basic face, forming an autoinhibited protein complex.82 AIM2-PYD has a death domain fold with a distinct charge distribution and hydrophobic patches; its α2 helix contains a highly conserved lysine residue that stabilizes the short α3 helix, and the AIM2 PYD can bind the AIM2 HIN domain or the ASC PYD through the overlapping surface near its α2 helix.34,81,83 Moreover, different AIM2 PYD domains yield distinct conformations around the α3 helix region, as the region is highly flexible and different environments make the pre-existing conformational substrates vary.84 Notably, this conformational switching is believed to be important for the autoinhibition of AIM2. Researchers found that the AIM2 HIN domain could recognize double-stranded DNA (dsDNA), such as bacteria and viruses.82 When DNA binds, the AIM2 PYD domain separates from the HIN domain, initiating downstream signaling.34,82 Additional evidence demonstrated that AIM2- and ASC-PYD filaments assemble bidirectionally, whereas recognition between AIM2 and the ASC protein occurs in a head-to-tail manner and requires at least one to be oligomeric.83 These works indicate that the interactions between PYD-HIN and PYD-PYD are essential for AIM2’s autoinhibition and inflammasome formation. Similar to NLRP3, the helical symmetry of the AIM2-PYD filament occurs via the filaments assembled between AIM2-PYD and downstream ASC-PYD, and activated AIM2 could also nucleate the PYD filaments of ASC and induce subsequent signaling cascades.21,85 It should be mentioned that some research suggests that AIM2-PYD does not act as an autoinhibitory factor; instead, it couples ligand binding with oligomerization to create a structural template.85 AIM2-PYD drives both dsDNA binding and filament formation, and the dsDNA-binding domain of AIM2 can both oligomerize and assist filament formation forming AIM2/DNA filaments.86 Additionally, the results of some cryo-EM structural analyses suggest the AIM2-PYD and ASC-PYD filaments may be distinct from one another.86 However, research on the structure of AIM2-PYD in an inactive state has been considered controversial and requires further clarification.
Structure of other inflammasomes
Human IFI16 has 4 isoforms, with isoform 1 being encoded by the IFI16 variant 1 (Gene ID: 3428). Isoform 1 contains three conserved domains, including a Pyrin domain, a HIN domain (HIN-200/IF120x domain), and a Neogenin C-terminus. Compared to the IFI16 isoform 1, isoform 2, isoform 3, and isoform 4 lack the Neogenin C-terminus, but have a provisional PTZ00449 domain. IFI16 also has two HIN domains (HINa and HINb) that are comprised of a few tightly packed oligosaccharide/nucleotide-binding (OB) fold subdomains. HINa binds to the DNA backbone via loop L45 of the OB2 fold, and HINb both induces interferon (IFN)-β and binds DNA.87,88,89,90,91,92 IFI16 recognizes DNA non-specifically through an electrostatic attraction between the sugar-phosphate backbone of dsDNA and the positively charged residues of its HIN domain.82 The isolated IFI16-HIN200 domains do not oligomerize, and the non-DNA-binding PYD drives filament assembly.85 Moreover, IFI16 contains a highly conserved multipartite nuclear localization signal (NLS). IFI16 has been shown to detect pathogenic nuclear DNA primarily inside the nucleus, supporting the need for a functional NLS.93
Human NLRC5 encodes several different isoforms (Gene ID: 84166). NLRC5 isoform 1 has the following conserved domains, including the atypical caspase recruitment domain, the NACHT domain, NLRC4_HD2, and the longest LRR domains, which include the LRR_RI and the two structural motifs LRR_RI and LRR_AMN.94 Compared to isoform 1, isoforms 2 to 7 and 10 have six conserved domains consistent with isoform 1, isoforms 8 and 9 have another protein phosphatase 1 regulatory subunit 42 (PPP1R42) domain, isoforms 11 to 20 lack an LRR_AMN1 domain, and isoform 21 lacks an LRR_AMN1 domain but contains a PP1R42 domain. NLRC5 is unique because it poses an unusually high number of LRRs and an atypical CARD domain. Homology modeling suggests that NLRC5 could form a homo-heptamer upon activation.94 Interestingly, human NLRC5 has intrinsic transcriptional activity within its N-terminal effector domain.95 Additionally, the NLRC5 N-terminal effector domain can interact with the downstream tandem CARD of the protein retinoic acid-inducible gene I (RIG-I).96 Structural analysis has also shown that NLRC5 belongs to the CARD subfamily and can be classified as an atypical CARD.97
Human NLRP6 is expressed as two isoforms, with isoform 1 being longer and containing four conserved domains, including a Pyrin DD found in ASC, a NATCH domain, and two LRR_RI domains (Gene ID: 171389). Alternatively, isoform 2 lacks an LRR_RI domain but has a NOD2_WH, an NLRC4_HD2, and a PPP1R42 domain. Upon stimulation by LPS, NLRP6 binds LPS directly, subsequently dimerizes and causes global conformational changes. Following a secondary stimulation by ATP, the NLRP6 homodimer forms a linear molecular platform that recruits ASC to create a higher-level molecular structure.98 The PYD of NLRP6 alone is capable of self-assembling into filamentous structures that can recruit the ASC adaptor via PYD-PYD interactions.99 With concentration-dependent assembly, full-length NLRP6 forms filaments containing the NBD and LRR domains that surround a PYD core.99
The NLRP7 gene is expressed as three isoforms, with isoform 3 being the longest and consisting of an N-terminal PYD, followed by a central NACHT domain and a C-terminal LRR domain as with all the NLRs (Gene ID: 199713). Additionally, there is a GTPase SAR1 family (Gem1) subdomain in the NACHT domain of isoform 3. NLRP7 isoforms 1 and 2 are shorter than isoform 3. Interestingly, the PYD domain of NLRP7 shows positive deviation from random coil chemical shift values, which indicates a highly α-helical structure.100 The NMR spectroscopic analysis demonstrates that NLRP7-PYD exhibits a six-α-helix bundle DD fold, which is different from other PYDs in that a hydrophobic cluster stabilizes helix α3 and loop α2-α3 in the NLRP7-PYD. Moreover, the electrostatic surfaces are different in NLRP7 and NLRP1 PYDs.101 Upon activation, the NACHT-associated domain and a small part of the LRR of one NLRP7 emerge from the protective LRR domain and interact with the formed oligomer of the NACHT domain from another NLRP7 molecule.102 Some missense mutations in NLRP7, such as L398R and R693W, decreased its oligomerization potential.102 Additionally, the NBD of NLRP7 has been shown to function as an ATP-binding domain with ATPase activity. NLRP7 inflammasome formation and activity require an intact nucleotide-binding Walker A motif in order for the NBD to effectively bind and hydrolyze nucleotides.103
Human NLRP9 consists of an N-terminal PYD and a central NACHT domain, directly followed by a C-terminal LRR domain (Gene ID: 338321).31 Structural analysis has shown that human NLRP9-PYD has an N-terminal loop that faces toward the helical bundle’s interior, and suggests the N-terminal loop of NLRP9-PYD might regulate the inflammasome’s assembly.104 In contrast, another study reported that the NLRP9-PYD is monomeric and unable to nucleate ASC specks or self-polymerize, suggesting NLRP9-PYD adopts a conformation that is compatible with filament formation.31 These findings indicate that the formation of the NLRP9 inflammasome may differ greatly from that of other inflammasomes.
Activation of the inflammasomes
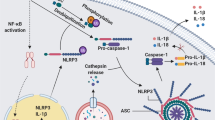
Upon stimulation by microbial ligands or other receptors, certain NLRs or PYHIN family members oligomerize and recruit additional components to build larger intracellular multi-protein complexes, also known as inflammasomes (Fig. 3). Although NLRP1 and NLRC4 can recruit caspase-1 directly through CARD–CARD interactions, most inflammasome sensors promote assembly by recruiting ASC via homotypic PYD‐PYD interactions (Fig. 2b).20,21 After recruitment, proximity-induced autoproteolytic cleavage of caspase-1 releases its catalytic subunits to form mature caspase1. Once active, caspase-1 then processes pro-IL‐1β and IL‐18 to induce the secretion of IL‐1β and IL‐18 and cleaves GSDMD to trigger pyroptosis.22,23

Representative pathways of the inflammasome activation. a Muramyl dipeptide, anthrax lethal toxin, Toxoplasma Godii, ultraviolet B irradiation, and double-stranded viral RNA can induce the cleavage of NLRP1, NLRP1 oligomerizes and leads to caspase-1 recruitment to form NLRP1 inflammasome. b Lipopolysaccharide, extracellular ATP, RNA virus, single-stranded viral RNA can activate the NLRP3, induce the ASC recruitment and following caspase-1 recruitment, and consequently form the NLRP3 inflammasome. c NAIP recognizes flagellin and interacts with NLRC4 to induce the ASC and caspase-1 recruitment and subsequent NLRC4 inflammasome formation. d Double-stranded DNA of parasite, bacterium, and DNA virus can be sensed by AIM2, then AIM2 oligomerized via its HIN domain, oligomerized AIM2 recruits ASC and caspase-1 respectively, and forms AIM2 inflammasome subsequently. e Inflammasomes cleave pro-caspase-1 to produce mature caspase-1 (also known as cleaved caspase-1 P10 and P20), cleaved caspase-1 cleaves GSDMD, pro-interleukin-1β, and pro-interleukin-18, cleaved GSDMD forms pyroptotic pore to execute pyroptosis, and interleukin-1β as well as interleukin-18 are released to extracellular space to regulate inflammation. The figure was created with the assistance of FIGDRAW
NLRP3 inflammasome activation
To date, our most complete description of inflammasome activation is through the NLRP3 inflammasome. There are two main pathways through which NLRP3 inflammasome activation occurs, including the canonical and noncanonical pathways. Several emerging studies have focused on the molecular mechanisms that drive NLRP3 inflammasome activation and how these circumstances vary depending on the type of host cell and stimulus involved.
Canonical NLRP3 inflammasome activation pathway
The canonical pathway of the NLRP3 inflammasome activation begins with the induction of NLRP3, caspase-1, and pro-IL-1β expression, subsequently leading to the complex assembly comprising NLRP3, ASC, and pro-caspase-1. It includes two steps, the priming and activation steps. Stimuli, including PAMPs and DAMPs, drive both the priming and activation steps of NLRP3 inflammasome activation. Throughout this process, the inflammasome functions as a platform for attracting the pro-inflammatory cytokines, such as IL-1β and IL-18, while also facilitating their processing and maturation. Additionally, the inflammasome triggers the GSDMD cleavage, leading to the release of its N-terminal fragments and the formation of pores that facilitate pyroptosis.
During the priming phase, PAMPs and DAMPs engage with PRRs, such as NLRs and Toll-like receptors (TLRs), and this interaction facilitates the transcription and expression of NLRP3, caspase-1, and pro-IL-1β. TLRs are membrane-bound receptors that recognize PAMPs. In turn, this recognition initiates the inflammatory cascade by promoting the activation of nuclear factor-κB (NF-κB) and stimulates NLRP3 and pro-IL-1β expressions. Additionally, adequate NLRP3 levels and specific post-translational modifications are vital for NLRP3 inflammasome activation.105 NLRP3 is typically ubiquitinated in a resting state. The priming signals then induce NLRP3 deubiquitination, and these modifications are required for NLRP3 activation. For example, BRCC3, a deubiquitinating enzyme, has been shown to remove ubiquitin moieties from NLRP3 by directly associating with the ubiquitinated NLRP3 LRR domain in various cells, including macrophages, 293 T, and NG5 cells.106 Abraxas brother 1 (ABRO1) also modulates NLRP3 deubiquitination and assists BRCC3 to activate NLRP3 inflammasome.107 Intriguingly, phosphorylation of NLRP3 at distinct amino acid sites yields the different effects. NLRP3 phosphorylation at Ser295 or Ser5 suppresses the assembly of the NLRP3 inflammasome platform, thereby inhibiting inflammasome activation.108 In contrast, phosphorylation of human NLRP3 at Ser198 is essential for NLRP3 deubiquitination and the subsequent NLRP3 inflammasome activation.109,110 Collectively, these findings suggest the priming step in NLRP3 inflammasome activation is highly regulated by post-translational modifications of NLRP3.
The NLRP3 inflammasome activation phase is regulated by many factors that ultimately help dictate how a given immune cell responds to the pathogen. For example, activation of the ATP-gated ion channel P2X purinoceptor 7 (P2X7) leads to K+ efflux and Ca2+ influx which has been shown to help trigger NLRP3 inflammasome activation by disrupting mitochondrial ion balance and subsequent mitochondrial reactive oxygen species (mROS) generation.111 Cl− efflux contributes to the NLRP3 inflammasome activation through a distinct different mechanism, whereby K+ efflux induces the oligomerization of NLRP3 while Cl− efflux promotes the polymerization of ASC.112 These findings demonstrate that ion flux plays a crucial role in NLRP3 inflammasome activation, indicating their modulation may serve as a potential therapeutic target. Dysregulation of various organelles is also involved in NLRP3 activation, encompassing disturbances in lysosomes, mitochondria dysfunction, and disintegration of the trans-Golgi apparatus. The lysosomotropic dipeptide Leu-Leu-Ome promotes lysosomal rupture and induces an ion exchange (K+ efflux and Ca2+ influx) to allow the activation of the NLRP3 inflammasome.113 Mitophagy clears the impaired mitochondria and suppresses the mROS release, which serves as the activator of NLRP3. In addition, NLRP3 activation facilitates the disassembly of the trans-Golgi network into dispersed vesicles, which recruit NLRP3, followed by the promotion of ASC polymerization and the subsequent downstream signaling that leads to NLRP3 activation.114 Cannabinoid receptor 1 (CB1R) has also been shown to help regulate activation of the NLRP3 inflammasome. After internalization, CB1R interacts with NLRP3, caspase 1, and GSDMD proteins to inhibit the degradation of NLRP3 inflammasome.115 However, whether CB1R is involved in the lysosome disruption is still unclear.
Non-canonical NLRP3 inflammasome activation pathway
The non-canonical NLRP3 inflammasome pathway involves the activation of “non-canonical” caspases, including caspase-4, caspase-5, and caspase-11. The non-canonical pathway is also activated by direct cytosolic stimuli and is most closely associated with its significance in inflammatory disorders. Unlike the canonical pathway, caspase-4 has been shown to act as the major caspase involved in non-canonical NLRP3 inflammasome activation and it has been demonstrated that Gram-negative bacterial infections induce the non-canonical pathway, which results in cellular damage and death via pyroptosis.116,117
Non-canonical activation of the NLRP3 inflammasome is induced by Gram-negative bacterial infections.116 PAMPs and DAMPs interact with TLRs to activate NF-κB and promote NLRP3 transcription. Recent advances have discovered that in mice caspase-11 (analogous to caspase-4 in humans) was activated in the LPS signaling pathway rather than caspase-1.118 Caspase-11 deficiency mice exhibited attenuated IL-1β production; however, caspase-11-/- macrophages exhibited normal IL-1β production in response to stimuli, suggesting that although LPS is involved in the canonical activation of the NLRP3 inflammasome,119 non-canonical inflammasome activation is dependent on caspase-11.116 LPS stimulates TLR4, and TLR4 signaling induces activation of mitogen-activated protein kinases (MAPKs), NF-κB, and interferon regulatory factors (IRFs).120 Subsequently, these events promote the transcription of IL-1β, IL-18, and NLRP3. Elevated IRF-3 and IRF-7 then form a complex which induces the expression of IFN-α/β.121 The binding of IFN-α/β to the IFN-α/β receptor results in activation of the JAK/STAT pathway and, consequently, upregulating the transcription of caspase-11.122,123 Active caspase-11 triggers pyroptosis by cleaving GSDMD, resulting in the release of HMGB1 and IL-1α.124 Additionally, researchers have identified a hypotonicity-induced NLRP3 activation mechanism, which demonstrated that low osmolarities can trigger a downregulation in intracellular Cl− concentrations that are sufficient to activate the NLRP3 inflammasome.125
However, it should be mentioned that certain details of the non-canonical pathway remain controversial. For instance, it was demonstrated that NLRP3 inflammasome activation can occur without the priming signal in human monocytes,126 and there is also evidence that a single stimulus can provide both the priming and activating signals.127 Advanced studies are needed to clarify this issue further. Ultimately, both the canonical and non-canonical pathways of NLRP3 inflammasome activation lead to pyroptosis through a mechanism that was triggered by GSDMD cleavage. Gasdermin E (GSDME) has also been identified as a downstream molecular of NLRP3-induced pyroptotic pathway.128 However, the levels of endogenous GSDME are relatively low and its function is still not fully understood. Thus, GSDMD has been recognized as a major executor that mediates pyroptotic cell death upon NLRP3 inflammasome activation.
NLRP1 inflammasome activation
Tschopp et al. discovered and described the first inflammasome-forming sensor, human NLRP1 in their landmark 2002 paper.3 Although it was the first to be discovered, the activation of NLRP1 has remained unclear despite the continued and ongoing research that has been conducted to elucidate its underlying mechanism. As the domain structure differs between NLRP1 and NLRP3, there are certain key differences between the components of NLRP3 and NLRP1 inflammasomes that we will discuss below.
Diverse bacterial and protozoan toxins can activate the NLRP1 inflammasome, including ultraviolet B irradiation, double-stranded viral RNA, viral proteases, the bacterial cell wall component muramyl dipeptide, and LeTx exposure.129,130 There are also several ways in which pathogens can activate the NLRP1 inflammasome. First of all, in response to enterovirus 3 C cysteine proteases or the anthrax lethal factor protease, cleavage of NLRP1 near its N-terminal PYRIN domain occurs, allowing the N-terminal NLRP1 to be sent to the proteasome for degradation131 and consequently inducing C-terminal CARD domain oligomerization and the activation of caspase-1.35 Additionally, long double-stranded RNA (dsRNA) or RNA-positive (+RNA) strands could bind to the NACHT-LRR domain of NLRP1 to activate NLRP1.130 Upon activation, NLRP1 oligomerizes and leads to caspase-1 activation and IL-1β secretion.132 Additionally, NLRP1 also cleaves pro-caspase-5, which could promote IL-1β in human keratinocytes.133 K+ efflux has also been implicated in NLRP1 activation.134 Furthermore, studies have also shown that 3C-like protease can inactivate the GSDMD, enabling NLRP1-induced caspase-3 activation to drive Gasdermin E-dependent pyroptosis.135
NLRC4 inflammasome activation
NLRC4 belongs to the NLRC family, which plays a vital role in the immune response to bacterial pathogens. Similar to other inflammasomes, transcriptional and post-transcriptional mechanisms tightly regulate NLRC4 activation.61 However, ligand binding and phosphorylation are the most well-described regulatory mechanisms of NLRC4 inflammasome activation. Regardless of the modifications involved, activating P53 through genotoxic stress or pro-inflammatory stimuli leads to the upregulation of NLRC4 expression.
Gram-negative bacteria with type III or IV secretion systems activate the NLRC4 inflammasome.136,137,138 Cytoplasmic injection of bacterial components from these types of bacteria may be able to trigger NLRC4 activation directly, and flagellin localization in the cytosol is sufficient to activate caspase-1 in an NLRC4-dependent manner.139,140 Other PAMPs may modulate NLRC4, as both flagellin-dependent and flagellin-independent mechanisms are involved in the activation of NLRC4 by P. aeruginosa.141,142 Since NLRC4 does not directly interact with an activating ligand, NLRC4 may sense cytosolic PAMPs through a common pathway, similar to the pathways proposed in NLRP3 activation. NLRC4 phosphorylation by PKCδ is essential for the NLRC4 inflammasome activation.143 Unlike NLRP3, high extracellular K+ does not inhibit NLRC4 activity, indicating NLRC4 is not an ionic flux sensor.144 Additionally, NLRC4 must collaborate with another NLR, NAIP, to protect against this pathogen.63,145 NAIPs are the upstream receptors that recognize bacterial ligands which then mediate NLRC4 inflammasome formation. NAIPs can interact with NLRC4 and induce NLRC4 oligomerization upon bacterial ligand binding. As NLRC4 lacks the PYD domain, NLRC4 may recruit a PYD-containing protein (such as an NLRP3) to react to bacterial infections.146 Moreover, NLRC4 contains a CARD domain, which suggests direct interactions with procaspase-1.34 ASC is not necessary for NLRC4-dependent caspase-1 activation in response to L. pneumophila. At the same time, ASC is required for the maximal response in bacterial-induced caspase-1 activation.147,148 These findings demonstrate that ASC and NAIP are crucial for NLRC4 inflammasome activation, although the exact mechanisms remain unclear and need further investigation.
AIM2 inflammasome activation
AIM2 is a PYHIN family member that plays a vital role in recognizing cytosolic DNA. As the first non-NLR family member to be identified as forming an inflammasome scaffold, AIM2 can recruit ASC and activate caspase-1-dependent IL-1β maturation. The AIM2 inflammasome protects against pathogens, like Francisella tularensis and Listeria monocytogene, by sensing cytosolic dsDNA.149,150 As mentioned above, oligomerization of the AIM2 inflammasome is mediated by binding between sites clustered on ligands and the C-terminal HIN domain of AIM2, but not by the central oligomerization domain (as was the case for the NACHT domain in NLRs). AIM2, ASC, and caspase-1 form the AIM2 inflammasome. As for NLRP3, AIM2 has a PYD domain that interacts with ASC through homotypic PYD-PYD interactions., which enables pro-caspase-1 recruitment via the ASC CARD domain. Subsequently, activation of caspase-1 promotes the maturation and secretion of pro-inflammatory cytokines (such as IL-1β and IL-18). Additionally, AIM2 drives a form of inflammatory signaling and cell death, known as PANoptosis, by regulating the innate immune sensors ZBP1 and pyrin.151 AIM2 has also been shown to have permissive ligand requirements, as bacteria and cytosolic dsDNA from viruses or the host can activate AIM2.152 As a result, it has been suggested that AIM2 is involved in self-DNA-induced autoimmune responses in systemic lupus erythematosus. Additional studies to further disambiguate the viral dsDNA and self-DNA pathways are needed.
IFI16, NLRC5, NLRP6, NLRP7, and NLRP9 inflammasome activation
IFI16 is another member of the PYHIN family that can form an atypical inflammasome. IFI16 expression has been found in myeloid precursor cells, mature lymphocytes, peripheral blood monocytes, T cells, and epithelial cells.153,154,155 Unlike AIM2, IFI16 is primarily found in the nuclei of resting cells and serves to recognize viruses that enter the nuclei. Several pathogens are known to be recognized by IFI16, including the Kaposi sarcoma-associated herpesvirus (KSHV) and the influenza A virus (IAV).156,157,158 IFI16 migrates to the cytoplasm from the nucleus upon activation, forming nuclear and cytosolic inflammasomes containing IFI16, ASC, and caspase-1.156 Subsequently, the IFI16 inflammasome induces caspase-1 activation and IL-1β cleavage. KSHV-induced IL-1β and IL-6 expressions are dependent on IFI16 and ASC expression,156 suggesting that the IFI16 inflammasome, but not other inflammasomes, initiates the responses to KSHV infection. Additionally, IFI16 binds to viral DNA and subsequently facilitates IFN-β production via a direct interaction between IFI16 and STING.159 It is evidenced that IFI16 is a unique PRR that plays dual roles in the cytoplasm and nucleus. Moreover, the existence of both nuclear and cytosolic inflammasomes indicates that the innate immune system applies a multifaceted approach to detect intracellular pathogens.
NLRC5, like NLRC4, is also vital in antibacterial defenses. NLRC5 is expressed both in the cytoplasm and nucleus of cells, and NLRC5 can regulate MHC class I gene expression. It is widely accepted that NLRC5 regulates gene expression within the nucleus, but its function within the cytoplasm is less clear and needs to be understood. In monocytes, NLRC5 plays a vital role in mediating caspase-1 activation and IL-1β secretion in response to infections from Escherichia coli, S. aureus, and Shigella flexneri, or upon stimulation by TLR ligands.29 Intriguingly, NLRP3 agonists can trigger IL-1β secretion via NLRC5. ASC and NLRP3 were also found to interact physically with NLRC5, with intact NACHT domains being required for NLRC5 to bind to NLRP3. The co-expression of ASC, pro-caspase-1, pro-IL-1β, NLRC5, and NLRP3 in HEK293T cells causes IL-1β cleavage.29 Interestingly, TLR ligands and NLRP3 agonists appear to have no effect on cytokine production in the absence of NLRC5, whereas the co-expression of NLRC5, pro-caspase-1, and pro-IL-1β without NLRP3 induces the cleavage of IL-1β.160 These data suggested that NLRC5 appears to be able to form a functional inflammasome, although it remains to be determined if NLRC5 can form an inflammasome complex independently.
NLRP6 inflammasomes have previously been reported to regulate intestinal microbiota in mice.15 There is also evidence that NLRP6 can form inflammasomes in vitro. For example, in a study where NLRP6 and ASC were expressed in 293T cells, NLRP6 was recruited into speck-like structures within the ASC. Moreover, transfecting plasmids encoding NLRP6, ASC, and pro-caspase-1 into COS-7L cells induces IL-1β secretion.161 Further evidence of NLRP6 inflammasomes comes from studies showing that NLRP6-deficient mice developed enhanced dextran sulfate sodium (DSS)-induced colitis, like that of ASC- and IL-18-deficient mice. Intriguingly, one study reported that housing wild-type mice and NLRP6-deficient mice together enhanced disease severity in wild-type mice, which was interpreted as evidence that microbiota can act as a driving factor of enhanced colitis in an NLRP6-dependent manner. In addition, IL-18 production was crucial for maintaining intestinal homeostasis. It was also hypothesized that unknown ligands might activate the NLRP6 inflammasome, ultimately leading to the IL-18 maturation, preventing dysbiosis, and regulating microbiota through an unknown mechanism. Further insight into the function of NLRP6 inflammasome in intestinal homeostasis was gained through studies on mucus production in mice challenged with an enteric pathogen.162 There was a significant decrease in mucus thickness in mice lacking NLRP6, ASC, or caspase-1, which increased bacterial adherence and dissemination in C. rodentium-infected mice. The NLRP6 inflammasome is clearly involved in protecting the intestinal barrier and regulating dysbiosis; however, how intestinal ligands activate the NLRP6 inflammasome remains unclear.
NLRP7 inflammasome activation was reportedly identified in human macrophages upon exposure to microbial acylated lipopeptides.16 Various bacterial acylated lipopeptides, as well as TLR2 agonists, could activate NLRP7-inducing caspase-1-mediated IL-1β production. Additionally, the activation of TLR2 in response to acylated lipopeptides is believed to be reliant on the maturation of IL-1β through NLRP7 and required for the transcription of chemokines, pro-IL-1β, and pro-IL-18. The transcription of these signaling molecules, in turn, acts as the priming step for NLRP7 inflammasome activation. By activating NLRP7, caspase-1 becomes enzymatically active, leading to IL-1β and IL-18 maturation, which subsequently restricts intracellular bacterial replication. However, caspase-1-independent IL-6 or tumor necrosis factor-α (TNF-α) secretions are not observed following NLRP7 inflammasome activation. Additionally, some in vitro studies have found that NLRP7 is a negative regulator of inflammation. For instance, NLRP7 directly interacts with pro-caspase-1 and inhibits IL-1β maturation in transiently transfected HEK293 cells.163 The same situation occurred in THP-1 cells, where NLRP7 levels were increased upon LPS or IL-1β stimulation. Specifically, NLRP7-expressing THP-1 cells secreted less IL-1β than empty vector-transfected cells when treated with LPS. Consistently, patients with a hydatidiform mole during pregnancy who have NLRP7 mutations and rare variants showed low levels of IL-1β and TNF secretion in response to LPS.164 Mechanistically, the PYD is critical for inhibiting IL-1β processing, such that protein-truncating mutations after the PYD abolish IL-1β inhibition. NLRP7 co-localizes with Golgi and microtubule-organizing centers in peripheral blood mononuclear cells, which indicates that NLRP7 also influences IL-1β and TNF secretion via cytokine trafficking in these cells. The exact role of NLRP7 in innate immunity remains unclear. Furthermore, it is unknown whether NLRP7 inflammasomes can be formed in other cell types or if other ligands can activate NLRP7. These questions may serve as exciting areas of research in the near future.
A recent study has suggested that NLRP9 could initiate inflammasome formation upon short dsRNA stimulation. NLRP9b acts as a sensor detecting rotavirus in the intestine. Interestingly, NLRP9b did not directly bind viral RNA; instead, the RNA helicase DHX9 acted as a direct RNA-binding protein that mediates viral recognition by NLRP9.17 However, the mechanism by which DHX9 differentiates between the viral and host RNA is still unclear. Interestingly, NLRP9b is mainly expressed in intestinal epithelial cells but not the neighboring immune cells. NLRP9b conditional KO mice showed higher susceptibility to rotavirus infection, suggesting that NLRP9b exerts a protective function in intestinal epithelial cells. Upon rotavirus infection, NLRP9b forms inflammasomes with ASC and caspase-1 to trigger the maturation of IL-18 and GSDMD-induced pyroptosis in mice.17 NLRP9b-, ASC-, or caspase1- deficient mice exhibit elevated viral loads and pathological symptoms than wild-type mice, suggesting that NLRP9b could initiate inflammasome formation upon rotavirus infection. Moreover, GSDMD-deficient mice are more vulnerable to rotavirus infection, suggesting that rotavirus clearance requires GSDMD. Human NLRP9 is also capable of binding to rotavirus RNA. However, it remains unclear whether human NLRP9 performs the same functions as the murine NLRP9. Further investigations are warranted to determine the exact molecular mechanisms underlying human NLRP9 inflammasome formation.
These studies provide a better understanding of innate immunity and inflammasomes, and suggest there may be new potential therapeutic targets for diseases associated with aberrant inflammasome activity. These studies also reveal that inflammasomes are expressed in various types of cells and respond to diverse pathogens in both the cytoplasm and nucleus. Variations in the mechanisms that regulate inflammasome activation reported by these studies could be attributed to several factors, including inherent differences in cell types, the expression levels of inflammasomes, and the types of stimuli used to challenge cells. Further investigations focused on the precise role that inflammasomes play in modulating the innate immune signaling pathways should be conducted in the relevant systems.
Roles of the inflammasomes in various diseases
There is a strong correlation between inflammasomes and various autoimmune and autoinflammatory diseases, including cardiovascular diseases, neurodegenerative diseases, and metabolic disorders (Fig. 4). Similarly, as our understanding of inflammasomes has grown over the past few decades, it has become increasingly clear that inflammasomes play either causative or contributing roles in the initiation and progression of various diseases. Here, we provide an overview of the potential roles those inflammasomes may play in different diseases.

Different inflammasomes contribute to diseases of different systems in the human body. The figure was created with the assistance of FIGDRAW
Cardiovascular disorders
Inflammation plays a key role in the development of cardiovascular disorders, and aberrant inflammasome activity has been implicated in several of these conditions, including atherosclerosis. Atherosclerosis is a chronic disease characterized by the progressive hardening or narrowing of arterial vessels that can lead to heart attacks and strokes.165,166 In atherosclerosis, high quantities of cholesterol and white blood cells clog the arterial wall, preventing oxygen-rich blood from reaching the organs.167,168,169 Compared to disease-free arterial tissues, atherosclerotic plaques contain higher levels of IL-18 and IL-18 receptors. Inflammasome activation leads to elevated IL-18 production, which may contribute to the pathology of atherosclerosis.170 For example, mice lacking Apolipoprotein E (ApoE) develop atherosclerosis spontaneously, and atherosclerotic plaques are more unstable when IL-18 levels are high, whereas IL-18 deficiency results in smaller lesions.171 Elevation of free fatty acids (FFAs) and low-density lipoprotein (LDL) in human blood can induce pro-IL-1β production via TLRs.172 The cell surface receptor CD36 promotes oxidized LDL (ox-LDL) internalization and cholesterol crystallization, and these cholesterol crystals activate the NLRP3 inflammasome in vitro via phagolysosomal damage.173 This data indicates that FFAs or LDL can provide the priming or activation signal for inflammasomes. In macrophages, cholesterol activates the NLRP3 inflammasome and mediates IL-1β release in a Cathepsin B-dependent manner.174 In LDL receptor knock-out mice, transplantation of bone marrow from NLRP3-/-, ASC-/-, or IL-1β-/- mice showed decreased atherosclerosis.170 Similarly, the size of atherosclerotic lesions in ApoE-deficient mice is significantly reduced by IL-1β inactivation.175,176 These findings are promising and suggest additional studies should be conducted to clarify the exact mechanism of inflammasome activation in atherosclerosis, as well as the contribution of IL-1β to atherogenesis.
Hypertension can cause myocardial hypertrophy and fibrosis, leading to the development of heart failure. Hypertension-induced cardiac or vascular upregulation of NLRP3 and IL-1β has been observed in different animal models, such as spontaneously hypertensive rats, decompensated right ventricular hypertrophy rats, and deoxycorticosterone acetate-induced hypertensive mice.163,177,178 Interestingly, a transverse aortic constriction (TAC) was also found to increase NLRP3 and caspase-1 activity in cardiomyocytes, but not in non-cardiomyocytes.179 This suggested that the original site of NLRP3 inflammasome activation may be in cardiomyocytes. However, it remains unclear how inflammasomes activate without ischemic damage or cell death. There is evidence that activation of the NLRP3 inflammasome is mediated by increased Ca2+/calmodulin-dependent protein kinase II δ (CaMKIIδ) activity in response to pressure overloads.179 Using cardiomyocyte-specific CaMKIIδ-KO (CKO) mice, researchers observed that caspase-1 activity was attenuated, and IL-1β and IL-18 levels were reduced in TAC-treated CKO mice.179 Additionally, diabetes mellitus may lead to diabetic cardiomyopathy, in which NLRP3 activation in a Cathepsin B-dependent manner could aggravate the condition by promoting pyroptosis.180 These effects may be intensified by glucose, which is a potential stimulus for NLRP3.181 Altogether, these findings suggest that therapies targeting the NLRP3 inflammasome may help to prevent cardiac remodeling and heart failure.
The NLRP3 inflammasome has also been implicated in the development of atrial fibrillation. In atrial cardiomyocytes from patients with atrial fibrillation, NLRP3 inflammasome activity was increased.182 Mice expressing constitutively active NLRP3 in their cardiomyocytes (cardiomyocyte-KI) showed spontaneous premature atrial contractions; using MCC950 to inhibit NLRP3 blunted the spontaneous premature atrial contractions.182 Cardiomyocyte-KI mice exhibited larger atria, electrical remodeling, and abnormal spontaneous Ca2+ release patterns from the sarcoplasmic reticulum, which were prevented by the knockdown of NLRP3 in cardiomyocytes. These findings suggest that targeting the NLRP3 inflammasome could be a new therapy for atrial fibrillation.
There is evidence that dilated cardiomyopathy is accompanied by an inflammatory component that plays an important role in its pathogenesis. For instance, there is a clinical correlation between circulating levels of NLRP3 inflammasome and cardiac function, as well as between the NT-pro BNP levels and the cumulative rehospitalization rate in patients with dilatated cardiomyopathy.183,184 NLRP3 activation also occurs in a time-dependent manner in response to ischemia.185 Ischemic cells release DAMPs and alarmins, which strongly stimulate the NLRP3 inflammasome. During the healing phase, ASC aggregates are most prevalent in cardiomyocytes and fibroblasts.186,187 Furthermore, patients with acute myocarditis have been found to have NLRP3 inflammasomes in their endomyocardium.188 CVB3, a common virus causing myocarditis, increases caspase-1, ASC, and IL-1β expression in infected mice by altering NLRP3 activation.189 Mechanistically, Cathepsin B mediates both inflammasome activation and pyroptosis in experimental CVB3-induced myocarditis.190
The NLRP3 inflammasome also regulates the initiation and propagation of another cardiovascular disorder: Venous thromboembolism (VTE). Elevated NRLP3 activity, indicated by high caspase-1, IL-1β, IL-6, or C-reactive protein levels, was observed in patients with VTE.191 Hypoxia and reduced blood flow induce NLRP3 activation and elevated levels of caspase-1 and IL-1β following experimental venous thrombosis.192 Experimental intervention studies have also found that genetic deletion of NLRP3, caspase-1, or GSDMD, and inhibition of caspase-1 or IL-1β has been shown to ameliorate venous thrombosis.193 Altogether, developing novel therapeutics against VTE may be possible by selectively targeting the NLRP3 inflammasome and maximizing the benefits of anticoagulation. In summary, there is considerable evidence that inflammasomes play either causative or contributing roles in the development of several cardiovascular diseases. This data suggests that inflammasomes, caspase-1, and IL-1β may be promising therapeutic targets for cardiovascular diseases.
Several commonly used medications like anti-tumor drugs (i.e. doxorubicin) and antipsychotic drugs are reported to have significant cardiotoxic effects.194,195 Moreover, a recent study found that Sirtuin 3 alleviates the doxorubicin-induced cardiotoxicity by inhibiting the activation of the NLRP3 inflammasomes.196 Antipsychotic cardiotoxicity was predominantly mediated by CB1R translocation-induced NLRP3 inflammasome stabilization and subsequent pyroptotic cell death.115 These findings indicate that inflammasome activation may act as a mediator of drug-induced toxicity. As such, targeting the inflammasome signaling pathway has the potential to relieve the cardiotoxic effects of anti-tumor or antipsychotic drugs.
Neurological disorders
NLRP3 is the first inflammasome to have been studied in the central nervous system (CNS), and while it is predominantly located in microglia, it is also expressed in neurons, astrocytes, and oligodendrocytes.197,198,199,200 NLRP3 inflammasomes play a crucial role in several cerebral pathologies, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and CNS infections.201 NLRP3 has two major isoforms, including the full-length NLRP3 and the one without exon 5.54 The full-length NLRP3 protein functions effectively, while the NLRP3 isoform without exon 5 is a non-functional variant that cannot be activated. The functional NLRP3 proteins are involved in neuroinflammation of neurodegenerative diseases. However, the exact functions of full-length NLRP3 and the exon 5-lacking in neurodegenerative disorders are still unclear. Other inflammasomes, including NLRP1, NLRC4, and AIM2, have also been implicated in some neurological disorders.202,203,204 AIM2 has been detected in most cells of the CNS, including neurons, microglia, and brain endothelial cells. On the other hand, NLRP1 inflammasomes have been observed predominantly in neurons, whereas NLRC4 expression has been found mainly in microglia and astrocytes. Furthermore, hallmarks of neurodegenerative disease such as amyloid- β (Aβ), α- synuclein (α- Syn), and transactive response DNA-binding protein 43 (TDP43) can act as immune stimulatory molecular patterns in the CNS.
AD is the most common neurodegenerative disorder.205 It is characterized by the formation of neuritic plaques and neurofibrillary tangles (NFTs) in the brain. Neuritic plaques are caused by the accumulation of Aβ, whereas NFTs are a result of hyperphosphorylated tau inside neurons.206 Aβ is primarily produced within neurons and is then released from the brain into the CSF and blood vessels.207 Upon exceeding a critical threshold, Aβ forms oligomers, fibrils, and deposits in neuritic plaques, which can act as DAMPs to activate NLRP3 inflammasomes.208,209 Fibrillar Aβ-induced microglial IL-1β release occurs in an NLRP3- and ASC-dependent manner.210,211 In addition, soluble and oligomeric Aβ peptides are equally as potent in inducing CD36-mediated NLRP3 inflammasome activation and IL-1β production.173 NLRP3 activation is further regulated by autophagy-mediated autophagy, as deficiencies in the cellular autophagy-related protein 7 (ATG7) were found to increase caspase-1 cleavage and IL-1β release in microglia.212 Another study found that cell media collected from Aβ-treated microglia was neurotoxic, and this effect was more pronounced in ATG7-deficient microglia, indicating that well-controlled microglial inflammasome activation could limit neuronal destruction. Studies have also shown that NLRP3 inflammasome functions in astrocytes, and astrocytes can release IL-1β in an ASC-dependent manner upon uptake of Aβ.213 However, the NLRC4 inflammasome was also found in astrocytes and could promote IL-1β maturation.214 Whether different inflammasomes act synergistically or independently needs further investigation. In an in vivo uptake assessment, amyloid precursor protein (APP)/presenilin 1 (PS1)/NLRP3-knockout mice (mice generated from the cross of APP/PS1 mice and NLRP3-knockout mice) showed evidence of Aβ phagocytosis and an increased phagocytic clearance capacity.215 Insulin-degrading enzyme (IDE) expression was also increased in brain lysates obtained from APP/PS1/NLRP3-knockout mice.215 IDE has been shown to degrade extracellular Aβ, indicating that NLRP3 plays a role in balancing the cerebral Aβ load. Additionally, synaptic loss occurs in AD brains, and blocking NLRP3-associated signaling protects against neuronal spine loss in APP/PS1 mice. Furthermore, the ASC speck formation is another feature of inflammasome activation, and upon release, the ASC specks bind quickly to Aβ peptides.216 Aβ was also shown to bind with ASC in brain samples from AD and APP/PS1 mice.216 Moreover, ASC specks can seed Aβ deposition in APP/PS1 mice.217 These findings suggest that the release of ASC specks may contribute to early Aβ deposition and the pathogenesis of AD. A recent study also found that susceptibility to inflammasome activation was correlated with a higher likelihood of cognitive deficits in AD patients.218 Additionally, AIM2 and NLRP1 inflammasomes in particular have been implicated in AD as Alzheimer’s patients have increased NLRP1 expression in their brains. Neurons in APP/PS1 mice showed an upregulation of NLRP1 levels.219 Also, NLRP1 reduced the number of apoptotic neurons but had no effect on overall Aβ deposition in this model. In addition, AIM2 is believed to play a vital role in AD pathogenesis, as microglial activation was attenuated, and IL-6 and IL-18 levels were increased when AIM2 was knocked out in 5XFAD mice. However, both open-field behavior and spatial memory performance were not improved,220 suggesting the AIM2 inflammasome may not directly affect cognition or memory formation.
Research suggests that α-Syn could also trigger inflammasome activation In PD. α-Syn can induce NLRP3 inflammasome activation and IL-1β release in both monocytes and microglia in a cathepsin B- and caspase-1-dependent manner.221,222 TLR2 inhibitors can block this α-Syn induced IL-1β release in human monocytes.223 This suggests that TLR2 may mediate the signaling pathway by which α-Syn induces NLRP3 inflammasome activation. However, additional data is needed to clarify this mechanism and its role in PD. In MPTP-induced PD mice and human microglia, dopamine inhibits the activation of microglial NLRP3 inflammasome through signaling via DRD1 and DRD2, leading to the ubiquitination and subsequent degradation of NLRP3.224 Similarly, inhibiting microglial NLRP3 inflammasome activation significantly reduced dopaminergic neurodegeneration and ameliorated motor deficits in the MPTP-treated mice, although the mechanism behind this remains unknown.225 Neuronal NLRP3 has also been implicated in PD pathogenesis. The parkin protein encoded by PRKN functions as an E3 ubiquitin ligase, and PRKN mutations lead to monogenic PD.226 In dopaminergic neurons, reducing parkin activity induces spontaneous activation of the NLRP3 inflammasomes, and parkin inhibits this activation by ubiquitinating NLRP3.227 These findings suggest that both neuronal and microglial NLRP3 inflammasomes play a role in the pathogenesis of PD; whether the two work synergistically remains to be determined.
ALS is a neurodegenerative disease that gradually destroys upper and lower motor neurons, leading to the atrophy and paralysis of voluntary muscles. Superoxide dismutase (SOD1) and TDP43 are key molecules at high risk of genetic mutations that cause familial ALS.209,228 There has been evidence that the NLRP3 inflammasome is activated in the brains and spinal cord of sporadic ALS patients as well as in ALS mouse models. Bioptic samples taken from the spinal cord of ALS patients showed an increase in NLRP3, ASC, caspase-1, and IL-1β, suggesting NLRP3 activation may be elevated in these patients.229 In addition, the SOD1 mutant mouse model of ALS exhibits an increased level of NLRP3 activation, as well as ASC speck formation and IL-1β maturation in its spinal cord.229,230 It was found that caspases-1 and IL-1 deficiencies restored the motor deficits associated with ALS in SOD1 mutant mice.231 Interestingly, one study found that NLRP3 was present in CD11b+ cells but not in Iba1+ cells in the spinal cord of SOD1 mice.232 This suggests that peripheral immune cells may also contribute to inflammation in neurodegenerative diseases. Furthermore, evidence also showed that NLRP3 inflammasome activation occurred in muscle and near neuromuscular junctions, which enhanced skeletal muscle degeneration in SOD1G93A mice.233 Altogether, there is still much to learn about the role of NLRP3 activation in ALS development.
MS is a common autoimmune disease in CNS characterized by oligodendrocyte attack and demyelination. It has been reported that MS patients have elevated caspase-1 and insulin-like growth factor 1 levels in their peripheral blood monocytes, brain tissues, and cerebrospinal fluid.234,235 In addition, a crucial function of NLRP3 in experimental autoimmune encephalomyelitis (EAE) is to prime CD4 + T cell migration through increasing the expression of chemotaxis-related protein, which indicates that the EAE animal model for MS involves the NLRP3 inflammasome.236 Neuromyelitis optica spectrum disorder (NMOSD) is another CNS autoimmune disease. MS and NMOSD share similar symptoms. Recent studies have identified that NLRP3 levels in the CSF were significantly increased in patients suffering from NMOSD or MS. In addition, NMOSD patients had higher CSF NLRP3 levels compared with MS patients.237 These findings indicate that levels of NLRP3 in CSF could be a potential diagnostic marker in NMOSD and MS.
Together, these findings support the notion that an increased susceptibility to aberrant inflammasome action and activity in various cell types of the CNS could make the brain more vulnerable to neurodegenerative changes, and subsequently promote the development of AD, PD, or other neurological diseases. There is a need for continued research in the development of drugs that target specific cell types or inflammasome signaling pathways. This research could provide valuable insights into how inflammasomes contribute to disease pathology in various neurodegenerative disorders. Such drugs may have significant therapeutic potential and should be further explored.
Respiratory disorders
Inflammasomes are implicated in the pathogenesis of several respiratory disorders, including asthma, chronic obstructive pulmonary disease (COPD), pulmonary infection, pulmonary fibrosis, and acute respiratory distress syndrome (ARDS). As a result, inflammasomes have become a significant focus for research aimed at developing new diagnostic biomarkers and therapeutic avenues in pulmonary diseases and other respiratory disorders.
Asthma is a chronic inflammatory disease of the airways that affects inflammatory cells and cytokines in the lungs.238 Exposure to industrial products, microbes, or other allergens induces reversible limitations in airflow into and out of the lungs, as well as airway hyper-responsiveness. Persistent airway inflammation causes structural changes in airway tissues, known as airway remodeling, resulting in nonreversible airway obstruction and progressive loss of lung function. There is increasing evidence that NLRP3 inflammasome plays a role in asthma. Researchers found upregulated expression of NLRP3 and of IL‐1β genes in phlegm obtained from the lungs of 127 asthmatic patients.239 The mechanism by which NLRP3 inflammasomes are activated in asthma is unclear. Chlamydia and Haemophilus infection, or OVA, titanium dioxide nanoparticles, and silica treatments stimulated the NLRP3, caspase-1, and IL‐1β expression, ultimately causing steroid-resistant neutrophil inflammation and hyperresponsiveness.240 Follistatin‐like 1 deficiency attenuates OVA‐induced mucus over secretion and airway mucin MUC5AC production, inhibits the NLRP3 and IL‐1β expression, inflammatory cytokines production, and inflammatory cell infiltration.241,242 Whether interventions on the NLPR3 inflammasome differ from other inflammation targets could serve as an additional avenue of research.
Activation of inflammasomes and alterations of their responses linked to the development of airway inflammation may also be seen in COPD.243 A comparison of COPD with smoking revealed elevated levels of NLRP3, Caspase-1, ASC, IL-1β, and IL-18 mRNA in peripheral blood mononuclear cells and bronchial tissues.244 However, the mRNA levels of NLRP3, Caspase-1, ASC, IL-1β, and IL-18 mRNA were higher in acute exacerbation of COPD (AECOPD) than those in COPD patients in the stable stage, suggesting a greater involvement of the NLRP3 inflammasome in AECOPD. Studies also found that cigarette smoke extract (CSE) stimulates the heat shock protein 60 expression and activates NLRP3 inflammasome through the TLR4-MyD88-NF-κB signal pathway.245 Smoking cessation is the most important COPD intervention for smokers. There is evidence that CSE induces pyroptosis via the ROS-NLRP3-caspase-1-GSDMD pathway in human bronchial epithelial cells.246 Particulate matter (PM2.5) also plays a role in lung injury: after PM exposure, Sirtuin1 (SIRT1) inhibits sterol regulatory element binding protein-1 (SREBP-1) and further decreases PIR and NLRP3 inflammasomes.247 In addition, the ROS-TRPM2-Ca2+-NLRP3 pathway also contributes to lung injury induced by PM 2.5.248 These studies indicate that the NLRP3 inflammasome can be activated via multiple pathways in lung injury, which provides a new therapeutic target for COPD.
Severe coronavirus disease 2019 (COVID-19) is a viral RNA infection that can cause persistent lung inflammation, dysregulation of cytokine production, sustained IFN response, as well as respiratory failure.249 Viruses could trigger the NLRP3 inflammasome. Postmortem study showed that patients with fatal COVID-19 were found to have abundant NLRP3, ASC, and caspase-1 in their lungs.250 As COVID-19 enters cells through the protein angiotensin-converting enzyme 2 (ACE2), dsRNA and ssRNA derived from the virus are recognized by TLR3 and TLR7, leading to elevated pro-IL-1β and pro-IL-18 levels, which are then cleaved into their mature forms once the NLRP3 inflammasome has been activated.251 NLRP3 can be directly activated by viral N protein.252 A correlation exists between levels of IL-1β, IL-18, lactate dehydrogenase, and COVID-19 severity in patients, which indicates that inflammasome activation and pyroptosis are involved in the pathology.253,254 COVID-19 patients have also consistently shown GSDMD in their serum.255 SARS-CoV-2 infected MISTRG6- human ACE2 (hACE2) humanized mouse model recapitulates the pathology, lung inflammation, dysregulation of cytokine production, sustained interferon response, as well as respiratory failure, with a human immune system.256,257 Infection and replication of SARS-CoV-2 in lung-resident macrophages play a crucial role in the development of the disease. When infected, human macrophages experience an inflammatory response that is controlled by CD16 and ACE2 receptors.256 This response includes the activation of inflammasomes, which leads to the release of IL-1 and IL-18, and pyroptosis. All of these factors contribute to the hyperinflammatory state of the lungs. However, inhibiting the NLRP3 inflammasome pathway can help reverse the chronic lung damage caused by this response. When inhibited with MCC950, the virus was released by infected macrophages, which indicated that the NLRP3 gene is activated to prevent SARS-CoV-2 infection.258 Together, early treatment targeting NLRP3 inflammasome could improve the prognosis of COVID-19.
Acute respiratory distress syndrome (ARDS) is an inflammatory disease that is characterized by diffuse alveolar injury hypoxemia, and acute respiratory failure.259 ARDS can lead to the development of pulmonary edema due to increased permeability of pulmonary microvascular endothelium, which impairs lung tissue ventilation. Caspase-1 and IL-18 promote ARDS development, and circulating IL-18 levels have been associated with disease severity and mortality.260 The recognition of LPS by TLR4 activates the NLRP3 inflammasome, as well as IL-1R1 expression on alveolar macrophage surfaces via the MyD88/NF-κB dependent pathway.261 LPS-TLR4 signals alveolar macrophages that increase ARDS by upregulating IL-1β-IL-1RI signaling. There is also evidence that NLRP3-mediated pyroptosis plays a role in ARDS pathogenesis. Extracellular histones induce alveolar macrophage pyroptosis via the NLRP3/caspase-1 pathway, which exacerbates lung inflammation in ARDS.262
The end result of inflammatory pulmonary diseases is fibrosis. The NLRP3 inflammasome mediates pulmonary fibrosis through the IL-1β-IL-1Rs-MyD88-NF-κB signaling pathway.263 Moreover, studies also found that NLRP3 inflammasome could transform lung endothelial cells into epithelial-mesenchymal transition, promoting pulmonary fibrosis. Caspase-1 and IL-1β play vital roles in pulmonary fibrogenesis. Caspases-1enzyme cleavages the pro-IL-1β and allows secretion of IL-1β.264 Elevated IL-1β has a profibrotic effect, and usually occurs in combination with higher expression of IL-1Rs in fibrogenesis.265 A study on mouse primary lung fibroblasts showed that the NLRP3 inflammasome increases IL-1β production, leading to lung fibrosis when induced by bleomycin.266 Collectively, these investigations have helped to clarify the role of inflammasomes in the development of pulmonary fibrosis and may lead to the discovery of new treatment targets for various respiratory disorders.
Digestive disorders
Inflammasomes play a role in a variety of digestive disorders and have become a popular topic of research. Research focused on inflammasomes offers a better understanding of the mechanisms of several digestive diseases and conditions, including certain bacterial and viral infections, fatty liver disease (FLD), pancreatitis, and inflammatory bowel disease (IBD).
To date, the only harmful pathogenic bacterium that has been found to survive in human gastric mucosa is helicobacter pylori (HP). Some research suggests that inflammasome activation may be a contributing factor in the severity of HP infections. For example, one study found that NLRP3 and GSDMD levels were significantly higher in the gastric tissues of HP-infected individuals compared with healthy controls.267 In the innate immune cell neutrophils, inflammasome activation is stimulated by HP, which triggers K+ efflux and ROS production, resulting in an increase in IL-1β secretion.268 Notably, the elevated IL-1β levels were abolished in NLRP3-deficient neutrophils, suggesting that activation of the NLRP3 inflammasome plays an important role in the inflammatory response to HP. Additionally, NLRP3 knock-down or knock-out prevented gastritis in HP-infected mice.269 These findings suggest that HP bacteria may manipulate the machinery regulating the NLRP3 inflammasome to suppress the immune response.
Chronic infection with viral hepatitis is of high prevalence worldwide. Hepatitis B virus (HBV) is a viral infection that attacks the liver and can cause chronic hepatitis B (CHB).270 HBV-related acute-on-chronic liver failure patients have been shown to have higher levels of NLRP3, caspase-1, IL-1β, and IL-18 in their liver tissues.271 Moreover, the NLRP3, ASC, and IL-1β levels in liver tissues of CHB patients were positively correlated with the concentrations of HBV-DNA.272 This data suggests that long-term HBV infection activates the NLRP3 signaling pathway and promotes the IL-1β and IL-18-mediated injury of liver tissues. Similarly, patients with chronic hepatitis C have significantly increased serum IL-1β levels.273 Mechanistically, the Hepatitis C virus (HCV) RNA induces MyD88-mediated TLR7 signaling, activates the NLRP3 inflammasome pathway, and consequently triggers IL-1β production.273 Upon HCV infection, ASC binds to NLRP3, causing fragmentation of the Golgi.274 As a result, HCV replication increases and chronic liver inflammation occurs. Apart from antiviral agents, inhibiting the NLRP3 inflammasome and its associated cytokines could be a viable therapeutic approach to reduce liver inflammation.
FLD is a hepatic disease that results in the progressive buildup of fat in the liver. Patients with FLD have high serum caspase-1 levels, and these levels closely correlate with disease severity.275 Excessive fat accumulation in dead hepatocytes activates macrophages via NLRP3 and caspase-1.276 Researchers have shown that inhibiting pyroptosis through targeting NLRP3 inflammasomes can alleviate liver inflammation.277,278 NLRP3 inflammasome is implicated in the development of FLD in mice, and diet-induced steatohepatitis is prevented in mice lacking NLRP3 inflammasome function.279 The inhibition of the NLRP3 inflammasome significantly reduced inflammation, lipid accumulation, and fibrosis in FLD.280,281 In contrast, there is evidence that NLRP3 deficiency plays a harmful role via increasing serum alanine transaminase (ALT) and aspartate transaminase (AST) levels. FLD model mice lacking NLRP3 displayed a higher triglyceride content, liver injury scores, and adipose tissue inflammation.282 More research is necessary to explain the contradictory results mentioned above. Long-term heavy drinking can cause alcoholic liver disease, and patients with severe liver damage had higher mRNA levels of NLRP3, IL-1β, IL-18, and caspase-1 compared to patients with milder liver damage.283 Long-term alcohol intake facilitated liver damage and promoted NLRP3, ASC, caspase-1, and IL-1β expression in wild-type mice.284 Chronic alcohol consumption can cause metabolic disorders characterized by excess production of uric acid and ATP, which can trigger NLRP3 inflammasome activation and mitochondrial damage.285 Aryl hydrocarbon receptor downregulation and activation of TXNIP are the primary mechanisms responsible for ethanol-induced NLRP3 activation in human macrophages.286 Taken together, these studies suggest that interventions aimed at inhibiting NLRP3 inflammasome activation could help abate alcoholic liver disease.
Pancreatitis is an inflammatory condition that can be broken down into three main types: acute pancreatitis, severe acute pancreatitis, and chronic pancreatitis. NLRP3 plays a crucial role in pancreatic tissue inflammation. NLRP3, caspase-1, pro-IL-1β, and pro-IL-18 activation were observed in a mouse model of acute pancreatitis.287 Additionally, Inhibition of P2X7R could reduce chronic pancreatic inflammation and fibrosis by attenuating NLRP3-mediated IL-1β and IL-18 secretions in a mouse model of chronic pancreatitis.288 The NLRP3 inflammasome is also believed to contribute to the development of fibrosis in patients with chronic pancreatitis. It activates pancreatic stellate cells (PSCs), which release a large amount of extracellular matrix (ECM).289 By inhibiting NLRP3, PSC, activation and ECM deposition can be reduced, ultimately relieving pancreatic fibrosis.290 Pancreatic cells recognize PAMPs and DAMPs in tissue damaged by acute pancreatitis, triggering NF-κB and NLRP3 inflammasome expression, caspase-1 maturation,291 and pyroptosis.287 In mice with severe acute pancreatitis, NLRP3 deficiency was also found to alleviate inflammatory complications.292 These studies suggest that inflammasome-targeted treatments could help alleviate or even treat many cases of pancreatitis.
Inflammatory bowel disease (IBD) is a chronic, recurring gastrointestinal disorder in which no structural or biochemical abnormalities occur. Ulcerative colitis (UC), Crohn’s disease (CD), infectious agents, and environmental factors are risk factors for IBD.293 UC and CD patients had increased levels of NLRP3 and IL-1β.294 Moreover, NLRP3 inflammasome and IL-1β levels are increased in IBD patients’ mucosa, and these levels closely correlate with disease severity.295 The development of intestinal inflammation was impaired by the deletion and inhibition of NLRP3, which indicates that IBD is promoted by overactivation of the NLRP3 inflammasome.296 In contrast, studies have demonstrated that the NLRP3 inflammasome regulates mucosal immune responses and intestinal homeostasis.297 Proliferation of intestinal endothelial cells requires NLRP3-induced IL-18.298 NLRP3 inflammasome activation-induced IL-1β and IL-18 protect against colitis and colitis-associated tumorigenesis in mice.299 Whether the NLRP3 inflammasome contributes to IBD in a beneficial or pathogenic way is a topic of debate. The inconsistent results may stem from variations in experimental protocols, the strains of mice utilized, or dose-response effects in microbiota. Additionally, certain genetic mutations in the NLRP3 inflammasome also appear to play a role in the pathology of IBD. NLRP3, encoded by RS772009059 (R779C), in 3 patients showed early-onset IBD.56 A positive correlation was also found between the incidence of severe diseases and NLRP3 inflammasome activity in macrophages when R779C is present in DSS-induced acute colitis models.56 More studies on how the NLRP3 inflammasome regulates inflammation will inspire new approaches to therapeutics for IBD.
Urogenital disorders
Inflammasomes have been implicated in the pathology of urogenital disorders, including renal, cystic, prostate, and ovarian diseases. Acute kidney injury (AKI) is characterized by a sudden decrease in glomerular filtration rate and an increase in waste product accumulation, which can lead to ischemia-reperfusion injury (IRI). NLRP3, IL-1β, and IL-18 levels are elevated in IRI.300 The NLRP3 inflammasome plays a vital role in AKI. NLRP3 inflammasome activation and mitochondrial damage were detected in IR-induced AKI model mice, and damaged mitochondria further activate the NLRP3 inflammasome via the mROS-TXNIP-NLRP3 pathway.301 NLRP3 deletion protected the kidney from further inflammatory damage and injury.302 Moreover, pyroptosis also occurred in tubular epithelial cells of renal IRI mice.303 Another study discovered that protection from IRI was observed in NLRP3 knockout mice, but not in ASC knockout or caspase-1 knockout mice. This suggests that NLRP3 may directly impact renal IRI’s tubular epithelial cells.304 Additionally, P2X7R deficiency attenuates NLRP3 inflammasome formation and kidney injury.305 Cathelicidin-related antimicrobial peptide (CRAMP) deficiency promotes inflammatory responses and apoptosis via NLRP3 inflammasome overexpression.306 It indicates that CRAMP plays a protective role in the kidney by inhibiting the NLRP3 inflammasome activation. Additionally, NLRP6 also has been implicated in AKI. NLRP6 expression is downregulated when nephrotoxic kidney injury occurs.307 NLRP6 deficiency is believed to exacerbate the severity of AKI by inhibiting the phosphorylation of ERK1/2 and p38 MAPK, and suppressing the nephroprotective gene Klotho expression.308 NLRC4 expression is increased after IRI. Treatment with RMT3-23, a neutralizing antibody against T cell immunoglobulin domain and mucin domain-containing molecule-3 (Tim-3), decreases NLRC4 expression in IRI.309 This study suggests that Tim-3 mediates NLRC4 inflammasome activation in AKI. It has been observed that the expression of NLRC5 is elevated in mice that have undergone IRI.310 NLRC5 deficiency suppresses oxidative stress and apoptosis by promoting the PIK3/Akt signaling pathway in HK-2 cells.310 Mechanistically, NLRC5 downregulates ERK1/2 and Akt signaling, exacerbating the inflammatory response and apoptosis in tubular epithelial cells. Therefore, these findings present a novel perspective on therapeutic implications in AKI patients.
The progression of renal disease is accompanied by tubulointerstitial inflammation and fibrosis. Evidence shows that NLRP3 plays a key role in the progression of unilateral ureteral obstruction nephropathy via the NLRP3 inflammasome pathway.311 NLRP3 deficiency suppresses tubular injury, tubulointerstitial inflammation, and fibrosis in NLRP3-knockout mice, and these observations are in line with the inhibition of caspase-1 activity as well as IL-1β/IL-18 production.312 In contrast, it was also reported that NLRP3 has a protective effect on early tubular injury. NLRP3 mRNA levels are elevated in renal tubular epithelial cells, attenuating renal injury by preserving renal integrity.313 These controversial results indicate that NLRP3 functions distinctly at the different stages of obstruction nephropathy. NLRP3 is targeted by drugs like aliskiren, fluorofenidone, and mefunidone, which decrease NLRP3 inflammasome activity and suppress the release of IL-1β in obstruction nephropathy.314
Inflammation facilitates systemic lupus erythematosus-induced lupus nephritis. NLRP3 inflammasome is found in podocytes and contributes to cellular injury and proteinuria during lupus nephritis development.315 NLRP3 inflammasome-mediated IL-1β and IL-18 production have been shown to be elevated in the renal tissue and podocytes of mice with renal impairments.316 Mechanistically, NLRP3 inflammasome activation is regulated by RIP3 in podocytes.317 In addition, activation of glycogen synthase kinase 3β and P2X7R activate the NLRP3/IL-1β pathway and subsequently aggravate the development and progression of lupus nephritis.318 The cancer treatment drug tris dipalladium could inhibit MAPK (ERK, JNK)-mediated NLRP3 activation and the autophagy/NLRP3 pathway, alleviating tubulointerstitial inflammation and restoring renal function.319 These data suggest that the NLRP3 inflammasome plays an important role in lupus nephritis pathophysiology.
IgA nephropathy is a chronic, progressive glomerulonephritis characterized by the deposition of the IgA immune complex in the glomerular mesangium. NLRP3 levels are significantly upregulated in patients with IgA nephropathy, as well as in the IgA nephropathy mouse model.320,321 The IgA immune complex activates the NLRP3 inflammasome, leading to mitochondrial dysfunction and mROS overproduction in macrophages. NLRP3 deficiency attenuates renal injury in IgA nephropathy.322 Furthermore, there is evidence that IgA induces podocyte NLRP3 expression as well as macrophage trans-differentiation, which leads to renal fibrosis in IgA nephropathy.320 Intriguingly, patients with IgA nephropathy have a worse prognosis when their NLRP3 mRNA expression is low.323 Reduced levels of NLRP3 mRNA and protein in the tubules may reflect a loss of the tubular epithelial phenotype and cell death. Further investigation needs to be done to clarify the exact role of NLRP3 inflammasome in IgA nephropathy.
The NLRP3 inflammasome and IL-1β release have been implicated in the development of urinary tract infections. CFT073, a strain of uropathogenic Escherichia coli (UPEC), increased caspase-1 activity and promoted IL-1β release from bladder epithelial cells.324 The increase in IL-1β release was initially suppressed and later induced by the biphasic effect of α-hemolysin. This was mediated by α-hemolysin through NLRP3 inflammasome activation in an NF-κB-independent manner. These findings suggest that α-hemolysin can modulate the NLRP3 inflammasome activity in bladder epithelial cells. Urine contains many substances that can be potentially harmful if not eliminated, and this is particularly evident with bladder stones. Bladder stones are hardened mineral clumps that are composed of calcium pyrophosphate (CPPD) and monosodium urate (MSU). It has been found that CPPD and MUS can trigger caspase-1 in urothelial cells. However, the activation of caspase-1 can be reduced by NAC and Verapamil, which are ROS scavengers and recognized as down-regulators of TXNIP.325 This data suggests that the CPPD and MSU cause NLRP3 inflammasome activation in bladder urothelium, and this mechanism is dependent on ROS generation and TXNIP expression.
The NLRP3 inflammasome has also been implicated in bladder outlet obstruction. Any number of conditions, such as bladder stones, can cause bladder outlet obstruction. During bladder outlet obstruction in the urothelium, NLRP3 is activated, eventually leading to fibrosis and decompensation. However, an NLRP3 inhibitor called Glyburide can block the inflammation and prevent dysfunction of bladder outlet obstruction in its early stages.326 This suggests that the NLRP3 inflammasome plays a critical role in bladder outlet obstruction. A blocked bladder outlet stimulates fibrosis, which is responsible for chronic bladder decompensation in this condition. Research has demonstrated that IL-1β receptor antagonists can prevent collagen production in the bladder caused by bladder outlet obstruction, indicating the involvement of the NLRP3/IL-1β pathway in fibrosis.327 Additionally, IL-1β was found to stimulate collagen expression in isolated urothelial cells. These data suggested that NLRP3-mediated IL-1β release triggers fibrosis during bladder outlet obstruction by driving collagen production in urothelial cells.
Prostatitis is an inflammatory disease which has both infectious and non-infectious causes. In a study using rats, it was discovered that a hormone imbalance can cause chronic non-bacterial prostatitis.328 This condition leads to an increase in expression levels of NLRP3, ASC, and Caspase-1, causing inflammasome activation and inflammatory responses. However, melatonin was found to be effective in suppressing prostate inflammation and pelvic pain, by inhibiting the NLRP3 inflammasome signaling pathway. Additionally, the activation of Sirt1 was observed in an experimental autoimmune prostatitis mouse model, further demonstrating the potential therapeutic benefits of melatonin. In another study, Trichomonas vaginalis stimulation was shown to increase the expression of NLRP3, ASC, Caspase-1, and IL-1β, while inhibition of NLRP3 and Caspase-1 decreased the T. vaginalis-induced IL-1β secretion in a prostate epithelial cell line (RWPE-1).329 These studies provide evidence that the NLRP3 inflammasome is associated with the development of prostatitis.
Polycystic ovary syndrome is a type of infertility mainly caused by hyperandrogenism. In polycystic ovary syndrome models, ovarian TLR4 expression, as well as serum anti-Müllerian hormone, testosterone, caspase1, IL-1β, and insulin levels were increased.330,331,332 Mechanistically, activation of NLRP3 in upregulated the 3β-hydroxysteroid dehydrogenase and androgen receptor (AR) expression and downregulated the follicle-stimulating hormone receptor expression, inhibition of NLRP3 suppressed the expression of ASC, GSDMD-C, and AR.331 These results indicate that activating the NLRP3 inflammasome is crucial for the progression of hyperandrogen-induced polycystic ovary syndrome.
The NLRP3 inflammasome plays a role in ovarian aging and female fertility. NLRP3, caspase-1, and IL-1β expression were increased in granulosa cells from mice with ovarian insufficiency. Additionally, NLRP3 expression increases in the ovary as wild-type mice age.333 Ablation of NLRP3 leads to improved pregnancy and survival rates, as well as hormone levels in the ovaries of these mice.333 These findings suggest that elevated NLRP3 inflammasome may contribute to age-related deficits in female fertility. Additionally, endometriosis is an estrogen-dependent chronic inflammatory syndrome. It can lead to infertility, and hormone-based treatments are usually used to treat it. Research suggests that the NLRP3 inflammasome could be involved in the development of endometriosis. Studies have shown that NLRP3 levels are higher in ovarian endometriosis samples, and using MCC950 to inhibit NLRP3 has been shown to decrease IL-1β concentrations in cyst-derived stromal cells.334 The results suggested that NLRP3/IL-1β is crucial for the pathogenesis of endometriosis.
Blood and lymphatic system disorders
A growing number of researchers are exploring the effect that aberrant inflammasome activation has on disorders of the blood and lymphatic system. To date, several studies suggest that inflammasomes play a role in the pathogenesis of leukemia, myeloproliferative neoplasms, myelodysplastic syndrome (MDS), lymphoma, and autoimmune diseases.
Leukemia refers to a group of clonal hematological diseases that affect the maturation and proliferation of myeloid cells and lymphocytes. Leukemia has both acute and chronic forms, including acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML). Studies have reported that dysregulated IL-1β secretion positively correlates with disease progression and poor prognosis in leukemia.335 Research has identified the KrasG12D mutation as a genetic risk factor for Leukemia. It has been observed that this mutation can activate the NLRP3 inflammasome, resulting in myeloproliferation and cytopenia. Nevertheless, experiments with KrasG12D murine models have demonstrated that NLRP3 deficiency can reverse these effects.336 Patients newly diagnosed with AML have also been found to have increased NLRP3 expression in their bone marrow mononuclear cells and their peripheral blood mononuclear cells (PBMCs).337 Glucocorticoids are often used to treat patients with ALL. NLRP3 and caspase-1 are significantly higher in ALL cells resistant to glucocorticoids, as caspase-1 cleaves the glucocorticoid receptor.338 In contrast, NLRP3 expression was significantly lower in CLL lymphocytes than in healthy donors, whereas P2X7R expression was higher.339 Aside from activating NLRP3 inflammasomes, P2X7R also inhibits apoptosis and promotes cell proliferation.339 NLRP3 downmodulation triggers P2X7R expression, which consequently leads to tumor growth. In addition, curcumin could induce the expression of AIM2, NLRC4, and IFI16 inflammasomes in leukemia cells U937, which subsequently activated caspase 1, promoted GSDMD cleavage, as well as induced pyroptosis.340 Altogether, targeting the inflammasomes could have therapeutic effects on leukemia.
Myeloproliferative neoplasms include polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Analysis of patients showed that the IL-1β levels were increased in all three different types of myeloproliferative neoplasms.341 Moreover, JAK2V617F positive macrophages produced greater IL-1β and IL-18, which promoted the production and activation of neutrophils and the entry of leukocytes into lesion.342 Additionally, AIM2, IL-1β, and caspase-1 were significantly increased in JAK2V617F positive cells.343 This data indicates that inflammasomes are vital in the pathogenesis of myeloproliferative neoplasms. MDS describes a group of malignant preleukemic HSC malignancies resulting from abnormal and ineffective hematopoiesis. The activation of the NLRP3 inflammasome was suggested as contributing to MDS. The alarmin S100A9 could induce ROS generation, which subsequently activates the NLRP3 inflammasome, leading to IL-1β secretion and pyroptosis.344
It has been reported that inflammasomes also play a role in lymphoma development. The NLRP3 inflammasomes are implicated in numerous cancers as a pro-tumorigenic factor. Patients with diffuse large B cell lymphoma (DLBCL) exhibit immunosuppression. This is characterized by significantly increased levels of IL-18 in lymphoma tissues, which is positively correlated with the expression of programmed death ligand 1 (PD-L1).345,346 PD-L1 is induced by NLRP3 inflammasome activation, and this reduces the proportion of cytotoxic T cells in DLBCL cell lines. However, in vivo blockade of the NLRP3 inflammasome inhibits lymphoma growth and suppresses anti-tumor immunity. This is achieved by decreasing the expression of PD-L1 in the tumor microenvironment and downregulating the proportion of PD-1/TIM-3-expressing T cells, regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages. It has been shown that the NLRP3 inflammasome regulates PD-L1 and immune cells to promote immunosuppression, while IL-18 negatively impacts anti-lymphoma immunity in vivo. Patients with Sjögren’s syndrome (SS) have hyperfunctioning P2X7R, which triggers acute inflammatory responses through the NLRP3 inflammasome.347 SS patients may develop mucosa-associated lymphoid tissue non-Hodgkin’s lymphoma (MALT-NHL). P2X7R, NLRP3, caspase-1, and IL-18 expression were higher in patients with germinative centers and autoantibody-positive individuals and were significantly higher in patients who developed a MALT-NHL during follow-up.347 Primary cutaneous T cell lymphomas most commonly occur as mycosis fungoides (MF), NLRP1 expression was increased in the early stages of MF, whereas caspase 1, IL-1β, and IL-18 levels were increased during both the early and late stages of MF formation.348 Intriguingly, in cutaneous T cell lymphoma (CTCL), unassembled NLRP3 can translocate to the malignant CD4 + T cells nucleus, and bind to human IL-4 promoter to regulate the IL-4 expression, IL-4 can inhibit the NLRP3 inflammasome assembly.349 It has been discovered that infections caused by Kaposi’s sarcoma-associated herpesvirus (KSHV) in B and endothelial cells are linked to the development of Kaposi’s sarcoma and primary effusion B-cell lymphomas, respectively. During primary infection of KSHV in endothelial cells, B cells in Kaposi’s sarcoma endothelial and primary effusion B-cell lymphomas can secrete IL-1β and IL-18 upon caspase-1 activation. It has been found that IFI16 interacts with ASC, but not AIM2 or NLRP3, and subsequently procaspase-1, to form an effective inflammasome in KHSV.350 Another study reported that mutations in IL-18 (rs1946518) and NFκB-94 ins/del (rs28362491) promoted the susceptibility to B-cell NHL.345,351 Additionally, allele G in IL-18 was shown to be remarkably associated with the risk of lymphoma.345 These findings show that specific inflammasomes play different roles in lymphoma development.
Patients with systemic lupus erythematosus (SLE) suffer from disordered Th1/Th2 and Treg/Th17 balances, evidence has demonstrated that NLRP3, NLRP1, NLRC4, and AIM2 were involved in the regulation of Th1, Tfh, and Th17 cell-mediated immune responses. Leptin contributes to SLE development by activating the NLRP3 inflammasome and promoting Th17 cell differentiation in lupus erythematosus mice.352 SLE is identified by the presence of anti-dsDNA antibodies, which can be measured using an antinuclear (ANA) test. Additionally, in human monocytes, anti-dsDNA antibodies can activate the NLRP3 inflammasome and cause the secretion of IL-1β, further contributing to the pathogenesis of SLE.353,354 Research has shown that AIM2 can encourage the production of B cells in patients with SLE.355 People with SLE tend to have higher levels of AIM2 mRNA in their liver, PBMCs, and spleen than healthy individuals. AIM2 has also been found to help prevent SLE-inhibiting DNA-induced IFN signals.356 However, some studies suggest that AIM2 may contribute to the development of Th17 cells in SLE. In mice, inhibiting AIM2 expression has been found to greatly improve SLE symptoms.355,357 While there is research on the roles of NLRP3 and AIM2 in SLE, the functions of other inflammasomes in the disease are not yet fully understood.
Rheumatoid Arthritis (RA) is an autoimmune disease characterized by immune dysregulation and joint inflammation. Patients with RA have elevated levels of NLRP3 and IL-1β secretion in their peripheral blood mononuclear cells (PBMC),358 as well as an increase in IL-18 in their bronchoalveolar lavage fluid (BALF) if they have RA-recurrent interstitial pneumonia (RA-UIP).359 NLRP3 deficiency has been found to inhibit Th17 cell differentiation in RA patients, indicating that NLRP3 promotes the differentiation of Th17 cells, which can exacerbate inflammation.360 Mechanistically, calcium-sensitive receptors (CaSR) in RA patients can activate the NLRP3 inflammasome, releasing IL-1 and promoting joint swelling.361 PTX3 and complement C1q promote NLRP3 inflammasome hyperactivation and scorching in RA patients.362 Additionally, researchers are increasingly studying AIM2 inflammasomes as cytoplasmic receptors in RA. It was observed that the levels of serum AIM2 were lower in RA patients compared to healthy controls; however, ASC, caspase-1, and IL-1β levels were higher.363 Individuals diagnosed with RA have elevated levels of mDNA in their plasma and synovial tissue compared to healthy individuals.364 They also have a higher chance of activating AIM2 inflammasomes. In addition, the adjuvant arthritis rat model showed activation of the NLRP1 inflammasome.365 Monocytes from RA patients were found to have an increased expression of NLRC4.366 However, more research is needed to determine the exact role of inflammasomes and their association with the susceptibility and severity of RA.
The use of inflammasome-targeting therapy has emerged as a possible therapeutic strategy for blood and lymphatic system disorders. The associations between blood and lymphatic system disorders and the inflammasomes are active research areas. It is imperative to conduct further research to fully understand the role of inflammasomes in these disorders and to develop effective treatments for them.
Other disorders
In addition to the disease mentioned above, other disorders are associated with abnormal inflammasome activity, including gout, diabetes, and obesity. Gout is a common disorder characterized by the MSU crystals depositing in the articular and non-articular structures. MSU crystals are a typical DAMP that induced the NLRP3 inflammasome activation, and the IL-1β release is vital in the initiation of inflammatory gout attacks or flares.367 MSU stimulation resulted in decreased knee neutrophil infiltration in mice lacking critical components of inflammasomes (NLRP3, ASC, or caspase-1).368 An endogenous DAMP, cold-inducible RNA-binding protein, activates MSU-stimulated neutrophil infiltration via an NLRP3/ASC/caspase-1/IL-1β/MyD88 pathway-mediated CXC-motif receptor 2-dependent process.369
Diabetes type 2 (T2D) is a chronic inflammatory disease characterized by insulin resistance, upregulated circulating TNF, interleukin, and adipokines levels. IL-1β hinders insulin sensitivity by phosphorylating insulin receptor substrate-1, leading to the insulin-induced PI3K-Akt signaling disruption in insulin-targeted cells.370 Studies have shown that individuals with T2D have higher levels of NLRP3 inflammasome activity in their myeloid cells than those without the condition.371 Research conducted on mice without NLRP3, ASC, or caspase-1 genes has revealed that they exhibit better glucose tolerance and insulin sensitivity when consuming high-fat diets.372,373 A 37-amino-acid peptide hormone, islet amyloid polypeptide, released by β-cells along with insulin, can form an amyloid structure in the pancreatic islets of T2D patients.374 It has been found that endocannabinoids can trigger the production of IL-1β through the NLRP3 inflammasome via the peripheral CB1 receptor, which can result in the death of pancreatic β-cells.375 In rat islets treated with anandamide, an endocannabinoid, levels of ASC and caspase-1 activation were increased, and IL-1β secretion was upregulated in mouse macrophage cell line RAW264 in an NLRP3-dependent manner. In addition, it has been found that saturated fatty acids can also trigger T2D by activating the NLRP3 inflammasome.376 The NLRP3 inflammasome can sense ceramide, which is a type of saturated fatty acid, leading to the activation of caspase-1 in mouse bone marrow-derived macrophages and epididymal adipose tissue explants.377 On the other hand, unsaturated fatty acids have been shown to enhance insulin sensitivity by decreasing the production of IL-1β.276
Obesity is an adipocyte hypertrophy characterized in part by immune cell infiltration-induced adipose tissue expansion. Obesity can increase multiple metabolic disorders. In obese patients, NLRP3 and ASC/PYCARD expression is increased.372 Research has shown that the inhibition of IL-1β, but not IL-18, can enhance adipogenic gene expression. This indicates that caspase-1 plays a role in regulating adipogenesis through IL-1β.378 Studies have also revealed that caspase-1 is essential for the formation of adipose tissue. In mice that lacked caspase-1, there was a decrease in adipocyte size, a reduction in fat mass, an increase in the expression of adipogenic genes, and an improvement in insulin sensitivity.379 In addition, NLRP3-, ASC-, and caspase-1-deficient mice were found to be protected against obesity induced by a high-fat diet.380 Researchers also have shown that mice lacking IL-18 developed obesity due to increased food intake.381 Further studies are needed to clarify the mechanism and accurate role of the inflammasomes and caspase-1 activation in adipocytes and the pathogenesis of obesity.
Inflammasome-targeted therapy
Different inflammasomes are increasingly recognized in various diseases, thus targeting inflammasome signaling pathways has the potential to develop new strategies for therapeutic intervention (Fig. 5). Currently, the FDA-approved drugs primarily target inflammasome-related pathways rather than directly targeting inflammasomes themselves (Table 1). Thus, more researches, however, are being conducted in the development and evaluation of inflammasome-targeting therapies (Table 2).

Inhibitors of the inflammasomes and inflammasome-related pathways. Distinct inhibitors target NLRP1, NLRP3, AIM2, ASC, caspase-1, interleukin-1β, interleukin-18, or GSDMD to inhibit the inflammasome activation and subsequent pyroptosis and cytokine release. The figure was created with the assistance of FIGDRAW
Sensor protein modulation
In recent years, there has been significant interest in developing inhibitors of inflammasome sensor proteins as potential therapeutics for inflammatory diseases. Cytosolic DPP9 binds to NLRP1 C terminus and inhibits inflammasome activation.382,383 Recent studies have revealed that the NLRP3 inhibitor MCC950 can effectively inhibit the activation of NLRP3 by directly targeting its NACHT domain.384 Specifically, MCC950 has been shown to block the Walker B motif within the NACHT domain of NLRP3, thus preventing ATP hydrolysis by NLRP3. Tranilast is a drug used for allergies that can prevent NLRP3 assembly by binding directly to its NACHT domain and preventing it from forming. Doing this also stops direct interactions between NLRP3 molecules and disrupts their natural interaction with ASC.385 Another drug, CY-09, interacts with the NLRP3 Walker A motif and removes ATP bound to NLRP3 without affecting NLRP1 or NLRC4.386 N-benzyl 5-(4-sulfamoylbenzylidene-2-thioxothiazolidin-4-one analogs), which are hybrids of CY-09, selectively inhibit the formation of oligomer specks of NLRP3 and ASC. This reduces the assembly of the NLRP3 inflammasome.387 Oridonin interacts with Cys279 of the NLRP3 NACHT domain via a covalent bond, inhibiting NLRP3-NEK7 interactions and attenuating the NLRP3 inflammasome activation.388 Tetrahydroquinoline, a synthesized compound, inhibits the NLRP3 inflammasome assembly and activation by binding to the NLRP3 NACHT domain and blocking ASC oligomerization.389 OLT1177, a β-sulfonyl nitrile compound, binds to NLRP3 directly to inhibit its ATPase activity, as well as to suppress caspase-1 activity and IL-1β production in monocytes from patients with cryopyrin-associated periodic syndrome.390 OLT1177 demonstrated excellent safety and tolerability in the phase I trial in heart failure and reduced ejection fraction in patients.391 β-Carotene binds to NLRP3’s PYD, suppressing NLRP3 inflammasome activation in macrophages induced by ATP, MSU crystals, and nigericin.392 4-Methylenedioxy-β-nitrostyrene binds to NACHT and LRR domains of NLRP3 to inhibit its ATPase activity.393 Lycorine disrupts the interaction of NLRP3 with ASC by targeting the PYD on Leu9, Leu50, and Thr53.394 Anticancer agent tivantinib directly inhibits the NLRP3 ATPase activity and the subsequent assembly of the NLRP3 inflammasome complex.395 Bay 11-7082 was demonstrated to inhibit the ASC pyroptosome and the NLRP3 inflammasome organization via cysteine alkylation in the ATPase region of NLRP3.396 However, Bay 11-7082 also suppresses the IKKβ kinase activity, resulting in the modulation of NLRP3 expression via NF-κB pathway.397 4-hydroxynonenal, a lipid peroxidation product, disrupts the interaction between NLRP3 and NEK7 via directly binding NLRP3.398 NIC-0102, an orally bioavailable proteasome inhibitor, promotes the NLRP3 polyubiquitination, disrupts the NLRP3-ASC interaction, and blocks ASC oligomerization, thus inhibiting activation of the NLRP3 inflammasome.399 INF39 suppresses the NEK7-NLRP3 interaction and, in turn, inhibits NLRP3-NLRP3 and NLRP3-ASC interactions, as well as ASC oligomerization and speckle formation.400
PYD-only protein POP3, an inhibitor of AIM2 inflammasomes, competes with ASC for AIM2 recruitment.401 Obovatol, a bisphenol chemical, inhibits the ASC pyroptosome formation and suppresses the NLRP3 and AIM2 inflammasome.402 RGFP966, a selective inhibitor of histone deacetylases 3, regulates the AIM2 spatiotemporally.403 DNA aptamers, small single-stranded DNA or RNA molecules, have the potential to bind to AIM2 and suppress its inflammasome activity.404 Roxadustat (FG-4592) inhibits AIM2 in a CD73-dependent manner.405
Recent research has found some products derived from medicinal plants or bioactive natural products can act as inhibitors of NLRP3 or AIM2 inflammasome. These will be valuable candidates for treating inflammasome-related diseases. Costunolide, the major active ingredient in the Chinese traditional medicinal herb Saussurea lappa, covalently binds to Cys598 in the NACHT domain of NLRP3 via the α-methylene-γ-butyrolactone motif in costunolide. This inhibits NLRP3 ATPase activity and NLRP3 inflammasome assembly.406 EFLA-945, an extract from red grapevine leaf, limits the DNA entry into THP-1-derived macrophages, thus inhibiting the AIM2 inflammasome activation.407 Ethanolic extracts derived from seeds of Cornus officinalis suppress AIM2 speck formation induced by dsDNA.408
ASC modulation
As ASC is the adaptor protein of canonical inflammasomes, targeting ASC could also regulate inflammasome activation. J114, a chemical compound, disrupted interactions of NLRP3 or AIM2 with ASC and suppressed ASC oligomerization.409 It has been found that Caffeic acid phenethyl ester (CAPE) can directly bind to ASC, thereby preventing NLRP3-ASC interactions that are triggered by MSU crystals.410 A study involving a mouse model of gouty arthritis induced by MSU crystals showed that oral administration of CAPE resulted in a decrease in caspase-1 activation and IL-1β release in foot tissue and air pouch exudate. VHHASC, a type of alpaca single-domain antibody, impairs interactions between ASC and CARD by leaving the PYD of ASC functional, and by stabilizing an intermediate filament of inflammasome activation.411 Lonidamine, a small-molecule glycolysis inhibitor, directly binds to ASC and inhibits its oligomerization.412 Dehydrocostus lactone, a main component of traditional Chinese medicine Saussurea lappa, blocked the ASC oligomerization.413 Sulforaphane may directly disrupt the formation of NLRP3 via suppressing ASC or Caspase-1.414 Brevilin A, a natural ingredient derived from Centipeda minima, decreases the NLRP3 inflammasome activation by blocking the ASC oligomerization.415 1,2,4-trimethoxybenzene, an active ingredient obtained from essential oils, inhibited ASC oligomerization as well as protein-protein interaction between NLRP3 and ASC.416
Caspase modulation
Recently, the pharmaceutical industry has focused on developing inhibitors of caspase-1 protease, a component of all canonical inflammasomes. A substance called VX-765, also known as belnacasan, can help lessen the severity of AD by selectively inhibiting caspase-1.417 This is achieved through the covalent modification of catalytic cysteine residues. In addition, VX-765 has been found to improve cognitive impairments associated with AD and preserve ventricular functions, thereby ameliorating myocardial infarction in mice. Unfortunately, long-term exposure to VX-765 in animals has been shown to cause hepatotoxicity, preventing further development of the substance.417 Sennoside A, an ingredient in dietary supplements and weight-loss medicines, inhibits the enzymatic activity of caspase-1 to downregulate the NLRP3 and AIM2 inflammasome-involved inflammation dependent on P2X7.418 Pralnacasan, an orally absorbed nonpeptide compound, can inhibit caspase-1.419 Pralnacasan was shown to reduce joint damage in a collagenase-induced osteoarthritis mouse model, suggesting it could be used to treat osteoarthritis.419 Pralnacasan has also been shown to attenuate dextran sulfate sodium-induced murine colitis with little to no side effects.420
IL-1/IL-18 modulators
IL-1β and IL-18 are the major inflammasome-activated inflammatory cytokines and play a significant role in the pathogenesis of several diseases. The biological anti-IL-1 agents have been approved for clinical application. Anakinra is a type of medication used to treat inflammatory diseases. It works as a recombinant interleukin-1 receptor antagonist.421 Patients with RA who are treated with anakinra experience a decrease in disease activity.422 A small randomized trial conducted at multiple centers found that anakinra significantly improved inflammatory and glycemic parameters in patients with both RA and T2D (NCT02236481).423 Canakinumab, which is a monoclonal antibody that targets human anti-IL-1β, has been approved by the US FDA and the European Medicines Agency as a treatment for cryopyrin-associated periodic syndromes.424 In a randomized, double-blind trial, canakinumab was found to be effective in reducing the recurrence rate of cardiovascular events compared to control group.425 Notably, anakinra requires more frequent injections and has a shorter half-life of only 4–6 h. Canakinumab, on the other hand, has a longer half-life of 26 days and has been shown in a randomized, double-blind clinical trial to provide better treatment response and increased safety for patients with active RA (NCT00784628). Rilonacept, an IL-1 inhibitor (IL-1 Trap) that is mainly used for the treatment of gout in children, was recently approved for a phase 3 trial in recurrent pericarditis.426,427 Gevokizumab, an IL-1β binding protein, possesses unique allosteric modulating properties.428 Patients with inflammatory and other diseases may benefit from gevokizumab.
IL-18 blockers are currently under investigation through clinical trials as well. One of these blockers is GSK1070806, which is a recombinant human IL-18 neutralizing antibody.429 A phase 1 clinical study was conducted on healthy and obese males with normal immune systems to determine the safety, efficacy, and antibody metabolism rate of GSK1070806. The study showed that GSK1070806 was generally well-tolerated, with positive results.430
GSDMD modulators
GSDMD is the principal mediator of pyroptosis and directly triggers pyroptosis when inflammasomes are activated, making it a potential target for treating inflammasome-related diseases. Disulfiram is a drug that has been approved by the FDA for treating alcoholism. It works as an inhibitor of pore formation that is induced by GSDMD. This drug also blocks pore formation by covalently modifying Cys191/Cys192 in human/mouse GSDMD.431 When mice are treated with disulfiram, they show reduced pyroptosis, cytokine release, and LPS-induced septic death. It has been discovered that Necrosulfonamide can prevent the clustering of GSDMD by attaching to the Cys191 amino acid in GSDMD.432 This, in turn, reduces the incidence of neuronal pyroptosis triggered by Aβ in vivo. Additionally, Dimethyl fumarate can also impede GSDMD clustering and oligomerization.433 Further research is required to investigate the potential of GSDMD inhibitors for treating various ailments.
Other modulators
In addition to the methods mentioned, other substances can affect inflammasomes by targeting the signaling pathways related to inflammasome activation. One of these substances, JC2-11, is a type of benzylideneacetophenone derivative that has shown promise as a potential pan-inflammasome inhibitor. Early research suggests that it can prevent caspase-1 cleavage, IL-1β release, GSDMD cleavage, and the production of mROS.433 Licochalcone B, a component of licorice, binds to NEK7 to suppress NLRP3 inflammasome activation by decreasing the interaction between NLRP3 and NEK7.434 Furthermore, pristimerin and ginsenoside Rg3 prevent the interaction between NEK7 and NLRP3, leading to reduced interaction between NLRP3 and ASC, decreased ASC oligomerization, and ultimately, reduced speckle formation.435,436 Methyl gallate inhibits the NLRP3 inflammasome assembly by blocking the ROS over-generation and NLRP3 oligomerization.437 5-androstenediol promotes NF-κB signaling and suppresses inflammasome-mediated pyroptosis.438 Riboflavin downregulates mROS production and mDNA release, thus inhibiting the NLRP3 inflammasome assembly.439 Dihydroartemisinin induces autophagy to inhibit the AIM2 inflammasome and NF-κB/HIF-1α/VEGF pathways.440 ML365, the most potent TWIK2 channel blocker, inhibits ATP-induced NLRP3 inflammasome.441 Curcumin down-regulates NLRP1, caspase-1, GSDMD, and IL-1β in oxygen-glucose deprivation and middle cerebral artery occlusion models.442 Clinical trials found that N‐acetylcysteine, an antioxidant and anti‐inflammatory reagent, can reduce macrophagic expression of NLRP3, decrease IFN-γ production in NK cells, and subsequently reduce the synthesis and release of IL‐18.443 Glucocorticoids are widely used in treating COPD and AECOPD. A new anti-inflammatory drug, 17-oxo-DHA, when combined with hormones can inhibit the activation of the NLRP3 inflammasome and the release of mature IL-1β.444 Despite attempts to treat COPD patients with therapies targeting inflammasome-related effectors at moderate to severe stages, some randomized clinical trials have not shown significant benefits. For example, when COPD patients were infused with a human anti-IL-1β monoclonal antibody called canakinumab for 45 weeks, their forced expiratory volume in 1 s and forced vital capacity did not improve when compared to a placebo group. These results suggest that targeting inflammasome-related effectors may not have the desired effects. Therefore, large-scale, well-designed studies are necessary to draw a definitive conclusion. There are also some therapeutic candidates that are capable of inhibiting inflammasome activation but their mechanism is not currently understood. Fimasartan, phenolic and quinonemethide nor-triterpenes, dihydrotanshinone I, aryl sulfonamide derivatives, and tertiary sulfonylurea compounds could inhibt the NLRP3 or AIM2 inflammasome activation.445,446,447,448,449
Conclusions
Inflammasomes play an irreplaceable role in multiple diseases. Understanding the mechanisms of activation and assembly of different inflammasomes and their functions in various conditions will provide valuable insights for effective intervention strategies in the future. This review introduced different inflammasomes, their structures, activation mechanisms, how they impact various diseases, and the potential therapeutic targets and intervention strategies. Inflammasomes are multiprotein complexes that play a vital role in regulating inflammatory and immune responses. Various types of inflammasomes have been identified, each characterized by different protein components. Understanding the distinct architectures of these inflammasomes is essential for elucidating their specific functions and developing targeted therapies for inflammatory diseases. Advanced structural studies on different inflammasomes have provided us with a brand-new perspective and approach to studying the mechanisms of inflammation activation. Inflammasome activation is associated with a conformation change of different sensor proteins, and the inflammasome assembly via homotypic interactions. Different inflammasomes are activated by specific stimuli, which trigger their assembly and the activation of caspase-1, leading to the maturation of proinflammatory cytokines like IL-1β and IL-18. The canonical pathway triggers inflammasome activation in response to a two-step process. The first step involves the activation of PRRs which detect PAMPs or DAMPs and activate NF-κB to induce transcription of NLRP3 and proinflammatory cytokines. Once NLRP3 has been upregulated, the second step involves the assembly of the inflammasome complex and subsequent caspase-1 activation. The non-canonical pathway of inflammasome activation is triggered by the activation of caspase-4 or -5 (caspase-11 in mice) in response to cytosolic LPS from Gram-negative bacteria. Caspase-4/5 cleaves GSDMD, leading to the release of proinflammatory cytokines and pyroptosis. Alternative pathways include the activation of inflammasomes by other PRRs like RIG-I-like receptors (RLRs) or DNA sensors like AIM2. These pathways involve different inflammasome components and mechanisms for their activation. Understanding the different inflammasome activation pathways helps develop potential therapies for multiple diseases. New research has made important strides in comprehending how certain inflammasomes contribute to various human ailments. This knowledge can be useful in discovering fresh treatments for metabolic, neurodegenerative, and inflammatory conditions. It is essential to identify the roles of specific inflammasomes in systemic diseases, so that targeted therapeutic approaches can be developed to regulate their activities and reduce inflammation. The identification of therapeutic targets and the creation of inflammasome-targeted strategies have yielded encouraging results in both lab and clinical trials.
There are still many unanswered questions in the research field of inflammasomes. The activation of inflammasomes has different pathways, including canonical and non-canonical, with multiple processes involved. The intricate process of inflammasome activation also involves multiple signaling pathways. Understanding these mechanisms fully remains a challenge. Although there are some inhibitors available, the development of highly selective and potent inhibitors targeting specific inflammasome components is still in progress. Additionally, the regulatory mechanisms controlling inflammasome activity and its interplay with other immune pathways, such as autophagy and cytokine signaling, are not fully understood. Animal models used to study inflammasome-related diseases may not fully replicate human physiology, making it difficult to apply findings from animal models to human clinical settings. Moreover, it can be difficult to determine the specific contributions of different inflammasomes in disease states due to their overlapping functions. While inflammasomes have been linked to various diseases, the use of inflammasome-targeted therapies in clinical practice is currently limited. Further research is required to bridge this gap and enhance our knowledge of inflammasome biology. This could lead to the development of safe and effective treatments that modulate inflammasome activity in disease settings. More investigations are necessary to fully understand the role of inflammasomes in disease pathogenesis and to address any controversies. As research in this area continues, we can expect to see better clinical outcomes and a greater understanding of the relationship between inflammation and human diseases.
References
Taguchi, T. & Mukai, K. Innate immunity signalling and membrane trafficking. Curr. Opin. Cell Biol. 59, 1–7 (2019).
Hanazawa, S. et al. Functional role of interleukin 1 in periodontal disease: induction of interleukin 1 production by Bacteroides gingivalis lipopolysaccharide in peritoneal macrophages from C3H/HeN and C3H/HeJ mice. Infect. Immun. 50, 262–270 (1985).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Watson, R. W., Rotstein, O. D., Parodo, J., Bitar, R. & Marshall, J. C. The IL-1 beta-converting enzyme (caspase-1) inhibits apoptosis of inflammatory neutrophils through activation of IL-1 beta. J. Immunol. 161, 957–962 (1998).
Dinarello, C. A. Unraveling the NALP-3/IL-1beta inflammasome: a big lesson from a small mutation. Immunity 20, 243–244 (2004).
Inohara, N. et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J. Biol. Chem. 274, 14560–14567 (1999).
Ogura, Y. et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 276, 4812–4818 (2001).
Wagner, R. N., Proell, M., Kufer, T. A. & Schwarzenbacher, R. Evaluation of Nod-like receptor (NLR) effector domain interactions. PLoS One 4, e4931 (2009).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Cartwright, N. et al. Selective NOD1 agonists cause shock and organ injury/dysfunction in vivo. Am. J. Respir. Crit. Care Med. 175, 595–603 (2007).
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M. & Schwarzenbacher, R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One 3, e2119 (2008).
Sutterwala, F. S. et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med 204, 3235–3245 (2007).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Khare, S. et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36, 464–476 (2012).
Zhu, S. et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017).
Maharana, J. Elucidating the interfaces involved in CARD-CARD interactions mediated by NLRP1 and Caspase-1 using molecular dynamics simulation. J. Mol. Graph Model 80, 7–14 (2018).
Mayor, A., Martinon, F., De Smedt, T., Petrilli, V. & Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 8, 497–503 (2007).
de Alba, E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 284, 32932–32941 (2009).
Lu, A. et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014).
Kostura, M. J. et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc. Natl. Acad. Sci. USA 86, 5227–5231 (1989).
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298 (2015).
Albrecht, M., Choubey, D. & Lengauer, T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem. Biophys. Res Commun. 327, 679–687 (2005).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Veeranki, S., Duan, X., Panchanathan, R., Liu, H. & Choubey, D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS One 6, e27040 (2011).
Damiano, J. S., Newman, R. M. & Reed, J. C. Multiple roles of CLAN (caspase-associated recruitment domain, leucine-rich repeat, and NAIP CIIA HET-E, and TP1-containing protein) in the mammalian innate immune response. J. Immunol. 173, 6338–6345 (2004).
Choubey, D. et al. Interferon-inducible p200-family proteins as novel sensors of cytoplasmic DNA: role in inflammation and autoimmunity. J. Interferon Cytokine Res 30, 371–380 (2010).
Davis, B. K. et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 186, 1333–1337 (2011).
Kempster, S. L. et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G253–263, (2011).
Marleaux, M., Anand, K., Latz, E. & Geyer, M. Crystal structure of the human NLRP9 pyrin domain suggests a distinct mode of inflammasome assembly. FEBS Lett. 594, 2383–2395 (2020).
Hiller, S. et al. NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure 11, 1199–1205 (2003).
Finger, J. N. et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 287, 25030–25037 (2012).
Jin, T., Curry, J., Smith, P., Jiang, J. & Xiao, T. S. Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins 81, 1266–1270 (2013).
Robert Hollingsworth, L. et al. Mechanism of filament formation in UPA-promoted CARD8 and NLRP1 inflammasomes. Nat. Commun. 12, 189 (2021).
Gong, Q. et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat. Commun. 12, 188 (2021).
Xu, Z. et al. Homotypic CARD-CARD interaction is critical for the activation of NLRP1 inflammasome. Cell Death Dis. 12, 57 (2021).
D’Osualdo, A. et al. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One 6, e27396 (2011).
Huang, M. et al. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 592, 773–777 (2021).
Moecking, J. et al. NLRP1 variant M1184V decreases inflammasome activation in the context of DPP9 inhibition and asthma severity. J. Allergy Clin. Immunol. 147, 2134–2145.e2120 (2021).
Moecking, J. et al. Inflammasome sensor NLRP1 disease variant M1184V promotes autoproteolysis and DPP9 complex formation by stabilizing the FIIND domain. J. Biol. Chem. 298, 102645 (2022).
Rahman, T. et al. NLRP3 sensing of diverse inflammatory stimuli requires distinct structural features. Front Immunol. 11, 1828 (2020).
Sahoo, B. R. et al. A conformational analysis of mouse Nalp3 domain structures by molecular dynamics simulations, and binding site analysis. Mol. Biosyst. 10, 1104–1116 (2014).
Brinkschulte, R. et al. ATP-binding and hydrolysis of human NLRP3. Commun. Biol. 5, 1176 (2022).
Samson, J. M. et al. Computational Modeling of NLRP3 Identifies Enhanced ATP Binding and Multimerization in Cryopyrin-Associated Periodic Syndromes. Front Immunol. 11, 584364 (2020).
Bae, J. Y. & Park, H. H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536 (2011).
Oroz, J., Barrera-Vilarmau, S., Alfonso, C., Rivas, G. & de Alba, E. ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces. J. Biol. Chem. 291, 19487–19501 (2016).
Hochheiser, I. V. & Geyer, M. An assay for the seeding of homotypic pyrin domain filament transitions. Methods Mol. Biol. 2523, 197–207 (2022).
Isazadeh, M. et al. Split-luciferase complementary assay of NLRP3 PYD-PYD interaction indicates inflammasome formation during inflammation. Anal. Biochem 638, 114510 (2022).
Hochheiser, I. V. et al. Directionality of PYD filament growth determined by the transition of NLRP3 nucleation seeds to ASC elongation. Sci. Adv. 8, eabn7583 (2022).
Tapia-Abellan, A. et al. Sensing low intracellular potassium by NLRP3 results in a stable open structure that promotes inflammasome activation. Sci. Adv. 7, eabf4468 (2021).
Andreeva, L. et al. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 184, 6299–6312.e6222 (2021).
Ohto, U. et al. Structural basis for the oligomerization-mediated regulation of NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 119, e2121353119 (2022).
Hoss, F. et al. Alternative splicing regulates stochastic NLRP3 activity. Nat. Commun. 10, 3238 (2019).
Ehtesham, N. et al. Three functional variants in the NLRP3 gene are associated with susceptibility and clinical characteristics of systemic lupus erythematosus. Lupus 30, 1273–1282 (2021).
Zhou, L. et al. Excessive deubiquitination of NLRP3-R779C variant contributes to very-early-onset inflammatory bowel disease development. J. Allergy Clin. Immunol. 147, 267–279 (2021).
Dessing, M. C. et al. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci. Rep. 6, 36315 (2016).
Ozturk, K. H. & Unal, G. O. Novel splice‑site variants c.393G>A, c.278_2A>G in exon 2 and Q705K variant in exon 3 of NLRP3 gene are associated with bipolar I disorder. Mol. Med. Rep. 26, 293 (2022).
Chauhan, D. et al. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep. 23, e54277 (2022).
Hu, Z. et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 (2013).
Poyet, J. L. et al. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313 (2001).
Yang, J. et al. Sequence determinants of specific pattern-recognition of bacterial ligands by the NAIP-NLRC4 inflammasome. Cell Discov. 4, 22 (2018).
Yang, X. et al. Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res. 28, 35–47 (2018).
Paidimuddala, B. et al. Mechanism of NAIP-NLRC4 inflammasome activation revealed by cryo-EM structure of unliganded NAIP5. Nat. Struct. Mol. Biol. 30, 159–166 (2023).
Tenthorey, J. L., Kofoed, E. M., Daugherty, M. D., Malik, H. S. & Vance, R. E. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol. Cell 54, 17–29 (2014).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Zhang, L. et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 (2015).
Hu, Z. et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 (2015).
Halff, E. F. et al. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 (2012).
Diebolder, C. A., Halff, E. F., Koster, A. J., Huizinga, E. G. & Koning, R. I. Cryoelectron tomography of the NAIP5/NLRC4 inflammasome: Implications for NLR activation. Structure 23, 2349–2357 (2015).
Li, Y. et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl Acad. Sci. USA 115, 10845–10852 (2018).
Lu, A. et al. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 23, 416–425 (2016).
Matyszewski, M. et al. Cryo-EM structure of the NLRC4(CARD) filament provides insights into how symmetric and asymmetric supramolecular structures drive inflammasome assembly. J. Biol. Chem. 293, 20240–20248 (2018).
Wang, L. et al. A novel mutation in the NBD domain of NLRC4 causes mild autoinflammation with recurrent urticaria. Front Immunol. 12, 674808 (2021).
Canna, S. W. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet 46, 1140–1146 (2014).
Chear, C. T. et al. A novel de novo NLRC4 mutation reinforces the likely pathogenicity of specific LRR domain mutation. Clin. Immunol. 211, 108328 (2020).
Ionescu, D. et al. First description of late-onset autoinflammatory disease due to somatic NLRC4 mosaicism. Arthritis Rheumatol. 74, 692–699 (2022).
Moghaddas, F. et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)-LRR oligomerization interface. J. Allergy Clin. Immunol. 142, 1956–1967.e1956 (2018).
Steiner, A. et al. Recessive NLRC4-autoinflammatory disease reveals an ulcerative colitis locus. J. Clin. Immunol. 42, 325–335 (2022).
Raghawan, A. K., Ramaswamy, R., Radha, V. & Swarup, G. HSC70 regulates cold-induced caspase-1 hyperactivation by an autoinflammation-causing mutant of cytoplasmic immune receptor NLRC4. Proc. Natl. Acad. Sci. USA 116, 21694–21703 (2019).
Lu, A., Kabaleeswaran, V., Fu, T., Magupalli, V. G. & Wu, H. Crystal structure of the F27G AIM2 PYD mutant and similarities of its self-association to DED/DED interactions. J. Mol. Biol. 426, 1420–1427 (2014).
Jin, T. et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 (2012).
Matyszewski, M. et al. Distinct axial and lateral interactions within homologous filaments dictate the signaling specificity and order of the AIM2-ASC inflammasome. Nat. Commun. 12, 2735 (2021).
Wang, H., Yang, L. & Niu, X. Conformation switching of AIM2 PYD domain revealed by NMR relaxation and MD simulation. Biochem Biophys. Res Commun. 473, 636–641 (2016).
Morrone, S. R. et al. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 6, 7827 (2015).
Lu, A. et al. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 1, 15013 (2015).
Liao, J. C. et al. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure 19, 418–429 (2011).
Ni, X. et al. New insights into the structural basis of DNA recognition by HINa and HINb domains of IFI16. J. Mol. Cell Biol. 8, 51–61 (2016).
Fan, X. et al. Structural mechanism of DNA recognition by the p204 HIN domain. Nucleic Acids Res. 49, 2959–2972 (2021).
Trapani, J. A., Dawson, M., Apostolidis, V. A. & Browne, K. A. Genomic organization of IFI16, an interferon-inducible gene whose expression is associated with human myeloid cell differentiation: correlation of predicted protein domains with exon organization. Immunogenetics 40, 415–424 (1994).
Liu, Z., Zheng, X., Wang, Y. & Song, H. Bacterial expression of the HINab domain of IFI16: purification, characterization of DNA binding activity, and co-crystallization with viral dsDNA. Protein Expr. Purif. 102, 13–19 (2014).
Tian, Y. & Yin, Q. Structural analysis of the HIN1 domain of interferon-inducible protein 204. Acta Crystallogr F. Struct. Biol. Commun. 75, 455–460 (2019).
Li, T., Diner, B. A., Chen, J. & Cristea, I. M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 109, 10558–10563 (2012).
Motyan, J. A., Bagossi, P., Benko, S. & Tozser, J. A molecular model of the full-length human NOD-like receptor family CARD domain containing 5 (NLRC5) protein. BMC Bioinforma. 14, 275 (2013).
Neerincx, A. et al. The N-terminal domain of NLRC5 confers transcriptional activity for MHC class I and II gene expression. J. Immunol. 193, 3090–3100 (2014).
Cui, J. et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141, 483–496 (2010).
Gutte, P. G., Jurt, S., Grutter, M. G. & Zerbe, O. Unusual structural features revealed by the solution NMR structure of the NLRC5 caspase recruitment domain. Biochemistry 53, 3106–3117 (2014).
Leng, F. et al. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci. Rep. 10, 198 (2020).
Shen, C. et al. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc. Natl. Acad. Sci. USA 116, 2052–2057 (2019).
de Sa Pinheiro, A. et al. Backbone and sidechain (1)H, (15)N and (13)C assignments of the NLRP7 pyrin domain. Biomol. NMR Assign. 3, 207–209 (2009).
Pinheiro, A. S. et al. Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 285, 27402–27410 (2010).
Singer, H. et al. NLRP7 inter-domain interactions: the NACHT-associated domain is the physical mediator for oligomeric assembly. Mol. Hum. Reprod. 20, 990–1001 (2014).
Radian, A. D., Khare, S., Chu, L. H., Dorfleutner, A. & Stehlik, C. ATP binding by NLRP7 is required for inflammasome activation in response to bacterial lipopeptides. Mol. Immunol. 67, 294–302 (2015).
Ha, H. J. & Park, H. H. Crystal structure of the human NLRP9 pyrin domain reveals a bent N-terminal loop that may regulate inflammasome assembly. FEBS Lett. 594, 2396–2405 (2020).
Chen, S. et al. Novel Role for Tranilast in Regulating NLRP3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J. Am. Heart Assoc. 9, e015513 (2020).
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Ren, G. et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 38, e100376 (2019).
Mezzasoma, L., Talesa, V. N., Romani, R. & Bellezza, I. ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP axis by interfering with canonical, non-canonical, and alternative routes of inflammasome activation in human THP1 cells. Int. J. Mol. Sci. 22, 24 (2020).
Zhang, Z. et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 214, 2671–2693 (2017).
Song, N. et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 68, 185–197.e186 (2017).
Yaron, J. R. et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 6, e1954 (2015).
Green, J. P. et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. USA 115, E9371–E9380 (2018).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Chen, J. & Chen, Z. J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018).
Li, L. et al. CB1R-stabilized NLRP3 inflammasome drives antipsychotics cardiotoxicity. Signal Transduct. Target Ther. 7, 190 (2022).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Yamamoto, M. et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells 9, 1055–1067 (2004).
Aachoui, Y. et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978 (2013).
Tong, W. et al. Resveratrol inhibits LPS-induced inflammation through suppressing the signaling cascades of TLR4-NF-kappaB/MAPKs/IRF3. Exp. Ther. Med 19, 1824–1834 (2020).
Simoes, D. C. M., Paschalidis, N., Kourepini, E. & Panoutsakopoulou, V. An integrin axis induces IFN-beta production in plasmacytoid dendritic cells. J. Cell Biol. 221, e202102055 (2022).
Igbe, I. et al. Dietary quercetin potentiates the antiproliferative effect of interferon-alpha in hepatocellular carcinoma cells through activation of JAK/STAT pathway signaling by inhibition of SHP2 phosphatase. Oncotarget 8, 113734–113748 (2017).
Flood, B. et al. Caspase-11 regulates the tumour suppressor function of STAT1 in a murine model of colitis-associated carcinogenesis. Oncogene 38, 2658–2674 (2019).
Aizawa, E. et al. GSDME-dependent incomplete pyroptosis permits selective IL-1alpha release under caspase-1 inhibition. iScience 23, 101070 (2020).
Compan, V. et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500 (2012).
Gritsenko, A. et al. Priming is dispensable for NLRP3 inflammasome activation in human monocytes in vitro. Front Immunol. 11, 565924 (2020).
Ming, S. L. et al. The human-specific STING agonist G10 activates Type I interferon and the NLRP3 inflammasome in porcine cells. Front Immunol. 11, 575818 (2020).
Zhou, B. & Abbott, D. W. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 35, 108998 (2021).
Robinson, K. S. et al. ZAKalpha-driven ribotoxic stress response activates the human NLRP1 inflammasome. Science 377, 328–335 (2022).
Langorgen, J., Williksen, J. H., Johannesen, K. & Erichsen, H. G. [Tissue adhesives versus conventional skin closure]. Tidsskr. Nor. Laegeforen 107, 2944–2945 (1987).
Chui, A. J. et al. N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85 (2019).
Saresella, M. et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 11, 23 (2016).
Zwicker, S. et al. Th17 micro-milieu regulates NLRP1-dependent caspase-5 activity in skin autoinflammation. PLoS One 12, e0175153 (2017).
Zhang, B. et al. Chronic glucocorticoid exposure activates BK-NLRP1 signal involving in hippocampal neuron damage. J. Neuroinflammation 14, 139 (2017).
Planes, R. et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 82, 2385–2400.e2389 (2022).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Reyes Ruiz, V. M. et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 114, 13242–13247 (2017).
Miao, E. A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 107, 3076–3080 (2010).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Lage, S. L. et al. Cytosolic flagellin-induced lysosomal pathway regulates inflammasome-dependent and -independent macrophage responses. Proc. Natl. Acad. Sci. USA 110, E3321–E3330 (2013).
Gram, A. M. et al. Salmonella flagellin activates NAIP/NLRC4 and canonical NLRP3 inflammasomes in human macrophages. J. Immunol. 206, 631–640 (2021).
Pereira, M. S. et al. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J. Immunol. 187, 6447–6455 (2011).
Mohamed, M. F. et al. CrkII/Abl phosphorylation cascade is critical for NLRC4 inflammasome activity and is blocked by Pseudomonas aeruginosa ExoT. Nat. Commun. 13, 1295 (2022).
Arlehamn, C. S., Petrilli, V., Gross, O., Tschopp, J. & Evans, T. J. The role of potassium in inflammasome activation by bacteria. J. Biol. Chem. 285, 10508–10518 (2010).
Naseer, N. et al. Human NAIP/NLRC4 and NLRP3 inflammasomes detect Salmonella type III secretion system activities to restrict intracellular bacterial replication. PLoS Pathog. 18, e1009718 (2022).
Eibl, C. et al. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51, 7330–7341 (2012).
Case, C. L. & Roy, C. R. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila. mBio 2, (2011).
Van Opdenbosch, N. et al. Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep. 21, 3427–3444 (2017).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Lee, S. et al. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597, 415–419 (2021).
Zhou, Q. et al. Pseudorabies virus infection activates the TLR-NF-kappaB axis and AIM2 inflammasome to enhance inflammatory responses in mice. J. Virol. 97, e0000323 (2023).
Dawson, M. J., Elwood, N. J., Johnstone, R. W. & Trapani, J. A. The IFN-inducible nucleoprotein IFI 16 is expressed in cells of the monocyte lineage, but is rapidly and markedly down-regulated in other myeloid precursor populations. J. Leukoc. Biol. 64, 546–554 (1998).
Gariglio, M. et al. Immunohistochemical expression analysis of the human interferon-inducible gene IFI16, a member of the HIN200 family, not restricted to hematopoietic cells. J. Interferon Cytokine Res. 22, 815–821 (2002).
Wei, W. et al. Expression of IFI 16 in epithelial cells and lymphoid tissues. Histochem Cell Biol. 119, 45–54 (2003).
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Roy, A. et al. Nuclear innate immune DNA sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 90, 8822–8841 (2016).
Jiang, Z. et al. IFI16 directly senses viral RNA and enhances RIG-I transcription and activation to restrict influenza virus infection. Nat. Microbiol 6, 932–945 (2021).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
Kumar, H. et al. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J. Immunol. 186, 994–1000 (2011).
Janowski, A. M., Kolb, R., Zhang, W. & Sutterwala, F. S. Beneficial and detrimental roles of NLRs in carcinogenesis. Front Immunol. 4, 370 (2013).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Kinoshita, T., Wang, Y., Hasegawa, M., Imamura, R. & Suda, T. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1beta secretion. J. Biol. Chem. 280, 21720–21725 (2005).
Messaed, C. et al. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J. Biol. Chem. 286, 43313–43323 (2011).
Luo, Y. et al. Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res 27, 352–372 (2017).
Liu, W. et al. Erythroid lineage Jak2V617F expression promotes atherosclerosis through erythrophagocytosis and macrophage ferroptosis. J. Clin. Invest 132, e155724 (2022).
Yvan-Charvet, L. et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Invest 117, 3900–3908 (2007).
Buono, C. et al. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 102, 1596–1601 (2005).
Zhang, Z. et al. Evidence that cathelicidin peptide LL-37 may act as a functional ligand for CXCR2 on human neutrophils. Eur. J. Immunol. 39, 3181–3194 (2009).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Elhage, R. et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res 59, 234–240 (2003).
Chang, P. Y., Chang, S. F., Chang, T. Y., Su, H. M. & Lu, S. C. Synergistic effects of electronegative-LDL- and palmitic-acid-triggered IL-1beta production in macrophages via LOX-1- and voltage-gated-potassium-channel-dependent pathways. J. Nutr. Biochem 97, 108767 (2021).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 (2013).
Grace, N. D. Prevention of recurrent variceal bleeding–is surgical rescue the answer? Ann. Intern Med. 112, 242–244, (1990).
Kirii, H. et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb. Vasc. Biol. 23, 656–660 (2003).
Alexander, M. R. et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Invest 122, 70–79 (2012).
Sun, H. J. et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 8, e3074 (2017).
Al-Qazazi, R. et al. Macrophage-NLRP3 Activation Promotes Right Ventricle Failure in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 206, 608–624 (2022).
Suetomi, T. et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/calmodulin-dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 138, 2530–2544 (2018).
Liu, C. et al. Cathepsin B deteriorates diabetic cardiomyopathy induced by streptozotocin via promoting NLRP3-mediated pyroptosis. Mol. Ther. Nucleic Acids 30, 198–207 (2022).
Zhang, Y. et al. Gut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc Res 118, 785–797 (2022).
Yao, C. et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242 (2018).
Zeng, C. et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 34, 101523 (2020).
Sun, X. et al. Colchicine ameliorates dilated cardiomyopathy Via SIRT2-mediated suppression of NLRP3 inflammasome activation. J. Am. Heart Assoc. 11, e025266 (2022).
Toldo, S. et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J. Cardiol. 209, 215–220 (2016).
Wang, S. H. et al. GSK-3beta-mediated activation of NLRP3 inflammasome leads to pyroptosis and apoptosis of rat cardiomyocytes and fibroblasts. Eur. J. Pharm. 920, 174830 (2022).
de Almeida Oliveira, N. C. et al. Multicellular regulation of miR-196a-5p and miR-425-5 from adipose stem cell-derived exosomes and cardiac repair. Clin. Sci. (Lond.) 136, 1281–1301 (2022).
Bao, J., Sun, T., Yue, Y. & Xiong, S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 114, 41–48 (2019).
Liu, X. et al. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res Cardiol. 117, 40 (2022).
Wang, Y. et al. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLoS Pathog. 14, e1006872 (2018).
Chu, C. et al. miR-513c-5p suppression aggravates pyroptosis of endothelial cell in deep venous thrombosis by promoting caspase-1. Front Cell Dev. Biol. 10, 838785 (2022).
Folco, E. J., Sukhova, G. K., Quillard, T. & Libby, P. Moderate hypoxia potentiates interleukin-1beta production in activated human macrophages. Circ. Res 115, 875–883 (2014).
Zhang, Y. et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. 5, 2619–2623 (2021).
Kong, C. Y. et al. Underlying the mechanisms of doxorubicin-induced acute cardiotoxicity: Oxidative stress and cell death. Int. J. Biol. Sci. 18, 760–770 (2022).
Li, X. Q., Tang, X. R. & Li, L. L. Antipsychotics cardiotoxicity: What’s known and what’s next. World J. Psychiatry 11, 736–753 (2021).
Sun, Z. et al. SIRT3 attenuates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome via autophagy. Biochem Pharm. 207, 115354 (2023).
Li, S. et al. Microglial NLRP3 inflammasome activates neurotoxic astrocytes in depression-like mice. Cell Rep. 41, 111532 (2022).
Gustin, A. et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10, e0130624 (2015).
Johann, S. et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63, 2260–2273 (2015).
Liu, D. et al. Melatonin Attenuates White Matter Injury via Reducing Oligodendrocyte Apoptosis After Subarachnoid Hemorrhage in Mice. Turk. Neurosurg. 30, 685–692 (2020).
Yao, J., Wang, Z., Song, W. & Zhang, Y. Targeting NLRP3 inflammasome for neurodegenerative disorders. Mol Psychiatry, https://doi.org/10.1038/s41380-023-02239-0, (2023).
Yan, J. et al. CCR5 activation promotes NLRP1-dependent neuronal pyroptosis via CCR5/PKA/CREB pathway after intracerebral hemorrhage. Stroke 52, 4021–4032 (2021).
Wang, L. Q. et al. LncRNA-Fendrr protects against the ubiquitination and degradation of NLRC4 protein through HERC2 to regulate the pyroptosis of microglia. Mol. Med 27, 39 (2021).
Ma, C. et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 218, e20201796 (2021).
Zhang, Y., Chen, H., Li, R., Sterling, K. & Song, W. Amyloid beta-based therapy for Alzheimer’s disease: challenges, successes and future. Signal Transduct. Target Ther. 8, 248 (2023).
Moloney, C. M., Lowe, V. J. & Murray, M. E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimers Dement 17, 1554–1574 (2021).
Head, E. et al. Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J. Alzheimers Dis. 20, 637–646 (2010).
Jie, F. et al. Stigmasterol attenuates inflammatory response of microglia via NF-kappaB and NLRP3 signaling by AMPK activation. Biomed. Pharmacother. 153, 113317 (2022).
Luciunaite, A. et al. Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem 155, 650–661 (2020).
Yang, Y. et al. Sulforaphane attenuates microglia-mediated neuronal damage by down-regulating the ROS/autophagy/NLRP3 signal axis in fibrillar Abeta-activated microglia. Brain Res 1801, 148206 (2023).
Schnaars, M., Beckert, H. & Halle, A. Assessing beta-amyloid-induced NLRP3 inflammasome activation in primary microglia. Methods Mol. Biol. 1040, 1–8 (2013).
Chaturvedi, S., Naseem, Z., El-Khamisy, S. F. & Wahajuddin, M. Nanomedicines targeting the inflammasome as a promising therapeutic approach for cell senescence. Semin Cancer Biol. 86, 46–53 (2022).
Couturier, J. et al. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflammation 13, 20 (2016).
Liu, L. & Chan, C. IPAF inflammasome is involved in interleukin-1beta production from astrocytes, induced by palmitate; implications for Alzheimer’s Disease. Neurobiol. Aging 35, 309–321 (2014).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013).
Friker, L. L. et al. beta-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep. 30, 3743–3754.e3746 (2020).
Hulse, J. & Bhaskar, K. Crosstalk between the NLRP3 inflammasome/ASC speck and amyloid protein aggregates drives disease progression in Alzheimer’s and Parkinson’s Disease. Front Mol. Neurosci. 15, 805169 (2022).
Spanic, E., Langer Horvat, L., Ilic, K., Hof, P. R. & Simic, G. NLRP1 Inflammasome activation in the hippocampal formation in Alzheimer’s disease: Correlation with neuropathological changes and unbiasedly estimated neuronal loss. Cells 11, 2223 (2022).
Tan, M. S. et al. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 5, e1382 (2014).
Wu, P. J., Hung, Y. F., Liu, H. Y. & Hsueh, Y. P. Deletion of the inflammasome sensor Aim2 mitigates abeta deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer Disease Mouse Model. Neuroimmunomodulation 24, 29–39 (2017).
Pike, A. F. et al. alpha-Synuclein evokes NLRP3 inflammasome-mediated IL-1beta secretion from primary human microglia. Glia 69, 1413–1428 (2021).
Freeman, D. et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8, e62143 (2013).
Konstantin Nissen, S. et al. Changes in CD163+, CD11b+, and CCR2+ peripheral monocytes relate to Parkinson’s disease and cognition. Brain Behav. Immun. 101, 182–193 (2022).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
Lee, E. et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 26, 213–228 (2019).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Panicker, N. et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 110, 2422–2437.e2429 (2022).
Coyne, A. N. et al. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 13, eabe1923 (2021).
Debye, B. et al. Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol. 28, 14–27 (2018).
Bellezza, I. et al. Peroxynitrite activates the NLRP3 inflammasome cascade in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol. Neurobiol. 55, 2350–2361 (2018).
Meissner, F., Molawi, K. & Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 107, 13046–13050 (2010).
Cihankaya, H., Theiss, C. & Matschke, V. Little helpers or mean rogue-role of microglia in animal models of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22, 993 (2021).
Moreno-Garcia, L. et al. Inflammasome in ALS skeletal muscle: NLRP3 as a potential biomarker. Int. J. Mol. Sci. 22, 2523 (2021).
McKenzie, B. A. et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 115, E6065–E6074 (2018).
Huang, W. X., Huang, P. & Hillert, J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult. Scler. 10, 482–487 (2004).
Inoue, M., Williams, K. L., Gunn, M. D. & Shinohara, M. L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 109, 10480–10485 (2012).
Peng, Y. et al. NLRP3 level in cerebrospinal fluid of patients with neuromyelitis optica spectrum disorders: Increased levels and association with disease severity. Mult. Scler. Relat. Disord. 39, 101888 (2019).
Еlmahdy, M. K., Abdelaziz, R. R., Elmahdi, H. S. & Suddek, G. M. Effect of Agmatine on a mouse model of allergic airway inflammation: A comparative study. Autoimmunity 55, 608–619 (2022).
Wood, L. G. et al. Saturated fatty acids, obesity, and the nucleotide oligomerization domain-like receptor protein 3 (NLRP3) inflammasome in asthmatic patients. J. Allergy Clin. Immunol. 143, 305–315 (2019).
Kim, R. Y. et al. Role for NLRP3 inflammasome-mediated, IL-1beta-dependent responses in severe, steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 196, 283–297 (2017).
Wang, Y. et al. FSTL1 aggravates OVA-induced inflammatory responses by activating the NLRP3/IL-1beta signaling pathway in mice and macrophages. Inflamm. Res 70, 777–787 (2021).
Chaly, Y. et al. Follistatin-like protein 1 enhances NLRP3 inflammasome-mediated IL-1beta secretion from monocytes and macrophages. Eur. J. Immunol. 44, 1467–1479 (2014).
Wang, H. et al. NLRP3 inflammasome involves in the acute exacerbation of patients with chronic obstructive pulmonary disease. Inflammation 41, 1321–1333 (2018).
Mo, R., Zhang, J., Chen, Y. & Ding, Y. Nicotine promotes chronic obstructive pulmonary disease via inducing pyroptosis activation in bronchial epithelial cells. Mol. Med. Rep. 25, 92 (2022).
Buscetta, M. et al. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J. 34, 1819–1832 (2020).
Zhang, M. Y. et al. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 269, 119090 (2021).
Tien, C. P. et al. Ambient particulate matter attenuates Sirtuin1 and augments SREBP1-PIR axis to induce human pulmonary fibroblast inflammation: molecular mechanism of microenvironment associated with COPD. Aging (Albany NY) 11, 4654–4671 (2019).
Wang, C. et al. Oxidative stress activates the TRPM2-Ca(2+)-NLRP3 axis to promote PM(2.5)-induced lung injury of mice. Biomed. Pharmacother. 130, 110481 (2020).
Sanche, S. et al. High contagiousness and rapid spread of severe acute respiratory syndrome Coronavirus 2. Emerg. Infect. Dis. 26, 1470–1477 (2020).
Gholaminejhad, M. et al. Formation and activity of NLRP3 inflammasome and histopathological changes in the lung of corpses with COVID-19. J. Mol. Histol. 53, 883–890 (2022).
Bortolotti, D. et al. TLR3 and TLR7 RNA sensor activation during SARS-CoV-2 infection. Microorganisms 9, 1820 (2021).
Pan, P. et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 12, 4664 (2021).
Yong, S. J. et al. Inflammatory and vascular biomarkers in post-COVID-19 syndrome: A systematic review and meta-analysis of over 20 biomarkers. Rev. Med. Virol. 33, e2424 (2023).
Abd El-Ghani, S. E. et al. Serum interleukin 1beta and sP-selectin as biomarkers of inflammation and thrombosis, could they be predictors of disease severity in COVID 19 Egyptian patients? (a cross-sectional study). Thromb. J. 20, 77 (2022).
Suzuki, S. et al. Serum gasdermin D levels are associated with the chest computed tomography findings and severity of COVID-19. Respir. Investig. 60, 750–761 (2022).
Sefik, E. et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 606, 585–593 (2022).
Sefik, E. et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. bioRxiv, the preprint server for biology, https://doi.org/10.1101/2021.09.27.461948 (2022).
Albornoz, E. A. et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psych. advance online publication, https://doi.org/10.1038/s41380-022-01831-0 (2022).
Radermacher, P., Maggiore, S. M. & Mercat, A. Fifty years of research in ARDS. Gas exchange in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med 196, 964–984 (2017).
Zhang, T. et al. CaMK4 promotes acute lung injury through NLRP3 inflammasome activation in type II alveolar epithelial cell. Front Immunol. 13, 890710 (2022).
He, X. et al. TLR4-upregulated IL-1beta and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci. Rep. 6, 31663 (2016).
Jiang, P. et al. Extracellular histones aggravate inflammation in ARDS by promoting alveolar macrophage pyroptosis. Mol. Immunol. 135, 53–61 (2021).
Song, C. et al. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP3 inflammasome and IL-1beta/IL-1R1/MyD88/NF-kappaB pathway. J. Cell Mol. Med. 20, 2064–2077 (2016).
Galliher-Beckley, A. J., Lan, L. Q., Aono, S., Wang, L. & Shi, J. Caspase-1 activation and mature interleukin-1beta release are uncoupled events in monocytes. World J. Biol. Chem. 4, 30–34 (2013).
Kolb, M., Margetts, P. J., Anthony, D. C., Pitossi, F. & Gauldie, J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest 107, 1529–1536 (2001).
Liu, W., Han, X., Li, Q., Sun, L. & Wang, J. Iguratimod ameliorates bleomycin-induced pulmonary fibrosis by inhibiting the EMT process and NLRP3 inflammasome activation. Biomed. Pharmacother. 153, 113460 (2022).
Yang, W. et al. Pyroptosis impacts the prognosis and treatment response in gastric cancer via immune system modulation. Am. J. Cancer Res. 12, 1511–1534 (2022).
Jang, A. R. et al. Unveiling the crucial role of type IV secretion system and motility of helicobacter pylori in IL-1beta production via NLRP3 inflammasome activation in neutrophils. Front Immunol. 11, 1121 (2020).
Pachathundikandi, S. K., Blaser, N., Bruns, H. & Backert, S. Helicobacter pylori Avoids the Critical Activation of NLRP3 inflammasome-mediated production of oncogenic mature IL-1beta in human immune cells. Cancers (Basel) 12, 803 (2020).
Pan, X. et al. Human hepatocytes express absent in melanoma 2 and respond to hepatitis B virus with interleukin-18 expression. Virus Genes 52, 445–452 (2016).
Li, Z. & Jiang, J. The NLRP3 inflammasome mediates liver failure by activating procaspase-1 and pro-IL-1 beta and regulating downstream CD40-CD40L signaling. J. Int. Med. Res. 49, 3000605211036845 (2021).
Xie, W. H. et al. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm. Res 69, 683–696 (2020).
Negash, A. A. et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 9, e1003330 (2013).
Daussy, C. F. et al. The Inflammasome Components NLRP3 and ASC act in concert with IRGM to rearrange the golgi apparatus during Hepatitis C virus infection. J. Virol 95, e00826–20 (2021).
Dixon, L. J., Berk, M., Thapaliya, S., Papouchado, B. G. & Feldstein, A. E. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest 92, 713–723 (2012).
L’Homme, L. et al. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J. Lipid Res. 54, 2998–3008 (2013).
Li, Q. et al. Irisin alleviates LPS-induced liver injury and inflammation through inhibition of NLRP3 inflammasome and NF-kappaB signaling. J. Recept Signal Transduct. Res 41, 294–303 (2021).
Ruan, S. et al. Antcin A alleviates pyroptosis and inflammatory response in Kupffercells of non-alcoholic fatty liver disease by targeting NLRP3. Int Immunopharmacol. 100, 108126 (2021).
Pierantonelli, I. et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 7, 12200 (2017).
Gallego, P., Castejon-Vega, B., Del Campo, J. A. & Cordero, M. D. The Absence of NLRP3-inflammasome modulates hepatic fibrosis progression, lipid metabolism, and inflammation in KO NLRP3 mice during aging. Cells 9, 2148 (2020).
Yen, I. C. et al. 4-Acetylantroquinonol B ameliorates nonalcoholic steatohepatitis by suppression of ER stress and NLRP3 inflammasome activation. Biomed. Pharmacother. 138, 111504 (2021).
Rampanelli, E. et al. Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation. Sci. Rep. 7, 2861 (2017).
Lippai, D. et al. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J. Leukoc. Biol. 94, 171–182 (2013).
DeSantis, D. A. et al. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm. 2013, 751374 (2013).
Iracheta-Vellve, A. et al. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 63, 1147–1155 (2015).
Kim, S. K., Choe, J. Y. & Park, K. Y. Ethanol augments monosodium urate-induced NLRP3 inflammasome activation via regulation of AhR and TXNIP in human macrophages. Yonsei Med. J. 61, 533–541 (2020).
Wang, J. et al. Cathepsin B aggravates acute pancreatitis by activating the NLRP3 inflammasome and promoting the caspase-1-induced pyroptosis. Int Immunopharmacol. 94, 107496 (2021).
Zhang, G. X. et al. P2X7R blockade prevents NLRP3 inflammasome activation and pancreatic fibrosis in a mouse model of chronic pancreatitis. Pancreas 46, 1327–1335 (2017).
Cui, L. et al. Saikosaponin A inhibits the activation of pancreatic stellate cells by suppressing autophagy and the NLRP3 inflammasome via the AMPK/mTOR pathway. Biomed. Pharmacother. 128, 110216 (2020).
Li, F. et al. Pachymic acid alleviates experimental pancreatic fibrosis through repressing NLRP3 inflammasome activation. Biosci. Biotechnol. Biochem. 86, 1497–1505 (2022).
Hoque, R. et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141, 358–369 (2011).
Sendler, M. et al. NLRP3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology 158, 253–269.e214 (2020).
Ananthakrishnan, A. N. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. (N. Y) 9, 367–374 (2013).
Ranson, N. et al. NLRP3-Dependent and -Independent Processing of Interleukin (IL)-1beta in Active Ulcerative Colitis. Int. J. Mol. Sci. 20, 57 (2018).
Wang, S. L. et al. Inhibition of NLRP3 attenuates sodium dextran sulfate-induced inflammatory bowel disease through gut microbiota regulation. Biomed. J. 46, 100580 (2023).
He, R. et al. L-Fucose ameliorates DSS-induced acute colitis via inhibiting macrophage M1 polarization and inhibiting NLRP3 inflammasome and NF-kB activation. Int Immunopharmacol. 73, 379–388 (2019).
Hirota, S. A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Zaki, M. H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Zaki, M. H., Lamkanfi, M. & Kanneganti, T. D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 (2011).
Bai, Y. J. et al. Effects of IL-1beta and IL-18 induced by NLRP3 inflammasome activation on myocardial reperfusion injury after PCI. Eur. Rev. Med. Pharm. Sci. 23, 10101–10106 (2019).
Wen, Y. et al. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int J. Biochem Cell Biol. 98, 43–53 (2018).
Gong, W. et al. NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am. J. Physiol. Ren. Physiol. 310, F1081–1088, (2016).
Xiao, C. et al. Tisp40 induces tubular epithelial cell GSDMD-mediated pyroptosis in renal ischemia-reperfusion injury via NF-kappaB signaling. Front Physiol. 11, 906 (2020).
Kim, H. J. et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharm. Exp. Ther. 346, 465–472 (2013).
Qian, Y. et al. P2X7 receptor signaling promotes inflammation in renal parenchymal cells suffering from ischemia-reperfusion injury. Cell Death Dis. 12, 132 (2021).
Pan, L. L. et al. Cathelicidin-related antimicrobial peptide protects against ischaemia reperfusion-induced acute kidney injury in mice. Br. J. Pharm. 177, 2726–2742 (2020).
Valino-Rivas, L. et al. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol. Dial. Transpl. 35, 587–598 (2020).
Cuarental, L. et al. The transcription factor Fosl1 preserves Klotho expression and protects from acute kidney injury. Kidney Int 103, 686–701 (2023).
Guo, Y., Zhang, J., Lai, X., Chen, M. & Guo, Y. Tim-3 exacerbates kidney ischaemia/reperfusion injury through the TLR-4/NF-kappaB signalling pathway and an NLR-C4 inflammasome activation. Clin. Exp. Immunol. 193, 113–129 (2018).
Li, Q. et al. NLRC5 deficiency protects against acute kidney injury in mice by mediating carcinoembryonic antigen-related cell adhesion molecule 1 signaling. Kidney Int 94, 551–566 (2018).
Lichtnekert, J. et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS One 6, e26778 (2011).
Wu, M. et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell Endocrinol. 478, 115–125 (2018).
Pulskens, W. P. et al. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One 9, e85775 (2014).
Wang, W. et al. Aliskiren restores renal AQP2 expression during unilateral ureteral obstruction by inhibiting the inflammasome. Am. J. Physiol. Ren. Physiol. 308, F910–922, (2015).
Fu, R. et al. Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis. Arthritis Rheumatol. 69, 1636–1646 (2017).
Lv, F. et al. CD36 aggravates podocyte injury by activating NLRP3 inflammasome and inhibiting autophagy in lupus nephritis. Cell Death Dis. 13, 729 (2022).
Hu, H., Li, M., Chen, B., Guo, C. & Yang, N. Activation of necroptosis pathway in podocyte contributes to the pathogenesis of focal segmental glomerular sclerosis. Clin. Exp. Nephrol. 26, 1055–1066 (2022).
Zhao, J. et al. Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice. Arthritis Rheumatol. 67, 1036–1044 (2015).
Wu, C. Y. et al. Tris DBA Ameliorates Accelerated and Severe Lupus Nephritis in Mice by Activating Regulatory T Cells and Autophagy and Inhibiting the NLRP3 Inflammasome. J. Immunol. 204, 1448–1461 (2020).
Peng, W., Pei, G. Q., Tang, Y., Tan, L. & Qin, W. IgA1 deposition may induce NLRP3 expression and macrophage transdifferentiation of podocyte in IgA nephropathy. J. Transl. Med 17, 406 (2019).
Yang, S. M. et al. Antroquinonol mitigates an accelerated and progressive IgA nephropathy model in mice by activating the Nrf2 pathway and inhibiting T cells and NLRP3 inflammasome. Free Radic. Biol. Med 61, 285–297 (2013).
Tsai, Y. L. et al. NLRP3 inflammasome: Pathogenic role and potential therapeutic target for IgA nephropathy. Sci. Rep. 7, 41123 (2017).
Chun, J. et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci. Rep. 6, 24667 (2016).
Demirel, I. et al. Activation of the NLRP3 inflammasome pathway by uropathogenic escherichia coli is virulence factor-dependent and influences colonization of bladder epithelial cells. Front Cell Infect. Microbiol 8, 81 (2018).
Harper, S. N., Leidig, P. D., Hughes, F. M. Jr., Jin, H. & Purves, J. T. Calcium pyrophosphate and monosodium urate activate The NLRP3 inflammasome within bladder urothelium via reactive oxygen species and TXNIP. Res Rep. Urol. 11, 319–325 (2019).
Hughes, F. M. Jr. et al. The NLRP3 inflammasome mediates inflammation produced by bladder outlet obstruction. J. Urol. 195, 1598–1605 (2016).
Hughes, F. M. Jr., Sexton, S. J., Jin, H., Govada, V. & Purves, J. T. Bladder fibrosis during outlet obstruction is triggered through the NLRP3 inflammasome and the production of IL-1beta. Am. J. Physiol. Ren. Physiol. 313, F603–F610 (2017).
Lu, J. et al. Rapamycin‑induced autophagy attenuates hormone‑imbalance‑induced chronic non‑bacterial prostatitis in rats via the inhibition of NLRP3 inflammasome‑mediated inflammation. Mol. Med Rep. 19, 221–230 (2019).
Gu, N. Y. et al. Trichomonas vaginalis induces IL-1beta production in a human prostate epithelial cell line by activating the NLRP3 inflammasome via reactive oxygen species and potassium ion efflux. Prostate 76, 885–896 (2016).
Lai, Y. et al. Elevated levels of follicular fatty acids induce ovarian inflammation via ERK1/2 and inflammasome activation in PCOS. J. Clin. Endocrinol. Metab. 107, 2307–2317 (2022).
Wang, D. et al. Exposure to hyperandrogen drives ovarian dysfunction and fibrosis by activating the NLRP3 inflammasome in mice. Sci. Total Environ. 745, 141049 (2020).
Refaie, M. M. M., El-Hussieny, M. & Abdelraheem, W. M. Diacerein ameliorates induced polycystic ovary in female rats via modulation of inflammasome/caspase1/IL1beta and Bax/Bcl2 pathways. Naunyn Schmiedebergs Arch. Pharm. 395, 295–304 (2022).
Navarro-Pando, J. M. et al. Inhibition of the NLRP3 inflammasome prevents ovarian aging. Sci. Adv. 7, eabc7409 (2021).
Murakami, M. et al. Effectiveness of NLRP3 Inhibitor as a Non-Hormonal Treatment for ovarian endometriosis. Reprod. Biol. Endocrinol. 20, 58 (2022).
Carey, A. et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 18, 3204–3218 (2017).
Hamarsheh, S. et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 11, 1659 (2020).
Jia, Y. et al. Aberrant NLRP3 inflammasome associated with aryl hydrocarbon receptor potentially contributes to the imbalance of T-helper cells in patients with acute myeloid leukemia. Oncol. Lett. 14, 7031–7044 (2017).
Paugh, S. et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 47, 607–614 (2015).
Salaro, E. et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci. Rep. 6, 26280 (2016).
Zhou, Y. et al. Curcumin activates NLRC4, AIM2, and IFI16 inflammasomes and induces pyroptosis by up-regulated ISG3 transcript factor in acute myeloid leukemia cell lines. Cancer Biol. Ther. 23, 328–335 (2022).
Cominal, J. G. et al. Bone Marrow Soluble Mediator Signatures of Patients With Philadelphia Chromosome-Negative Myeloproliferative Neoplasms. Front Oncol. 11, 665037 (2021).
Rai, S. et al. Inhibition of interleukin-1beta reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat. Commun. 13, 5346 (2022).
Liew, E. L. et al. Identification of AIM2 as a downstream target of JAK2V617F. Exp. Hematol. Oncol. 5, 2 (2015).
Shi, L. et al. Cellular senescence induced by S100A9 in mesenchymal stromal cells through NLRP3 inflammasome activation. Aging (Albany NY) 11, 9626–9642 (2019).
Zhao, X. et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 8, 108571–108583 (2017).
Lu, F. et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 497, 178–189 (2021).
Baldini, C., Santini, E., Rossi, C., Donati, V. & Solini, A. The P2X7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin’s lymphoma in Sjogren’s syndrome: a prospective, observational, single-centre study. J. Intern Med. 282, 175–186 (2017).
Bostan, E., Gokoz, O. & Atakan, N. The role of NLRP1 and NLRP3 inflammasomes in the etiopathogeneses of pityriasis lichenoides chronica and mycosis fungoides: an immunohistochemical study. Arch. Dermatol Res. 315, 231–239 (2023).
Huanosta-Murillo, E. et al. NLRP3 regulates IL-4 expression in TOX(+) CD4(+) T cells of cutaneous T cell lymphoma to potentially promote disease progression. Front Immunol. 12, 668369 (2021).
Singh, V. V. et al. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 87, 4417–4431 (2013).
Liu, Z. H. et al. Genetic Polymorphisms in NLRP3 Inflammasome-Associated Genes in Patients with B-Cell Non-Hodgkin’s Lymphoma. J. Inflamm. Res. 14, 5687–5697 (2021).
Yu, Y. et al. Leptin facilitates the differentiation of Th17 cells from MRL/Mp-Fas lpr lupus mice by activating NLRP3 inflammasome. Innate Immun. 26, 294–300 (2020).
Shin, M. S. et al. Self double-stranded (ds)DNA induces IL-1beta production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J. Immunol. 190, 1407–1415 (2013).
Zhang, H. et al. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J. Transl. Med. 14, 156 (2016).
Yang, M. et al. AIM2 deficiency in B cells ameliorates systemic lupus erythematosus by regulating Blimp-1-Bcl-6 axis-mediated B-cell differentiation. Signal Transduct. Target Ther. 6, 341 (2021).
Leite, J. A. et al. The DNA sensor AIM2 protects against streptozotocin-induced type 1 diabetes by regulating intestinal homeostasis via the IL-18 Pathway. Cells 9, 959 (2020).
Wu, H. et al. The IL-21-TET2-AIM2-c-MAF pathway drives the T follicular helper cell response in lupus-like disease. Clin. Transl. Med 12, e781 (2022).
Choulaki, C. et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther. 17, 257 (2015).
Meloni, F. et al. Cytokine profile of bronchoalveolar lavage in systemic sclerosis with interstitial lung disease: comparison with usual interstitial pneumonia. Ann. Rheum. Dis. 63, 892–894 (2004).
Zhao, C., Gu, Y., Zeng, X. & Wang, J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin. Immunol. 197, 154–160 (2018).
D’Espessailles, A., Mora, Y. A., Fuentes, C. & Cifuentes, M. Calcium-sensing receptor activates the NLRP3 inflammasome in LS14 preadipocytes mediated by ERK1/2 signaling. J. Cell Physiol. 233, 6232–6240 (2018).
Wu, X. Y. et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J. Autoimmun. 106, 102336 (2020).
Chen, Y. et al. Expression of AIM2 in rheumatoid arthritis and its role on fibroblast-like synoviocytes. Mediat. Inflamm. 2020, 1693730 (2020).
Hajizadeh, S., DeGroot, J., TeKoppele, J. M., Tarkowski, A. & Collins, L. V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. 5, R234–R240 (2003).
Li, F. et al. Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation 37, 146–153 (2014).
Delgado-Arevalo, C. et al. NLRC4-mediated activation of CD1c+ DC contributes to perpetuation of synovitis in rheumatoid arthritis. JCI Insight 7, e152886 (2022).
Zhou, Y. et al. Lipoxin A4 attenuates MSU-crystal-induced NLRP3 inflammasome activation through suppressing Nrf2 thereby increasing TXNRD2. Front Immunol. 13, 1060441 (2022).
Amaral, F. A. et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 64, 474–484 (2012).
Fujita, Y. et al. Cold-inducible RNA-binding protein (CIRP) potentiates uric acid-induced IL-1beta production. Arthritis Res. Ther. 23, 128 (2021).
Gonzalez-Cabello, R., Perl, A., Kalmar, L. & Gergely, P. Short-term stimulation of lymphocyte proliferation by indomethacin in vitro and in vivo. Acta Physiol. Hung. 70, 25–30 (1987).
Lee, H. M. et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62, 194–204 (2013).
Youm, Y. H. et al. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 152, 4039–4045 (2011).
Chiazza, F. et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol. Med 21, 1025–1037 (2016).
Tomita, T. Islet amyloid polypeptide in pancreatic islets from type 2 diabetic subjects. Islets 4, 223–232 (2012).
Jourdan, T. et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med 19, 1132–1140 (2013).
Gianfrancesco, M. A. et al. Saturated fatty acids induce NLRP3 activation in human macrophages through K(+) efflux resulting from phospholipid saturation and Na, K-ATPase disruption. Biochim Biophys. Acta Mol. Cell Biol. Lipids 1864, 1017–1030 (2019).
Camell, C. D. et al. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J. Biol. Chem. 290, 29402–29413 (2015).
Stienstra, R. et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 12, 593–605 (2010).
van Diepen, J. A. et al. Caspase-1 deficiency in mice reduces intestinal triglyceride absorption and hepatic triglyceride secretion. J. Lipid Res. 54, 448–456 (2013).
Stienstra, R. et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 108, 15324–15329 (2011).
Zorrilla, E. P. & Conti, B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav. Immun. 37, 45–53 (2014).
Hollingsworth, L. R. et al. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 592, 778–783 (2021).
Sharif, H. et al. Dipeptidyl peptidase 9 sets a threshold for CARD8 inflammasome formation by sequestering its active C-terminal fragment. Immunity 54, 1392–1404.e1310 (2021).
Coll, R. C. et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 15, 556–559 (2019).
Zhuang, T. et al. Tranilast directly targets NLRP3 to protect melanocytes from keratinocyte-derived il-1beta under oxidative stress. Front Cell Dev. Biol. 8, 588 (2020).
Jiang, H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219–3238 (2017).
Zuo, D. et al. Design and synthesis of an N-benzyl 5-(4-sulfamoylbenzylidene-2-thioxothiazolidin-4-one scaffold as a novel NLRP3 inflammasome inhibitor. Bioorg. Med. Chem. Lett 65, 128693 (2022).
He, H. et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 9, 2550 (2018).
Dai, Z. et al. Development of novel tetrahydroquinoline inhibitors of NLRP3 inflammasome for potential treatment of DSS-induced mouse colitis. J. Med Chem. 64, 871–889 (2021).
Marchetti, C. et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl Acad. Sci. USA 115, E1530–E1539 (2018).
Wohlford, G. F. et al. Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc Pharm. 77, 49–60 (2020).
Yang, G. et al. Direct binding to NLRP3 pyrin domain as a novel strategy to prevent NLRP3-driven inflammation and gouty arthritis. Arthritis Rheumatol. 72, 1192–1202 (2020).
He, Y. et al. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 289, 1142–1150 (2014).
Liang, Q. et al. Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharm. Res. 158, 104884 (2020).
Huang, Y. et al. Tivantinib alleviates inflammatory diseases by directly targeting NLRP3. iScience 26, 106062 (2023).
Juliana, C. et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802 (2010).
Lee, J., Rhee, M. H., Kim, E. & Cho, J. Y. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat. Inflamm. 2012, 416036 (2012).
Hsu, C. G. et al. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 29, 1790–1803 (2022).
Wu, X. et al. Discovery of a novel oral proteasome inhibitor to block NLRP3 inflammasome activation with anti-inflammation Activity. J. Med Chem. 65, 11985–12001 (2022).
Shi, Y., Lv, Q., Zheng, M., Sun, H. & Shi, F. NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int Immunopharmacol. 92, 107358 (2021).
Khare, S. et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol. 15, 343–353 (2014).
Kim, J. et al. Obovatol inhibits NLRP3, AIM2, and non-canonical inflammasome activation. Phytomedicine 63, 153019 (2019).
Zhang, M. J. et al. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 34, 648–662 (2020).
Ni, X., Castanares, M., Mukherjee, A. & Lupold, S. E. Nucleic acid aptamers: clinical applications and promising new horizons. Curr. Med. Chem. 18, 4206–4214 (2011).
Yang, H. et al. Roxadustat (FG-4592) protects against ischaemia-induced acute kidney injury via improving CD73 and decreasing AIM2 inflammasome activation. Nephrol. Dial. Transpl. 38, 858–875 (2023).
Xu, H. et al. Costunolide covalently targets NACHT domain of NLRP3 to inhibit inflammasome activation and alleviate NLRP3-driven inflammatory diseases. Acta Pharm. Sin. B 13, 678–693 (2023).
Chung, I. C. et al. EFLA 945 restricts AIM2 inflammasome activation by preventing DNA entry for psoriasis treatment. Cytokine 127, 154951 (2020).
Lee, S. B. et al. Cornus officinalis seed extract inhibits AIM2-inflammasome activation and attenuates imiquimod-induced psoriasis-like skin inflammation. Int. J. Mol. Sci. 24, 5653 (2023).
Jiao, Y. et al. Discovery of a novel and potent inhibitor with differential species-specific effects against NLRP3 and AIM2 inflammasome-dependent pyroptosis. Eur. J. Med Chem. 232, 114194 (2022).
Lee, H. E. et al. Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: a novel strategy to treat acute gout. Sci. Rep. 6, 38622 (2016).
Schmidt, F. I. et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 213, 771–790 (2016).
Chen, C. et al. Directly targeting ASC by lonidamine alleviates inflammasome-driven diseases. J. Neuroinflammation 19, 315 (2022).
Chen, Y. et al. Dehydrocostus lactone inhibits NLRP3 inflammasome activation by blocking ASC oligomerization and prevents LPS-mediated inflammation in vivo. Cell Immunol. 349, 104046 (2020).
Li, Y. et al. Pharmacological Effects and Mechanisms of Chinese Medicines Modulating NLRP3 Inflammasomes in Ischemic Cardio/Cerebral Vascular Disease. Am. J. Chin. Med. 46, 1727–1741 (2018).
Qin, Q. et al. Brevilin A inhibits NLRP3 inflammasome activation in vivo and in vitro by acting on the upstream of NLRP3-induced ASC oligomerization. Mol. Immunol. 135, 116–126 (2021).
Liao, Y. J. et al. Correction: 1,2,4-Trimethoxybenzene selectively inhibits NLRP3 inflammasome activation and attenuates experimental autoimmune encephalomyelitis. Acta Pharm. Sin. 43, 504 (2022).
Flores, J., Fillion, M. L. & LeBlanc, A. C. Caspase-1 inhibition improves cognition without significantly altering amyloid and inflammation in aged Alzheimer disease mice. Cell Death Dis. 13, 864 (2022).
Wu, J. et al. Sennoside A is a novel inhibitor targeting caspase-1. Food Funct. 13, 9782–9795 (2022).
Rudolphi, K., Gerwin, N., Verzijl, N., van der Kraan, P. & van den Berg, W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cartil. 11, 738–746 (2003).
Loher, F. et al. The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharm. Exp. Ther. 308, 583–590 (2004).
Bami, S. et al. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 67, e28581 (2020).
Ruscitti, P., Berardicurti, O., Cipriani, P. & Giacomelli, R. Benefits of anakinra versus TNF inhibitors in rheumatoid arthritis and type 2 diabetes: long-term findings from participants furtherly followed-up in the TRACK study, a multicentre, open-label, randomised, controlled trial. Clin. Exp. Rheumatol. 39, 403–406 (2021).
Ruscitti, P. et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PLoS Med. 16, e1002901 (2019).
Hosono, K., Kato, C. & Sasajima, T. Real-world safety and effectiveness of canakinumab in patients with cryopyrin-associated periodic fever syndrome: a long-term observational study in Japan. Clin. Exp. Rheumatol. 40, 1543–1553 (2022).
Everett, B. M. et al. Inhibition of interleukin-1beta and reduction in atherothrombotic cardiovascular events in the CANTOS trial. J. Am. Coll. Cardiol. 76, 1660–1670 (2020).
Schumacher, H. R. Jr. et al. Rilonacept (interleukin-1 trap) for prevention of gout flares during initiation of uric acid-lowering therapy: results from a phase III randomized, double-blind, placebo-controlled, confirmatory efficacy study. Arthritis Care Res (Hoboken) 64, 1462–1470 (2012).
Klein, A. L. et al. Phase 3 trial of interleukin-1 trap rilonacept in recurrent pericarditis. N. Engl. J. Med. 384, 31–41 (2021).
Issafras, H., Corbin, J. A., Goldfine, I. D. & Roell, M. K. Detailed mechanistic analysis of gevokizumab, an allosteric anti-IL-1beta antibody with differential receptor-modulating properties. J. Pharm. Exp. Ther. 348, 202–215 (2014).
Wlodek, E. et al. A pilot study evaluating GSK1070806 inhibition of interleukin-18 in renal transplant delayed graft function. PLoS One 16, e0247972 (2021).
Mistry, P. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin- 18 mAb GSK1070806 in healthy and obese subjects. Int. J. Clin. Pharm. Ther. 52, 867–879 (2014).
Hu, J. J. et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 21, 736–745 (2020).
Han, C. et al. New mechanism of nerve injury in Alzheimer’s disease: beta-amyloid-induced neuronal pyroptosis. J. Cell Mol. Med 24, 8078–8090 (2020).
Lv, T. et al. Targeting of GSDMD sensitizes HCC to anti-PD-1 by activating cGAS pathway and downregulating PD-L1 expression. J. Immunother. Cancer 10, e004763 (2022).
Li, Q. et al. Licochalcone B specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep. 23, e53499 (2022).
Zhao, Q. et al. Pristimerin protects against inflammation and metabolic disorder in mice through inhibition of NLRP3 inflammasome activation. Acta Pharm. Sin. 42, 975–986 (2021).
Shi, Y. et al. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 34, 208–221 (2020).
Liu, P. et al. Methyl gallate improves hyperuricemia nephropathy mice through inhibiting NLRP3 pathway. Front Pharm. 12, 759040 (2021).
Wu, T. et al. 5-Androstenediol prevents radiation injury in mice by promoting NF-kappaB signaling and inhibiting AIM2 inflammasome activation. Biomed. Pharmacother. 121, 109597 (2020).
Ahn, H. & Lee, G. S. Riboflavin, vitamin B2, attenuates NLRP3, NLRC4, AIM2, and non-canonical inflammasomes by the inhibition of caspase-1 activity. Sci. Rep. 10, 19091 (2020).
Wang, Y., Li, Z., Teng, M. & Liu, J. Dihydroartemisinin inhibits activation of the AIM2 inflammasome pathway and NF-kappaB/HIF-1alpha/VEGF pathway by inducing autophagy in A431 human cutaneous squamous cell carcinoma cells. Int J. Med. Sci. 18, 2705–2715 (2021).
Wu, X. Y. et al. ML365 inhibits TWIK2 channel to block ATP-induced NLRP3 inflammasome. Acta Pharm. Sin. 43, 992–1000 (2022).
Huang, L. et al. Curcumin alleviates cerebral ischemia-reperfusion injury by inhibiting NLRP1-dependent neuronal pyroptosis. Curr. Neurovasc Res 18, 189–196 (2021).
Liu, X., Lu, X. & Hu, Z. N-acetylcysteine (NAC) inhibits synthesis of IL-18 in macrophage by suppressing NLRP3 expression to reduce the production of IFN-gamma from NK cells. Comput Math. Methods Med. 2021, 7596343 (2021).
Cipollina, C. et al. 17-oxo-DHA displays additive anti-inflammatory effects with fluticasone propionate and inhibits the NLRP3 inflammasome. Sci. Rep. 6, 37625 (2016).
Yang, X. et al. Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp. Neurol. 310, 22–32 (2018).
Gonzalez-Cofrade, L. et al. Phenolic and quinone methide nor-triterpenes as selective NLRP3 inflammasome inhibitors. Bioorg. Chem. 132, 106362 (2023).
Wei, Z. et al. Dihydrotanshinone I specifically inhibits NLRP3 inflammasome activation and protects against septic shock in vivo. Front Pharm. 12, 750815 (2021).
Albanese, V. et al. Novel aryl sulfonamide derivatives as NLRP3 inflammasome inhibitors for the potential treatment of cancer. J. Med. Chem. 66, 5223–5241 (2023).
Yokochi, E., Kohno, S. & Ohata, K. Pharmacological studies on the clathrate compound of mobenzoxamine with beta-cyclodextrin. (I). Effects on the digestive system]. Nihon Yakurigaku Zasshi 92, 297–310 (1988).
Acknowledgements
This work was supported by the Key Laboratory of Alzheimer’s Disease of Zhejiang Province (ZJAD-2021004), the National Natural Science Foundation of China (82201576), Beijing Hospitals Authority Youth Programme (QML20210804) and Beijing Medical Research 2021-8 (YZ); and the National Natural Science Foundation of China (82150710557, 82230043 and 82293642) to WS. WS was the Canada Research Chair in Alzheimer’s Disease.
Author information
Authors and Affiliations
Contributions
JY and WS conceived and designed this project. JY and KS wrote the draft of the manuscript. JY, KS, ZW and YZ did the literature search and review. WS and YZ revised the manuscript and supervised the project. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yao, J., Sterling, K., Wang, Z. et al. The role of inflammasomes in human diseases and their potential as therapeutic targets. Sig Transduct Target Ther 9, 10 (2024). https://doi.org/10.1038/s41392-023-01687-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01687-y
- Springer Nature Limited
This article is cited by
-
Elucidating the role of Pyroptosis in papillary thyroid cancer: prognostic, immunological, and therapeutic perspectives
Cancer Cell International (2024)
-
The effect of colchicine on cancer risk in patients with immune-mediated inflammatory diseases: a time-dependent study based on the Taiwan’s National Health Insurance Research Database
European Journal of Medical Research (2024)
-
The STING agonist IMSA101 enhances chimeric antigen receptor T cell function by inducing IL-18 secretion
Nature Communications (2024)