
Abstract
Autophagy is a conserved lysosomal degradation pathway where cellular components are dynamically degraded and re-processed to maintain physical homeostasis. However, the physiological effect of autophagy appears to be multifaced. On the one hand, autophagy functions as a cytoprotective mechanism, protecting against multiple diseases, especially tumor, cardiovascular disorders, and neurodegenerative and infectious disease. Conversely, autophagy may also play a detrimental role via pro-survival effects on cancer cells or cell-killing effects on normal body cells. During disorder onset and progression, the expression levels of autophagy-related regulators and proteins encoded by autophagy-related genes (ATGs) are abnormally regulated, giving rise to imbalanced autophagy flux. However, the detailed mechanisms and molecular events of this process are quite complex. Epigenetic, including DNA methylation, histone modifications and miRNAs, and post-translational modifications, including ubiquitination, phosphorylation and acetylation, precisely manipulate gene expression and protein function, and are strongly correlated with the occurrence and development of multiple diseases. There is substantial evidence that autophagy-relevant regulators and machineries are subjected to epigenetic and post-translational modulation, resulting in alterations in autophagy levels, which subsequently induces disease or affects the therapeutic effectiveness to agents. In this review, we focus on the regulatory mechanisms mediated by epigenetic and post-translational modifications in disease-related autophagy to unveil potential therapeutic targets. In addition, the effect of autophagy on the therapeutic effectiveness of epigenetic drugs or drugs targeting post-translational modification have also been discussed, providing insights into the combination with autophagy activators or inhibitors in the treatment of clinical diseases.
Similar content being viewed by others
Introduction
Autophagy is stimulated by cellular or environmental stresses through the formation of autophagosomes to clear damaged organelles, protein aggregates, and intracellular pathogens. Autophagy is a distinct biological process that exerts significant roles in determining the cellular status and fate. Autophagy orchestrated by ATGs is a conserved lysosomal degradation pathway where cellular components are dynamically degraded and re-processed to maintain physical homeostasis. However, the physiological effect of autophagy tends to be multifaced. On the one side, autophagy supports stress adaption and targets pernicious cargos, such as damaged protein aggregates, dysfunctional organelles, and invasive pathogens.1,2 This cytoprotective mechanism protects against multiple diseases, especially tumor, cardiovascular disorder, and neurodegenerative and infectious disease. Conversely, autophagy can also display a detrimental role via pro-survival effects on cancer cells or cell-killing effects on normal body cells.3
Epigenetic and post-translational modifications, respectively, manipulate gene expression at transcriptional and post-transcriptional level, and affect protein activity, function and degradation at post-translational level. These modifications are highly critical for normal cellular development and maintenance of tissue-specific gene and protein expression, a dysregulation of which will cause abnormal genes and protein expression signature and malignant phenotype transformation that induces disease occurrence and progression.4,5 The epigenetic and post-translational regulation of autophagy is very complex and plays different roles in human disease. Under certain circumstances, autophagy-related regulators and components, such as TFs, ATG proteins and signaling effector proteins undergo epigenetic and post-translational regulation, leading to autophagy activation or inhibition. Therefore, the clinical outcome of autophagy in different diseases is still a mystery and whether autophagy represses and promotes disease development remains deep investigation.
Considering the scarcity of knowledge in terms of role of epigenetic and post-translational alteration of autophagy in human diseases, the present review attempts to understand and summarize the effects of epigenetic and post-translational modifications of autophagy-related regulators and components in human disease. It will be conductive for uncovering the underlying regulatory mechanisms of disease-related autophagy, seeking for new targets for better diagnosis and treatment. Meanwhile, due to the prospective exploration and application of epigenetic drugs and some drugs targeting post-translational modifications, the different effects of autophagy following these drugs treatment in human diseases are also reviewed here, providing insights into the combined application with autophagy activators or inhibitors.
Autophagy and its main regulatory mechanism
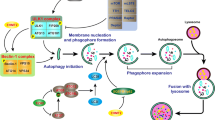
The basic autophagy process, orchestrated by series of autophagy-related regulators and ATG-encoded downstream effector proteins, can be divided into four predominant steps: autophagy initiation (containing ULK1 complex-ULK1/FIP200/ATG13), vesicle nucleation, elongation and autophagosome formation (containing Beclin1-VPS34 complex, ATG5/ATG12/ATG7/ATG10/ATG16L, LC3/ATG3/ATG7/ATG4 and p62, OPTN, NIX), autophagosome-lysosome fusion (containing Rab GTPases and N-ethylmaleimidesensitive factor attachment protein receptors (SNAREs)), and cargo degradation.6
The autophagy process is induced by cellular or environmental stimulus, such as nutrient deprivation, metabolic and energetic stresses, pathogen invasion or oxidant stresses. mTOR subtly senses intra- and extra-cellular nutrient and stress levels, and its phosphorylation and activation suppress the autophagy process by blocking ULK1 complex activity. PI3K/AKT/mTOR and AMPK/mTOR are two quite significant upstream signaling pathways of mTOR in autophagy regulation, which is implicated in tumor progression, myocardial dysfunction, neurodegeneration and metabolic disorders.7,8 Moreover, Wnt signaling pathway is associated with autophagy repression, and is involved in embryogenesis and differentiation. During nutrient deprivation, β-catenin and Dishevelled proteins are targeted for autophagic degradation by LC3. When Wnt signaling is activated, β-catenin acts as a corepressor of p62 to inhibit autophagy. In contrast, another key Wnt signaling protein, GSK3β, negatively regulates the Wnt pathway and has been shown to induce autophagy by phosphorylation of the TSC complex.9 Under ER stress, the unfolded protein response (UPR) has been found to activate ATGs expression and subsequent autophagy, which is mediated by ATF4-CHOP and TRAF2-IRE1-JNK signaling pathway. ATF4 and CHOP transcriptionally regulates dozens of ATG genes and TRAF2-dependent activation of IRE1 and JNK results in Bcl2 phosphorylation, enables the dissociation of Beclin-1.10
Autophagy and diseases
Over the past few years, the relationship between autophagy dysregulation and multiple human diseases has been extensively investigated and appears to be quite complicated.2 Taking advantage of gene-knockout technology in vitro experiments and animal models, research studies have confirmed a set of autophagy-related genes to be closely linked to diseases, particularly tumor, inflammatory disease, neurodegenerative disease, cardiovascular disease, these autophagy-related genes play key roles in human diseases, even though autophagy-relevant detection in humans remains difficult in clinical tests (Fig. 1).

Autophagy and diseases. Autophagy dysregulation and tumor, inflammatory disease, neurodegenerative disease, cardiovascular disease
In terms of autophagy and tumor occurrence and progression, autophagy seems to be a ‘double-edged sword’ and its roles is mainly dependent on specific tumor types, stages, and environmental stresses. Great efforts have been made to decode their connecting mechanisms from different perspectives, including crosstalk between autophagy and tumor metabolic alteration,11 autophagy and antitumor or tumor-promoting immune responses.12 During the tumor initiation period, most models regard autophagy as an antitumor mechanism, and a few essential autophagy-related proteins and transcriptional factors, such as Beclin1, PARK2 (Parkin), and Forkhead box O (FOXO) family proteins, have been implicated as tumor suppressors that impede further malignant transformation.13,14,15 Nevertheless, along with tumor progression, autophagy can serve as pro-survival mechanism to assist tumor cells adaption to metabolic and energetic stresses.11,16,17 One typical case is the transformation of the roles of autophagy from antitumor autophagy to tumor-promoting autophagy within primary melanoma and metastatic melanoma.18 Since neurodegenerative diseases are characterized by the accumulation of abnormal protein condensates and aggregates (e.g., tau, APP, SOD1, α-synuclein, and polyglutamine proteins), defect of autophagic degradation can lead to clearance impediment and is one of the reasons for neurodegeneration. For instance, Beclin1 deficiency is prevalent in Alzheimer’s disease (AD) and Huntington disease.19,20 PICALM acts as an autophagy receptor that is able to interact with LC3 and target APP into autophagosomes, and PS-1 functions as an ER chaperone for the VOa1 subunit of the lysosomal v-ATPase and thus lysosomal PH maintenance, whereas PICALM and PS-1 mutations are frequently occurred in AD patients.21,22 In response to oxidative stress, Nrf2 induces autophagy receptor NDP52 to stimulate autophagy and remove aggregated tau proteins.23 ATP13A2 involved with lysosomal ATPase, is found mutated in autosomal recessive forms of early-onset Parkinsonism to accelerate α-synuclein accumulation.24 Autophagy is also involved in the regulation of diabetic complications, such as diabetic nephropathy, renal fibrosis and cardiomyopathy. mTOR/AMPK, TFEB/ZNSCAN3, SIRTs and FOXOs establish links between autophagy and diabetic complications.25,26
Epigenetic modifications and autophagy-related diseases
Epigenetic modifications including DNA methylation, histone modification and microRNAs play important roles in autophagy-related diseases. DNA methylation and covalent modifications of histone protein are dynamically orchestrated by a so-called series of epigenetic enzymes known as “writers”, “readers” and “erasers” to control and fine-tune large-scale gene transcription.27,28 microRNAs mainly via base pairing binds to gene mRNA to regulate gene post-transcriptional process.
DNA methylation regulates ATGs expression
DNA methylation refers to the addition of a methyl group at the 5-position cytosine (5-methylcytosine (5mC)) in CpG dinucleotides of the genome. This process is catalyzed by a family of DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, DNMT3B, and DNMT3L. Aberrant DNA methylation and demethylation events sustain autophagy level at an abnormal inactivation or activation status in multiple disease, such as tumor and nervous system lesion. A cohort of studies have discovered a series of disease-relevant DNA methylation events linked to autophagy deficiency and hyperactivation, conducive to exploring potent therapeutic targets and prospective epigenetic markers for prognosis. (Table 1).
It was reported that Helicobacter pylori-induced MAP1LC3Av1 methylation silencing leads to autophagy impairment and promotes gastric carcinogenesis.29 ATG4A is hypomethylated in ovarian tumor-initiating cells and predicts poor prognosis in ovarian cancer patients.30 Frequently hypermethylated ATG16L2 in childhood acute lymphoblastic leukemia is associated with a poor prognosis in response to imatinib treatment.31 Reduced Beclin1 promoter methylation due to DNMT3B inhibition increases autophagy and promotes tamoxifen resistance in breast cancer.32 High GABARAPL1 expression is a suitable prognostic marker in lymph node-positive breast cancer patients. DNA methylation and histone deacetylation cause its downregulation in breast cancer.33 Similarly, increased levels of methylated ATG2B, ATG4D, ATG9A, and ATG9B have also been observed in invasive ductal breast carcinomas. ATG2B methylation is positively correlated with tumor grade and stage, whereas ATG4D and ATG9A methylation is positively linked to lymph node involvement.34 Ten-eleven translocation (TET) proteins, such as 5mC oxidases, are crucial in active DNA demethylation through iterative methyl group oxidation, completing the conversion of 5mC into 5-hydroxymethylcytosine (5hmC). TET1 exerts its tumor suppressor function by regulating autophagy.35 Due to reduced TET1 expression in glioma cells, two ATGs, ATG13 and DNA damage-regulated autophagy modulator protein 1, display low 5hmC enrichment in their promoter region, leading to lower autophagy levels than in normal controls.36 Hypoxia can induce autophagy, hypoxia-induced interleukin-6 (IL6) acts as an autophagy initiator for tumor-promoting effects,37 hypoxia-induced astrocyte elevated gene-1 (AEG-1) regulates autophagy in the drug resistance of T-cell non-Hodgkin’s lymphoma,38 and hypoxia-inducible factor39 family proteins, which are hallmarks of hypoxia, activate autophagy through elevated ATG expression.40,41 Under hypoxic conditions, decreased BNIP methylation induces pro-survival autophagy in esophageal squamous cell carcinoma (ESCC),42 whereas HIF1A knockdown increases the methylation of ATG16L to limit medulloblastoma cell proliferation.43
Furthermore, autophagy-lysosome pathway (ALP) is frequently disrupted in Parkinson disease, leading to α-synuclein aggregation. It has identified aberrant methylation at 928 cytosines affecting 326 ALP genes in the appendix of individuals with PD and widespread hypermethylation that is also seen in the brain of individuals with PD.44 DNMT1 acts on neurodegeneration by modulating proteostasis-relevant intracellular processes. DNMT1 negatively impacts retrograde trafficking and autophagy, which involved in the clearance of aggregation-prone proteins by the aggresome-autophagy pathway. In Huntington disease, DNMT1 knockdown strengthens autophagy, and ameliorates Huntington-induced cytotoxicity.45 Decrease of autophagy activity is occurring upon aging and thought to contribute to aging-related diseases. It uncovers, upon autophagy induction, de novo DNMT3A-mediated DNA methylation on expression of LC3 results in reduced baseline autophagy activity.46 Meanwhile, ULK3-dependent activation of GLI1 contributes to upregulation of DNMT3A gene expression upon autophagy induction, thereby bringing additional understanding of the long-term effect of autophagy induction and a possible mechanism for its decline upon aging, pathological conditions, or in response to treatment interventions.47
Histone methylation
G9a/EHMT2
G9a/ Euchromatic histone lysine methyltransferase 1 (EHMT2) catalyzes methylation of histone 3 at lysine 9 (H3K9me1, H3K9me2, and H3K9me3) and is frequently overexpressed in various cancers, including glioma, gastric cancer, and lung cancer.48,49,50 G9a/EHMT2 overexpression was significantly associated with autophagy suppression. It can directly interact with the promoters of core autophagy genes, such as LC3B, TP53INP2/DOR, and WIPI1, increasing their histone methylation levels to reduce autophagy levels.51 In particular, in breast cancer cells, G9a cooperates with DNMT1 to collectively inhibit Beclin1 transcription.52 Chemical inhibition of G9a dissociates H3K9me2 from the Beclin1 promoter and recruits RNA polymerase II and nuclear factor kappa B (NF-κB) to its promoter for transcriptional activity. When G9a is inhibited, BNIP3 expression is transcriptionally activated, releasing Beclin1 from the B-cell lymphoma 2 (Bcl-2)/Beclin1 complex to trigger autophagy.53 In addition, mTOR is a direct target that is monomethylated by G9a at H3K9 and transcriptionally activated.49 It is reasonable to determine the connection between G9a expression, cellular nutritional status, and autophagy for mTOR’s role in regulating autophagy in response to integrated signals from changed nutrient and energy levels. In non-small cell lung cancer (NSCLC), EHMT2 inhibition leads to cancer cell death via autophagy induction, in which sterol regulatory element-binding protein 2 (SREBF2) expression is a crucial contributor.54 SREBF2 can communicate with the autophagy process by binding to the promoters of several autophagy genes, such as Map1lc3b, ATG4b, and ATG4d.55 Here, EHMT2 inhibition robustly decreases H3K9me1 and H3K9me2 at the promoter of the SREBF2 locus, resulting in upregulated SREBF2 expression and, consequently, autophagy-mediated cell death. Similarly, G9a is capable of H3K9me/me2 on the promoter of Beclin1 and P62 to transcriptionally inhibit autophagy flux, and then induces vascular smooth muscle cells death, indicating that G9a may be a potent therapeutic target for cardiovascular diseases including aortic dissection.56 In addition, Mtb phosphoribosyltransferase (MtbPRT) induces EHMT2-dependent H3K9me2/3 and reduces H3K9ac and H3K27ac by upregulating HDAC3 at the ATG5 and ATG7 promoter, which inhibit autophagy to promote Mtb survival57 (Fig. 2a).

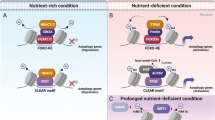
Histone methylation and autophagy regulation. a G9a negatively regulates core autophagy effectors and upstream autophagic regulators to influence autophagy level indirectly. b Under a nutrient-rich environment, SKP2 mediates CARM1 protein degradation, nutrient starvation activates AMPK-dependent FOXO3 phosphorylation, which transcriptionally represses SKP2, resulting in CARM1-mediated transcriptional activation of autophagy-related and lysosomal genes. c EZH2 repressively regulates autophagy via mTOR activation. EZH2 negatively controls TSC2/RHOA/DEPTOR gene transcription to elicit mTOR pathway, leading to autophagy inhibition. Additionally, EZH2 represses HMGA2 expression. HMGA2 can directly activate the MSI2 promoter region, which triggers autophagy via Beclin1 interactions. d DOT1L elevates LAMP5 expression via H3K79 methylation modification enhancements. LAMP5 directly interacts with ATG5 to interrupt an autophagy flux, protecting MLL chimeras from autophagy degradation. e JMJD2B promotes autophagy occurrence via histone demethylation at LC3B promoters, assisting in intracellular amino acid maintenance. f KDM1A negatively manipulates autophagy flux through SESN2- and CLU-dependent pathways or directly targeting p62. SESN2 inhibits mTORC1 activity, and CLU increases autophagosome biogenesis through MAP1LC3/LC3-ATG3 heterodimer stabilization
CARM1
CARM1 is responsible for histone H3 demethylation at arginine 17 (H3R17me2). The AMP-activated protein kinase (AMPK)-SKP2-CARM1 signaling pathway is involved in autophagy transcriptional regulation, and this has been found to pathologically associate with multiple diseases. CARM1 is normally degraded by S-phase kinase-associated protein (SKP) 2 (SKP2)-containing SKP1-cullin1-F-box protein (SCF) E3 ubiquitin ligase in the nucleus rather than in the cytoplasm in nutrient-rich environments. Conversely, nutrient starvation activates AMPK-dependent FOXO3 phosphorylation in the nucleus, transcriptionally repressing SKP2, which results in decreased CARM1 protein degradation. Upregulated CARM1 protein subsequently increases H3R17me2 on the promoters of autophagy-related and lysosomal genes, contributing to the transcriptional activation of these genes through the cooperative action of TFEB.58 In hepatocellular carcinoma (HCC), SKP2 levels were upregulated compared with corresponding normal liver tissues, which is statistically correlated with HCC progression.59 Moreover, during spinal cord injury, enhanced reactive oxygen species induce lysosomal dysfunction and then contributes to neuronal death, while TFE3 activation reverses outcomes of spinal cord injury, which is partly regulated by AMPK-SKP2-CARM1 signaling axis.60 In addition, C9orf72, linked with ALS-FTD, can promote the lysosomal degradation of CARM1, which in turn regulates autophagy-lysosome function and lipid metabolism.61 (Fig. 2b).
EZH2
EZH2, a catalytic subunit of polycomb repressive complex 2 (PRC2), mainly catalyzes the trimethylation of lysine 27 at histone H3 (H3K27me3) and is responsible for transcriptionally silencing target genes. On the one hand, EZH2 inhibition-triggered autophagy induces cell death of cancer cells62,63 and aortic vascular smooth muscle cells,64 liver injury65 and also helps bacterial elimination.57 In cancer cells, metastasis-associated 1 family member 2 (MTA2) first recognizes unmodified H3 with its SANT domain, then recruits EZH2 to a set of mTOR pathway-related genes, such as TSC2, RHOA, and DEPTOR, which are negative regulators of the mTOR signaling pathway, resulting in increased H3K27me3 accompanied by reduced H3K27ac at the promoters. As a result, the expression of mTOR pathway inhibitory genes is suppressed, which elicits mTOR activity and leads to autophagy inhibition.66 On the other hand, autophagy is triggered by EZH2 inhibition to serve as a possible pro-survival mechanism of residual tumor cells with high mobility group A2 (HMGA2) as the critical mediator.67 EZH2 is a repressive modulator of HMGA2. HMGA2 can directly activate the Musashi-2 (MSI2) promoter region, triggering autophagy via interactions with Beclin1, favoring cell proliferation.68 Recently, a few studies have revealed that a group of disease-related microRNAs cause alterations in autophagy levels by EZH2 regulation. For example, microRNA-638 and microRNA-92b increase autophagy and apoptosis and suppress cancer cell invasion and migration by targeting EZH2.69,70 miR-124 is capable of inducing autophagic cell death in tumors and alleviates autophagy-mediated diabetic peripheral neuropathy by directly targeting the EZH2-signal transducer and activator of transcription 3 (STAT3) signaling axis.71,72 In contrast, miR-101 directly interacts with EZH2 and downregulates its expression, resulting in autophagy inhibition73,74 (Fig. 2c).
DOT1L
DOTIL is an H3K79 histone methyltransferase. DOT1L expression is indispensable in mix-lineage leukemia (MLL) maintenance and progression by enhancing H3K79 methylation at its target gene loci, such as Meis1 and HOX gene clusters.75,76 The autophagic protein degradation pathway is repressed to sustain MLL fusion protein levels during MLL maintenance and progression. During this process, DOT1L targets the genetic locus of a lysosome-associated membrane protein family member (LAMP5) to enhance its H3K79 methylation modification to elevate its expression. Unlike other LAMP family members, such as LAMP1 and LAMP2, which act as enhancers in the autophagy pathway, LAMP5 directly interacts with ATG5, rather than Beclin1 or LC3, to interrupt an autophagy flux and protect MLL chimeras from autophagic degradation77 (Fig. 2d).
JMJD2B/ KDM4B
JMJD2B/KDM4B mainly reverses the tri- or demethylation (me3/2) of histone 3 at lysine 9. Epigenetic JMJD2B modifications stimulate autophagy levels and are linked to cancer cell proliferation. In CRC cells, JMJD2B is highly expressed and is accompanied by upregulated ATG signatures. Through direct demethylation at the promoters of LC3B, JMJD2B promotes autophagy to benefit protein degradation and recycling to maintain intracellular amino acids, which is critical for CRC cell survival under glucose deprivation conditions.78 Furthermore, in castration-resistant prostate cancer (CRPC), enhanced KDM4B depends on its demethylating activity to activate autophagy by regulating Wnt/β-catenin signaling, leading to CRPC cell proliferation79 (Fig. 2e).
LSD1/KDM1A
The histone demethylase LSD1/KDM1A manipulates autophagy directly regulating autophagy-related genes expressions, including ATGs, Beclin-1, LC3 and SQSTM1/p62, it also regulating the activities of some other autophagic signaling proteins such as p53, SESN2, mTORC1 and PTEN, which is implicated in tumors, vascular disorder and inflammation. KDM1A depletion triggers autophagy through the SESN2-dependent pathway.80,81 Mechanistically, KDM1A interacts with the promoter region of the SESN2 gene and represses its expression, whereas KDM1A inhibition or knockdown increases H3K4me2 levels, together with increased histone H3 acetylation and decreased H3K27me3, leading to SESN2 gene expression. SESN2, as a member of the SESN1/PA26-related protein family involved in cellular responses to different stress conditions, inhibits mTORC1 activity by regulating the Seh1-associated complex,82 leading to consequent autophagy induction.83,84 Another KDM1A regulatory approach in autophagy is that KDM1A, with MYCN, inhibits clusterin (CLU) transcription, which is an integral part of the autophagic process as it increases autophagosome biogenesis by stabilizing the MAP1LC3/LC3-ATG3 heterodimer.85,86 In addition, as for ovarian aging, LSD1 represses autophagy level by affecting H3K4me2 level and regulating the transcription of p62 to protect oocytes against preterm death87 (Fig. 2f).
Histone acetylation
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are essential epigenetic enzymes that mechanistically manipulate the acetylation of both histone and non-histone proteins in a dynamic manner. The transcriptional silencing or activation is typically associated with the modified status and site on histone. HDAC inhibition or overexpression regulates autophagy pathway-related genes expression, promoting autophagy-mediated tumor progression and tissue injury.28,88,89,90
Acetyl coenzyme A (acetyl-CoA) is an important donor for the acetylation of proteins, and acetyl-CoA synthetase 2 (ACSS2) manipulates its expression. Glucose deprivation assists AMPK-mediated ACSS2 phosphorylation and promotes its nuclear localization. In the nucleus, ACSS2 binds to TFEB and translocates to lysosomal and ATG promoters, where ACSS2 generates acetyl-CoA for histone H3 acetylation and gene expression, promoting lysosomal biogenesis, autophagy, cell survival, and brain tumorigenesis.91 In castration-resistant prostate cancer (CRPC), the Kruppel-like factor 5 (KLF5) protein and HDAC3 collectively bind to the Beclin1 promoter and suppress its transcription, leading to autophagy suppression and increased drug sensitivity to docetaxel. Moreover, coupling MYC and HDACs, especially class I HDACs, such as HDAC2, negatively epigenetically regulates autophagic and lysosomal function by concomitantly affecting histone acetylation and gene expression of TFEB, TFE3, and FOXH1 TFs, as well as autophagic and lysosomal elements. MYC occupies the promoters of lysosome and ATGs, resulting in TFEB, TFE3, and FOXH1 promoter dissociation.90 As a classic tumor inducer, MYC is frequently overexpressed and genetically mutated in numerous human cancers.92 Therefore, it is likely to support the notion that tumors rely on the HDAC/MYC-TF axis to sustain proliferation and growth by repressing autophagic and lysosomal systems. HDACs is supportive in autophagy-induced diabetic retinopathy and renal fibrosis. In the development of diabetic retinopathy, overexpressed histone HIST1H1C/H1.2, an important variant of the linker histone H1, upregulates SIRT1 and HDAC1 to maintain the deacetylation of H4K16, leads to ATG proteins expression, and then promotes autophagy, inflammation and cell toxicity.93 Small-chain fatty acids treatment-reduced HDAC2 upregulates H3K27ac on ULK1 promoter, reduces ULK1 expression and autophagy flux and thus alleviate diabetic renal fibrosis94 (Fig. 3).

Histone acetylation and autophagy regulation. a Glucose deprivation assists AMPK-mediated ACSS2 phosphorylation and promotes its nuclear localization. In the nucleus, ACSS2 binds to TFEB and translocates to lysosomal and autophagy gene promoters, where ACSS2 generates acetyl-CoA for histone H3 acetylation and gene expression. b KLF5 protein and HDAC3 bind to the Beclin1 gene promoter and suppress its transcription, leading to autophagy suppression. c Coupled with MYC, HDACs, especially class I HDACs, epigenetically negatively regulate autophagic and lysosomal function. d HIST1H1C/H1.2 upregulates SIRT1 and HDAC1 to maintain the deacetylation of H4K16, leads to ATG proteins expression and then promotes autophagy, inflammation and cell toxicity. e Small-chain fatty acids treatment-reduced HDAC2 upregulates H3K27ac on ULK1 promoter, reduces ULK1 expression and autophagy flux and thus alleviate diabetic renal fibrosis
MicroRNA
MicroRNA (miRNA) is a major class of small noncoding RNA, which is quite conserved in biological evolution, typically blocks gene expression at a post-transcriptional level. The role of some specific miRNAs in regulating disease-related autophagy pathway have been revealed, and they mainly directly target autophagy-related components or signaling effector proteins to induce or inhibit autophagy process. There are a set of miRNAs that share in more than one disease and represent their universal pathogenic roles (Fig. 4).

MicroRNA and autophagy regulation. In Behcet’s disease, HBV and mycobacterial infection, decreased miR-155 respectively inhibited TAB-AKT/mTOR-, SOCS1/Akt/mTOR- and Rheb/mTOR-dependent autophagy. miR-30a, miR-143 and miR-142-3p respectively targeted Beclin1/ATG5, ATG7/ATG2B and ATG5/ATG6 mRNA for degradation to inhibit autophagy and increase chemotherapy sensitiveness. miR-30a mediated autophagy suppression via targeting Beclin1 or ATG5 and alleviated arterial injury and airway fibrosis. Downregulated miR-142-3p could target ATG16L1, ATG4c to trigger autophagy and eliminate mycobacteria. miR-143 inhibited autophagy via targeting ATG7 or ATG2B, which induced Crohn’s disease or in other case, alleviated cardiac injury
miR-155-regulated autophagy implicates in viral hepatitis, Behcet’s disease and tuberculosis. miR-155 reinforces HBV replication by reinforcing the SOCS1/Akt/mTOR axis-stimulated autophagy.95 Decreased miR-155 in Behcet’s disease leads to defective autophagy in dendritic cells through Akt/mTOR pathway thereby stimulating cytokine production.96 After mycobacterial infection, enhanced miR-155 expression directly targets Rheb to accelerate autophagy process via inhibit Rheb/mTOR pathway for mycobacterial elimination.97 miR-30a-mediated autophagy suppression via targeting Beclin1 or ATG5 sensitizes tumor cells to chemotherapy,98,99,100 and alleviates airway fibrosis,101 arterial injury.102 Similarly, miR-142-3p could target ATG16L1, ATG4c or ATG5 to inhibit autophagy, promoting intracellular survival of mycobacterium tuberculosis or sensitizing hepatocellular carcinoma to sorafenib.103,104 Moreover, targeting ATG7, ATG2B or GABARAPL1, miR-143-inhibited autophagy promotes chemotherapy sensitivity of tumors, support cardiac progenitor cells survival and heart repair and increase inflammatory response in the development of Crohn’s disease.105,106,107,108
Post-translational regulation and autophagy-related diseases
After transcripts are translated into proteins, PTMs participate in further processing of the protein products by inducing their covalent linkage to new functional groups, such as phosphate, ubiquitin, methyl groups, and acetate, which are critical for protein activity, stability and folding, protein co-interaction, and function execution.109 Various PTMs on autophagy-related components, TFs, and upstream effectors have been identified as essential autophagy-regulating approaches leading to the activation and suppression of autophagy process. Here, we summarize PTMs of autophagy process in disease conditions, and their implications for disease progression and repression, expecting the discovery of potential new targets (Table 2).
Acetylation
HDAC- and HAT-mediated TF acetylation at the post-translational level is at least one mechanism by which autophagy is transcriptionally elicited/muted. Whether TF acetylation promotes or inhibits transcriptional activity remains controversial and is likely dependent on specific acetylation sites. For example, p53 acetylation enhances its transcriptional activity,110 while FOXO1 acetylation decreases its transcriptional activity.111 TFEB acetylation at K91, K103, K116, and K430 indicates increased transcriptional activity.112 Conversely, GCN5-catalyzed K274 and K279 acetylation disrupts their target gene binding.113 In cancer cells with HDAC inhibition, enhanced TFEB acetylation at K91, K103, K116, and K430 markedly transcriptionally activates the expression of its target genes related to autophagy and lysosomal biogenesis and induces cell death.112 GCN5-catalyzed K274 and K279 acetylation of TFEB impedes lysosome formation and further the clearance of Tau protein aggregates, exacerbating the neurodegenerative phenotypes.113
Tip60-mediated p53 acetylation at K120, collaborating with PIASy-mediated K386 sumoylation, functions as a death signal to promote p53 cellular accumulation and autophagy.114 In addition, TFAM knockdown in tumor cells increases NAD(+)/NADH ratio and leads to SIRT1 upregulation to deacetylate p53 at K382 sites, which disturbs autophagy via weakening transcriptional regulation of the PISD enhancer.115 IKKα plays a cytoprotective role when treating human hepatoma cells with the antitumor therapeutic reagent arsenite. Interestingly, it triggers a feedback loop of autophagy-dependent degradation through p53 acetylation and thus contributes to arsenite-induced apoptosis. During this process, checkpoint kinase 1 (CHK1) activation phosphorylates p53 and undergoes p300/CBP-mediated acetylation at K373 and/or K382, triggering autophagy and autophagy-mediated IKKα degradation.116 Furthermore, SIRT1-mediated p53 deacetylation promotes autophagy and alleviates sepsis-induced acute kidney injury.117
HDACIs can induce autophagy through FOXO1-dependent pathways, whereas autophagy inhibition markedly enhances HDACI-mediated cell death.118,119 Acetylated FOXO1 may be responsible for HDAC-mediated autophagy regulation. When oxidative stress or serum starvation occurs, cytoplasmic FOXO1 is acetylated at K262, K265, and K274 via SIRT2 dissociation, facilitating the interaction between cytosolic FOXO1 and ATG7 and the autophagic process, leading to tumor suppression in an autophagy-dependent manner.15,120 In response to Angiotensin II stimuli, HDAC4 expression is upregulated in vascular endothelial cells, which consequently deacetylates FOXO3a to transcriptionally activate autophagy, and therefore leads to vascular inflammation.121
The acetylation status of a set of fundamental autophagy-related machineries has been well identified and proven to be involved in autophagosome and autolysosome formation.122 Including Beclin1, LC3, p62, and STX17, these critical autophagic effectors function distinctively in the autophagy process, depending on their acetylation status. By affecting spatial structure-dependent interactions with other autophagic cofactors, acetylated and deacetylated proteins represent either non-autophagic (acetylated LC3 and Beclin1) or active autophagic forms (deacetylated LC3, acetylated p62 and STX17).123
Typically, Beclin1 acetylation is emblematic of autophagy repression. In particular, p300- and SIRT1-regulated Beclin1 acetylation at the K430 and K437 sites is associated with tumor development,124 sensitivity impairment to anticancer drugs125 and sepsis-induced acute kidney injury/cardiac dysfunction.126,127 SIRT1 or SIRT6 overexpression deacetylates Beclin1 and activates autophagy. Activated autophagy has been shown to promote the epithelial-mesenchymal transition (EMT) of cancer cells.128,129 This adverse effect takes advantage of the autophagic protein degradation pathway to accelerate epithelial-specific protein reduction. Similarly, LC3 can also be a direct substrate of SIRT1. By deacetylating LC3, the interplay between LC3 and ATG3 is strengthened, assisting autophagy-p62-mediated phosphatase and tensin homolog (PTEN) degradation and accelerating liver cancer occurrence.129 Autophagy prompted by stabilized SIRT1-induced ATG5 and ATG7 protein deacetylation causes chemoresistance in liver cancer cells and protect vascular endothelial cells from oxidant-induced cell injury.130
Adiposity is confirmed to be associated with sex difference, SIRT1 and autophagy. Mechanistically, estrogen receptor alpha induced SIRT1 expression, which then deacetylates and activates AKT and STAT3, resulting in suppression of autophagy and adipogenesis via mTOR-ULK1 and p55 cascades.131 Moreover, HDAC4-STAT1 signaling-regulating autophagy inhibition is partially responsible for podocyte injury during diabetic neuropathy, in which STAT1 is deacetylated by HDAC4 to promote its phosphorylation and activation, which then translocates into the nucleus to induce gene expression, leading to the induction of inflammation and apoptosis and the suppression of autophagy.132
Cytoskeleton proteins, such as α-tubulin and cortactin, are quite essential for autophagy vesicular trafficking and autophagy-lysosome fusion. HDAC6 promotes autophagy by recruiting a cortactin-dependent, actin-remodeling machinery, which in turn assembles an F-actin network that stimulates autophagosome-lysosome fusion and substrate degradation. HDAC6 deficiency or deregulation results in autophagy dysfunction, and thus retardation of protein aggregates degradation.133
ATP13A2 mutations cause Kufor-Rakeb syndrome in juvenile-onset atypical Parkinson’s disease. HDAC6 is recruited by ATP13A2 to lysosome and deacetylates cortactin and tubulin to promote autophagosome-lysosome fusion and protein aggregates degradation.134 HDAC6 upregulation after spinal cord injury induces microtubules deacetylation and reduced stability, leading to autophagy inhibition and injury occurrence.135 In prostate cancer, p62 increases HDAC6 levels and reduces the acetylation of α-tubulin and the stability of microtubules, causing autophagy flux impairment and EMT.136 Cockayne syndrome group B (CSB) protein directly interacts with HDAC6 and acetyltransferase MEC-17 to antagonize the deacetylation of α-tubulin by HDAC6, leading to autophagy restoration and rescuing loss of subcutaneous fat.137 (Fig. 5). It is noteworthy that, in addition to HDAC6-medited cytoskeleton proteins acetylation, SIRT2 has also involved in this process, which regulates α-tubulin and Tau acetylation to affect autophagy vesicular traffic and cargo clearance in several neurological disorders, including Parkinson’s disease and Alzheimer’s disease.138

Acetylation modification and autophagy regulation. a TFEB acetylation at K91, K103, K116, and K430 markedly activates the expression of its target genes related to autophagy and lysosomal biogenesis and induces cell death. b GCN5-catalyzed K274 and K279 acetylation of TFEB impedes lysosome formation and the clearance of Tau protein aggreagates. c Tip60-mediated p53 acetylation at K120 and K386 sumoylation function as a death signal to promote p53 cellular accumulation and autophagy. d P53 acetylation at K382 sites affects autophagy levels via the transcriptional regulation of the PISD enhancer. e P53 is phosphorylated by CHK1 activation and undergoes p300/CEP-mediated acetylation at the K373 and/or K382 sites, which triggers autophagy and autophagy-mediated IKKα degradation. f Cytoplasmic FOXO1 is acetylated at K262, K265, and K274 via SIRT2 dissociation, facilitating the interaction between cytosolic FOXO1 and ATG7 and the autophagic process. g Upregulated HDAC4 deacetylates FOXO3a to transcriptionally activate autophagy. h P300 and SIRT1/6-regulated acetylation of Beclin1 at the K430 and K437 sites. i SIRT1 overexpression deacetylates LC3, ATG5 and ATG7 and activates autophagy. j ERα-induced SIRT1 expression deacetylates and activates AKT and STAT3, resulting in suppression of autophagy via mTOR-ULK1 and p55 cascade. k HDAC4-STAT1 signaling-regulating autophagy inhibition. (l) HDAC6-mediated cytoskeleton proteins acetylation regulation
Ubiquitination
Ubiquitination is a reversible enzymatic process of ubiquitin linkage and removal catalyzed by a succession of ubiquitin ligases (including E1, E2, and E3) and deubiquitinating enzymes. Mono- or polyubiquitination, K48- or K63-linked ubiquitination, and various modified residues on autophagy-related components dictate whether to inhibit or activate autophagy139 (Fig. 6).

Ubiquitination and autophagy regulation. GCA triggers TRAF6 activity to induce K63-linked ULK1 ubiquitination. ULK1 deubiquitination by USP1 is required after TRAF6-induced ULK1 ubiquitination. Upon autophagy induction, ULK1 autophosphorylation facilitates its recruitment to KLHL20 for ubiquitination and proteolysis, which restrains the amplitude and duration of autophagy. TLR-mediated signaling can induce autophagy via the association of TRAF6 with the coiled-coil domain of Beclin1 and Beclin1 ubiquitination. A20 reduces the extent of K63-linked Beclin1 ubiquitination. PRDX1 binds to TRAF6 to inhibit K63-linked-ubiquitination of TRAF6, leading to reduced TRAF6 ubiquitin-ligase activity, which fails to activate Beclin1. KLHL20, UBE3C and TRABID reciprocally regulate K29/48-branched ubiquitination of VPS34, mediating its proteasome degradation. TRAF6 polyubiquitinates LC3B and promotes LC3B-ATG7 complex formation to drive selective autophagic CTNNB1 degradation. HACE1 ubiquitinates OPTN with K27 and K48 ubiquitin linkages and on multiple lysine residues of OPTN containing K193. OPTN ubiquitination at K193 promotes the interaction with p62 to activate autophagy. USP8 deubiquitinates p62 by removing the K11-linked ubiquitin chains from p62, and the deubiquitinated site is principally located at K420 site, which leads to p62 reduced degradation and autophagy flux. p62 can be also ubiquitinated by Keap1/Cullin3 to promote p62’s association with LC3 and autophagy
ULK1 ubiquitination
TNF receptor-associated factor 6 (TRAF6) and ubiquitin specific peptidase 1 (USP1) are ubiquitinases and deubiquitinases that deal with unc-51-like autophagy activating kinase 1 (ULK1) ubiquitin modifications. In imatinib-resistant chronic-phase chronic myeloid leukemia (CML), upregulated GCA triggers TRAF6 ubiquitinase activity to induce K63-linked ULK1 ubiquitination and reduces K48-linked ubiquitination. This results in ULK1 stabilization and pro-survival autophagy activation.140 For robust post-initiation autophagy, ULK1 deubiquitination by USP1 is required after TRAF6-induced ULK1 ubiquitination, promoting breast cancer cell survival and proliferation. Upon USP1 dysfunction or depletion, canonical autophagy cannot be fully activated; conversely, it turns out to be uncanonical autophagy, only using part of the autophagic machinery for cargo clearance. USP1 inhibition may potentiate the death of autophagy-competent cancer cells.141 In addition to be involved in autophagy initiation, ULK1 is also a critical autophagy brake. Upon autophagy induction, ULK1 autophosphorylation facilitates its recruitment to KLHL20 for ubiquitination and proteolysis, which restrains the amplitude and duration of autophagy. Impairment of KLHL20-mediated regulation of autophagy dynamics potentiates starvation-induced cell death and aggravates diabetes-associated muscle atrophy.142
TRAF6-Beclin1
TRAF6 ubiquitinates Beclin1 generally activated by K63-linked ubiquitination. Various upstream signaling molecules influence TRAF6-Beclin1. Toll-like receptor (TLR)-mediated signaling can induce autophagy via the association of TRAF6 with the coiled-coil domain of Beclin1 and ubiquitination of Beclin1. The deubiquitinating enzyme A20 reduces the extent of TLR signaling-induced K63-linked Beclin1 ubiquitination.143 Interestingly, autophagy induction contributes to TLR4- and TLR3-triggered progression of lung cancer cells through TRAF6 ubiquitination.144 In contrast, peroxiredoxin 1 (PRDX1), an antioxidant protein, counteracts TRAF6-dependent Beclin1 ubiquitination induced by TLR4 stimulation. Mechanistically, PRDX1 binds to the ring finger domain of TRAF6 to inhibit its own K63-linked-ubiquitination of TRAF6, leading to reduced TRAF6 ubiquitin ligase activity and Beclin1 inactivation.145 Similarly, p62 can block the TRAF6-Beclin1 combination and catalytic processes by competitively interacting with Beclin1.146 Another non-negligible example of TRAF6-Beclin1 axis regulation involves tripartite motif-containing 59 (TRIM59), a tripartite motif protein with potential oncogenic activity. In NSCLC cells, TRIM59 expression is reciprocally correlated with Beclin1 expression and basal autophagy levels. TRIM59 repress Beclin1 transcription by negatively regulating the NF-κB pathway, it also eliminates active ubiquitinated Beclin1 for TRIM59 functions as an E3 ligase that mediates K48-linked TRAF6 ubiquitination and promotes proteasomal TRAF6 degradation.147
VPS34 ubiquitination
Ubiquitin ligase UBE3C and deubiquitinating enzyme TRABID have been found to reciprocally regulate K29/K48-branched ubiquitination of VPS34. This ubiquitination enhances proteasome-dependent degradation of VPS34, thereby suppressing autophagosome formation and maturation. TRABID is downregulated during the pathogenesis of steatosis, which decreases VPS34 stability and results in lipid metabolism disturbance.148 KLHL20 ubiquitin ligase can be recruited to VPS34 complex to mediates VPS34, Becline1, ATG14 ubiquitination and degradation. Impairment of KLHL20-mediated regulation of autophagy dynamics aggravates diabetes-associated muscle atrophy.142
TRAF6-LC3B
Aberrant CTNNB1 signaling frequently occurs in cancers, particularly colorectal cancer (CRC). TRAF6 can polyubiquitinate LC3B and promote LC3B-ATG7 complex formation to drive selective autophagic CTNNB1 degradation, beneficial for inhibiting cancer metastasis.149 However, in most clinical CRC, TRAF6 is phosphorylated at Thr266 by glycogen synthase kinase 3 beta (GSK3β), leading to increased K48-linked polyubiquitination and degradation, thereby inhibiting autophagy-dependent CTNNB1 reduction.149
HACE1-OPTN
As a tumor suppressor, ubiquitin ligase HACE1 ubiquitinates optineurin (OPTN) with K27 and K48 ubiquitin linkages and on multiple lysine residues of OPTN containing K193. OPTN ubiquitination at K193 promotes p62 interactions to activate autophagy, accelerating damaged protein degradation and hampering tumorigenicity.139
p62 ubiquitination
Ubiquitin stress is an inducer of p62 ubiquitination catalyzed by E2 ubiquitin conjugating enzymes UBE2D2 and UBE2D3. Ubiquitylation of p62 disrupts dimerization of the UBA domain of p62, liberating its ability to recognize polyubiquitylated cargoes for selective autophagy.150 Moreover, p62 can be also ubiquitinated by Keap1/Cullin3, with the modified site at K420 within its UBA domain to promote p62’s association with LC3 and autophagy.151 SPOP mutation is frequently mutated in prostate cancer. Different from above ubiquitin-induced p62 degradation and autophagy flux, the gene encoding the E3 ubiquitin ligase substrate-binding adapter SPOP leads to non-degradative ubiquitination of p62 at K420 and thus autophagy suppression.152 Conversely, USP8 has been reported to directly deubiquitinate p62. It preferentially removes the K11-linked ubiquitin chains from p62, and the deubiquitinated site is principally located at K420 site, which leads to p62 reduced degradation and autophagy flux.153
Phosphorylation
Phosphorylation is one of the most ubiquitous PTMs and is antagonistically orchestrated by a set of protein kinase and protein phosphatase families. Protein phosphorylation is a fundamental step in signal transduction. Over the past few years, extensive efforts have been made to explore crisscross signaling transduction pathways to manipulate autophagy. Direct phosphorylation events of an array of autophagy-relevant components are fairly critical for autophagy regulation, some of which even influence multiple disease-related biological processes (Fig.7).

Phosphorylation and autophagy regulation. TOPK phosphorylates ULK1 at Ser469, Ser495, and Ser533, decreasing ULK1 activity and stability. LPS-activated p38 MAPK phosphorylates ULK1 at S757 and S504, preventing it from binding to ATG13, and reduced autophagy in microglia. GS3Kβ phosphorylates ULK1 at S405 and S415 sites, and thus promotes ULK1 and LC3B interaction. Activated ULK1 phosphorylates p62 and ATG14 to accelerate autophagy flux. Gadd45beta-MEKK4 pathway specifically directs p38 to autophagosomes, and p38 catalyzes phosphorylation of ATG5 at T75, resulting in an accumulation of autophagosomes through inhibition of lysosome fusion. ELP3 enhances PAK1 activity, thereby leading to ATG5 phosphorylation at T101. ATG4B is a direct MST4 substrate. MST4 phosphorylates ATG4B at S383 and contributes to increased autophagy activity. TRIM2 selectively ubiquitinates AXL to promote its autophagic degradation due to OPTN binding to the polyubiquitin chain of AXL. p85β mediates the phosphorylation of TRIM2 at the S443 residue and OPTN at S526, contributing to inhibited AXL degradation. Ras/PI3K/AKT/mTOR/CK1α/FOXO3 pathway limits an autophagy flux. CK1α/PTEN/AKT/FOXO3/ATG7 route excites autophagy. Polyubiquitination catalyzed by NEDD4-1 and phosphorylation by GSK3β and CK2 at the PTEN C-terminal reduce PTEN protein stability and inactivate phosphatase activity, activated PTEN blocks AKT phosphorylation at S473, subsequently inducing activated FOXO3a phosphorylation at S253. PP2A-DAPK1-Beclin1 and PP2A-AMPKα phosphorylation signaling pathways are involved in disease-related autophagy regulation
ULK1 phosphorylation
ULK1 protein contains quite rich phosphorylation modified sites, through which modulates autophagy inhibition or activation in response to various environmental conditions, engaging in different autophagy stages. In neuroinflammatory diseases, LPS-induced p38 MAPK activation phosphorylates ULK1 at S757 and S504, preventing it from binding to the downstream effector ATG13, and reduced autophagy in microglia.154 T-LAK cell-originated protein kinase (TOPK), an oncokinase, could directly bind to and phosphorylate ULK1 at Ser469, Ser495, and Ser533, decreasing ULK1 activity and stability. ULK1 Ser469, Ser495, and Ser533 mutations induce autophagy initiation and enhance the sensitivity of glioma cells to temozolomide (TMZ).155 High phosphorylation levels of ULK1 at S405 and S415 sites were observed in human pancreatic cancer cells. This phosphorylation situation is mediated by GSK3β and thus promote the interaction of ULK1 and MAP1LC3B to exhibit high levels of autophagy.156
ULK1, as a serine/threonine kinase, is also able to be implicated in the phosphorylation of downstream autophagic effector proteins. TBK1 coordinates with ULK1 to promote concerted phosphorylation of autophagy receptor SQSTM1/p62 at S351 site within the UBA domain. In amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD), mutant TBK1 reduces SQSTM1/p62 phosphorylation and compromises cargo binding and clearance, responsible for related neurotoxicity.157 It has found that ULK1 can regulate ATP14 phosphorylation, which displays a special role in the context of Huntington’s disease. ULK1 phosphorylates ATG14 at S29 in an mTOR-dependent manner, and this phosphorylation critically regulates ATG14-VPS lipid kinase activity to control autophagy level for the clearance of polyglutamine disease protein.158
ATG5 phosphorylation
ATG5 phosphorylation at T75 site is a representative marker of inhibitory autophagy. Gadd45beta-MEKK4-p38 pathway is one of the regulatory mechanisms responsible for this phosphorylation. Gadd45beta-MEKK4 pathway specifically directs p38 to autophagosomes, and then p38 catalyzes phosphorylation of ATG5 at T75, resulting in an accumulation of autophagosomes through inhibition of lysosome fusion.159 Furthermore, in brain tumor, the oncogenic role of PAK1 is mainly associated with autophagy. Under hypoxia condition, ELP3 enhances PAK1 activity, thereby leading to ATG5 phosphorylation at T101. This event not only protects ATG5 from ubiquitination-dependent degradation but also increases the affinity between the ATG12-ATG5 complex and ATG16L1.160
OPTN phosphorylation
TBK1-meidated phosphorylation of OPTN is functional in selective autophagy of damaged mitochondria and Salmonella. TBK1 phosphorylates OPTN’s UBAN domain at S473, thereby expanding the binding capacity of OPTN to diverse Ub chains. Meanwhile, phosphorylation of S177 and S513 promotes recruitment and retention of OPTN/TBK1 on ubiquitinated, damaged mitochondria.161 Moreover, TBK1 can also phosphorylate OPTN at S177 site, which helps to enhance LC3 binding affinity and autophagic clearance of cytosolic salmonella.162 p85β, encoded by PI3KR2, is the p85 regulatory subunit of PI3K and is frequently amplified in cancers. The receptor tyrosine kinase AXL is an upstream PI3K regulator. In multiple cancer cells, AXL, an indicator of malignant phenotypes and poor prognosis, is upregulated but is rarely detected as genetically aberrant.163,164 The Ubiquitinase TRIM2 selectively ubiquitinates AXL to promote autophagic degradation due to OPTN binding to the polyubiquitin chain of AXL. However, p85β mediates the phosphorylation of TRIM2 at residue S443 and OPTN at residue S526, thereby inhibiting AXL degradation and activating AXL oncogenic signaling.165 However, how p85β regulates TRIM2 and OPTN phosphorylation remains to be elucidated, and this will be more conducive to further understanding the autophagic control mechanism in the AXL/p85β context.
ATG4B phosphorylation
ATG4B is an intriguing ATG that has been reported to have tumorigenic functions and is regarded as a potential therapeutic target and prognostic marker.166,167 As a member of the mammalian sterile20-like (STE) serine/threonine kinase (STK) family, MST4 is prone to promoting tumor development through its phosphorylation activity (e.g., activating the tumor-promoting p-ERK pathway168). ATG4B is a direct substrate of MST4. MST4-directed ATG4B phosphorylation at S383, but not at other described phosphorylation sites, contributes to increased autophagy activity, consequently facilitating tumorigenesis and drug resistance to RT in glioblastomas.169 Interestingly, ATG4B phosphorylation can also be independent of autophagic flux to perform tumorigenic functions. Instead of affecting autophagy flux, AKT-mediated ATG4B phosphorylation at residue S34 aims to repress mitochondrial activity and enhance the Warburg effect in HCC cells through mitochondrial ATG4B translocation and subsequent inhibition of mitochondrial respiration-related F1Fo-ATP synthase.170
CK1α mediated phosphorylation
Casein kinase 1 alpha (CK1α) belongs to a family of highly conserved monomeric Ser/Thr kinases, engaging in various biological functions, including membrane trafficking, cell cycle, cell metabolism, apoptosis, and autophagy. Focusing on autophagy regulation, we observed that the effects of CK1α were two-sided.
In one scenario, the Ras/PI3K/AKT/mTOR/CK1α/FOXO3 pathway limits autophagic flux. Oncogenic Ras-driven cancer cells exhibit elevated basal autophagy to cope with the pressure of rapid growth. CK1α has been found to be a negative regulator in oncogenic RAS-induced autophagic levels. Theoretically, oncogenic Ras, via its downstream PI3K/AKT/mTOR effector pathway, increases CK1α protein abundance. Later, elevated CK1α additionally phosphorylates already S315 site-phosphorylated FOXO3a at S318 and S321 to destabilize FOXO3a. FOXO3a is a positive TF in ATG regulation, increasing the expression of LC3B, ATG12, BNIP3, and others. Thus, unstable and null FOXO3 induced by CK1α impedes the expression of a cohort of ATGs and limits the autophagic recycling of nutrients.171
In another scenario, the CK1α/PTEN/AKT/FOXO3/ATG7 pathway excites autophagy. Loss of PTEN function is a frequent event in cancers owing to post-transcriptional and PTMs. At the PTEN C-terminal, polyubiquitination catalyzed by NEDD4-1 is associated with reduced protein stability, and phosphorylation by GSK3β and CK2 is linked to inactive phosphatase activity. CK1α, as a PTEN-interacting partner, strongly interacts with PTEN to antagonize NEDD4-1 induced PTEN polyubiquitination and competitively abrogate PTEN phosphorylation, especially at T366 and S385, which are the main sites catalyzed by GSK3β and CK2. Concomitantly, functional PTEN reduces AKT phosphorylation at S473, a signal of AKT inhibition, followed by specific FOXO3a phosphorylation at S253. Finally, ATG7, but not LC3B or ATG5, is exclusively transactivated by FOXO3a to induce autophagy, leading to NSCLC cell growth suppression.172
PP2A-involved phosphorylation
Being involved in multiple autophagy-related signaling transduction pathways, protein phosphatase 2A (PP2A) is in a close correlation with the development of multiple diseases. PREP, a serine protease has been demonstrated to interact with PP2A and its endogenous inhibitor PME1 and activator PTPA, thus adjusting its activity and levels of PP2A. PREP inhibition represses PP2A phosphorylation and increases its activity, leading to DAPK1 and Beclin1 phosphorylation, and therefore autophagy induction. Several neurodegenerative diseases and cancers are connected with lower PP2A activity and PP2A activators may manifest great potential as drug therapy.173,174,175 After ischemia/reperfusion, increased PP2A and decreased AMPKα phosphorylation at T172 has been observed, attributing to acute kidney injury. Mechanistically, ischemia/reperfusion-triggered PP2A dephosphorylates T172-AMPKα, which decreases AMPK activity, suppresses ULK1-mediated autophagy, and thus impedes dysfunctional mitochondria clearance.176 Targeting PP2A-AMPK axis may be an effective therapeutic method to relieve cell injury due to abnormal autophagy dysfunction. As reported, GNMT deficiency in steatotic patients elevates the levels of methionine and SAMe, which accounts for PP2A methylation and its activity enhancement, leading to impaired autophagy flux and aggravating liver steatosis. Employing a methionine deficient diet normalizes the methylation capacity and PP2A methylation levels, benefiting autophagy restoration and lipid metabolism.177
Crosstalk between phosphorylation and ubiquitination in autophagy regulation
SMURF1 is a HECT-type E3 ubiquitin ligase that regulates various biological signaling networks and is involved in different diseases and disorders. However, studies on autophagy modulation with SMURF1 engagement and mediation are limited. Casein kinase (CSNK) isoforms have been identified as constitutive autophagy-regulating kinases.178 Recently, a fascinating study verified that SMURF1 could form a complex with UVRAG by binding to the PPxf motif, and catalyze K29- and K33-linked UVRAG polyubiquitination at residues K517 and K599.179 Ubiquitinated UVRAG, instead of inducing ubiquitination-mediated protein degradation, blocks RUBCN’s approach to the UVRAG-containing PIK3C3 complex, which inhibits the negative effects of RUBCN and promotes autophagosome maturation.179,180 Nevertheless, CSNK1A1-mediated UVRAG phosphorylation at S522 disrupts SMURF1-UVRAG binding and reverses UVRAG ubiquitination-mediated autophagosome maturation.179 UVRAG K29- and K33-linked ubiquitination or prevention of UVRAG (S522) phosphorylation facilitates autophagy-dependent EGFR degradation and inhibits HCC growth,179 indicating a potential therapeutic strategy targeting post-translationally modified autophagy-related proteins (Fig. 8a).

Crosstalk between phosphorylation and ubiquitination in autophagy regulation. SMURF1 forms a complex with UVRAG by binding to the PPxf motif and catalyzes K29- and K33-linked polyubiquitination of UVRAG at the K517 and K599 residues. Ubiquitinated UVRAG blocks RUBCN’s approach to the UVRAG-containing PIK3C3 complex, inhibiting the negative effect of RUBCN and promoting autophagosome maturation. CSNK1A1-mediated UVRAG phosphorylation at S522 disrupts the binding of SMURF1 to UVRAG and reverses UVRAG ubiquitination-mediated autophagosome maturation. CaMKII phosphorylates Beclin1 at S90 and subsequently increases TRAF6-catalyzed Beclin1 K63-ubiquitination, collectively contributing to autophagy induction for K63-linked ubiquitylated Id-1/2 degradation
Moreover, calcium/calmodulin-dependent protein kinase II (CaMKII) was found to influence post-translational autophagy regulation. CaMKII is a serine/threonine-protein kinase activated by Ca2+ and calmodulin (CaM). Notably, the Ca2 +/CaMKII-regulating signaling pathway that triggers or inhibits autophagy varies and is mainly dependent on specific cell types and contexts. For instance, the BAFF treatment-activated Ca2+-CaMKII-dependent Akt/mTOR signaling pathway inhibits autophagy and promotes cell proliferation and survival in normal and neoplastic B-lymphoid cells,181 whereas CaMKII directly targeting Beclin1 can induce autophagy to promote solid tumor cell differentiation. In the latter regulatory mechanism, CaMKII directly phosphorylates Beclin1 at S90 and subsequently increases TRAF6-catalyzed K63-ubiquitination of Beclin1, collectively contributing to autophagy induction for K63-linked ubiquitylated Id-1/2 degradation182 (Fig. 8b).
Epigenetic drugs and autophagy
Epigenetic compounds have become imperative tools for disease treatments, including treatment of cardiovascular disorder, infectious disease, and particularly tumor, which manifest potent therapeutic effects by eliciting apoptosis, cell cycle arrest, and differentiation (currently clinically applied epigenetic drugs are described and summarized elsewhere27). Although a few epigenetic drugs have showed great therapeutic effectiveness in clinical development and several have been officially approved by the FDA, the application of epigenetic drugs in disease treatment remains limited and still cell or disease type-dependent. Current studies have been investigating the potential therapeutic effectiveness of these drugs in multiple disease. However, it has been found that therapeutic effectiveness varies a lot in different contexts, and relevant molecular events are also vague and remain to be vividly elucidated. Autophagy is a multifaceted functional event that promotes cytoprotective or cell-damage, antitumor or tumorigenic activities. Indeed, numerous scientific studies have indicated the dual autophagic effects that is associated with diverse therapeutic consequences of epigenetic drug treatment, either serving as a synergistic mechanism to strengthen therapeutic response or an antagonistic mechanism to induce drug resistance and loss of effectiveness. Here, we summarize the possible molecular mechanisms by which autophagy distinctively affects the therapeutic consequences of epigenetic compounds, to provide novel insights into the combined utilization of epigenetic drugs and autophagy activators or inhibitors (Table 3).
DNMT inhibitors
Se-allylselenocysteine (ASC) can decrease global DNA methylation levels by downregulating DNMT1 expression.183 In human CRC, ASC induces autophagic cell death through increased PCDH17 expression and reduces hypermethylation of its promoter in human CRC. Another DNMT inhibitor, decitabine (DAC), is widely prescribed for treating acute myeloid leukemia (AML) and is recommended for patients with high-risk myelodysplastic syndrome (MDS). Sustained exposure to DAC treatment leads to apoptotic cell death in AML by promoting differentiation, senescence, and autophagy,184 which may be due to the unmethylated role of DAC in downregulating TIGAR.185 In high-risk MDS patients, FOXO3A reactivation is responsible for DAC-induced autophagy, cell cycle arrest, and apoptosis. However, unlike cancer cell-adverse autophagy induced by the aforementioned drugs, autophagy appears to serve as a pro-survival pathway in MDS patients with long-term exposure to 5-Azacitidine.186,187 The novel autophagy inhibitor ROC-325 augments the antileukemic activity of azacytidine.188
HDACIs
HDACIs can induce or inhibit autophagy. It has been reported that NF-kB hyperacetylation, p53 deficiency and acetylation, FOXO1 transcription, reactive oxygen species (ROS) accumulation, nuclear factor erythroid 2-related factor 2 (NRF2) and p21 upregulation, and mTOR inhibition might account for HDACI-induced autophagy.189
On the one hand, autophagy activation attenuates HDACI therapeutic effects and promotes cancer cell survival or sustained tissue damage. For example, suberoylanilide hydroxamic acid168 induces autophagy, accelerating mutated p53 degradation and assisting cancer cell survival.190 In breast cancer cells, SAHA targeting HDAC3 and HDAC6 elicits autophagy by downregulating survivin and XIAP gene transcription and inducing survivin protein acetylation and early nuclear translocation.191 Trichostatin A (TSA) strengthens autophagy levels by blocking the mTOR pathway and increasing FOXO1 transcriptional activity, whereas autophagy inhibition, such as using autophagy inhibitor chloroquine, can facilitate TSA-induced cell death and apoptosis.192,193 As an obligatory element, autophagy plays important role in pathological cardiac remodeling. TSA treatment suppresses autophagy via affecting HDAC1/2 activity and thus attenuates cardiac hypertrophy.194 Additionally, SIRT1/2 inhibition displays anticancer potential in a set of cancer types. However, in lung cancer cells, the SIRT1/2-specific inhibitor salermide upregulates pro-survival autophagy via HSPA5 acetylation and subsequent activation of activating transcription factor 4 (ATF4) and DNA damage inducible transcript 4 (DDIT4) to inhibit the mTOR signaling pathway.195
In contrast, autophagy accompanied by HDACIs manifests a synergistic therapeutic effect, such as enhancing cancer cell sensitivity to chemotherapy and alleviating cell injury. In gastric cancer, HDAC1/2 is overexpressed, displaying a positive correlation with poor prognosis. Valproic acid (VPA) treatment inhibits HDAC1/2 activity and induces autophagy through the HDAC1/PTEN/Akt signaling pathway, leading to increased apoptosis.196 VPA, combined with the mTOR inhibitor temsirolimus, synergistically inhibits cell growth and triggers autophagic cell death in the context of Burkitt leukemia/lymphoma.197 In addition, VPA-induced autophagy attenuates sepsis-induced myocardial dysfunction via HDAC1/3 inhibition and PTEN/AKT/mTOR pathway.198 Spermidine, a natural compound, induces autophagy by inhibiting EP300, which is primarily related to its cytoplasmic effects.199 In HCC treatment, spermidine promotes HDAC4 nuclear translocation to elevate cytoplasmic MAP1S acetylation, leading to autophagy induction and tumor development inhibition. Furthermore, sulforaphane, a natural histone deacetylase inhibitor, induced autophagy in triple-negative breast cancer. The underlying mechanism is associated with HDAC6 repression, which results in membrane translocation and PTEN acetylation.200 A novel HDACI, TMU-35435, has also been reported to suppress HDAC6 and cause misfolded protein aggregation, inducing autophagic cell death and enhancing radiation sensitivity in triple-negative breast cancer treatment.201 Additionally, MHY2256, a new synthetic histone deacetylase inhibitor, induces apoptosis and autophagic cell death via p53 acetylation and SIRT1 inhibition.202 Moreover, SAHA and TSA-induced autophagy via AMPK/mTOR pathway protects kidney injury from cisplatin treatment in proximal tubular cells.203 Interestingly, TSA can induce autophagy process in macrophage, modulate its M2 phenotype transformation to reduce sepsis-induced organ injury.204 These evidences demonstrating accessible treatment methods combining HDACIs and autophagy activators.
Bromodomain and extra-terminal domain inhibitors
The BET family of proteins are epigenetic readers that recognize acetylated lysine residues on histone and non-histone proteins. Bromodomain-containing protein 4 (BRD4), a member of the BET family, has recently been shown to be involved in ATG expression transcriptional regulation. Under nutrient-sufficient conditions, BRD4 recognizes KAT8/hMOF-resultant histone H4K16 acetylation targets on the promoter regions of autophagy and lysosome genes. Subsequently, G9a is recruited to catalyze H3K9 demethylation and represses autophagy gene transcription. Upon nutrient deprivation, the histone deacetylase SIRT1 is released from its inhibitory molecule, CCAR2/DBC1, in an AMPK-dependent manner. Active SIRT1 then deacetylates H4K16, which in turn dissociates BRD4 from the promoter regions and leads to active autophagic transcription programs.205,206
As a BRD4 inhibitor, JQ1 has been shown to promote functional recovery from spinal cord injury by activating autophagy in an AMPK/mTOR/ULK1 pathway-dependent manner.207 Similarly, upregulation of BRD4 in diabetic mouse hearts inhibits PINK1/Parkin-mediated mitophagy, resulting in accumulation of damaged mitochondria and subsequent impairment of cardiac structure and function. JQ1 activates mitophagy via PINK1/Parkin, helping improve mitochondrial function and ameliorate diabetic cardiomyopathy.208 Also, JQ1 is able to induce ferroptosis, where ferritinophagy (a process characterized by increased autophagy and iron levels) is required to enhance cell death.209 In this process, JQ1-mediated autophagy degrades ferritin heavy chain 1 (FTH1), leading to enhanced intracellular iron and ROS levels. Upon autophagy inhibition, ferroptosis, at least in part, is limited in the context of JQ1 treatment. Furthermore, other molecular agents, such as selective BRD4/HDAC dual inhibitor compound 17c (inducing BRD4-AMPK-mediated autophagic cell death and suppressing the IL6-JAK-STAT signaling pathway that is related to drug resistance) and a novel small-molecule BRD4 inhibitor 9 f (FL-411) have been developed and discovered as potential BRD4 inhibitors that can induce autophagic cell death in different cancer models.210 However, in AML, JQ1-activated autophagy, which is AMPK-ULK1-mediated and mTOR signaling-independent, results in JQ1 resistance in AML stem cells.211 Additionally, Akt/mTOR-mediated JQ1-induced autophagy causes JQ1 resistance in ovarian cancer cells.212
Histone methylase inhibitors
G9a inhibition can mechanistically trigger autophagy. In several cancer models, including multiple myelomas and glioblastomas, the G9a inhibitor BIX01294 targeting G9a induces cell proliferation inhibition and autophagy-associated apoptosis by inactivating the mTOR/4EBP1 pathway and transcriptionally reducing c-MYC levels.213,214,215 A genome-wide study illustrated that the interaction between MYC and G9a, with the MYC MBII region as the binding point, affects MYC’s capacity to drive transcriptional repression and tumorigenesis. Inhibiting G9a catalytic activity preferentially hinders MYC binding to repressed genes and shifts repressed loci with H3K9me2 enrichment to active epigenetic states.216 Abnormal G9a expression is also a survival advantage of head and neck squamous cell carcinoma. Instead of apoptosis or necrosis induction, pharmacological G9a suppression activates autophagic cell death mainly through the DUSP4-dependent ERK-mTOR dephosphorylation pathway.50 In addition, Two other potential antitumor agents, SH003 and kaempferol, have also been reported to efficiently inhibit G9a and induce autophagic cell death. The autophagy-associated tumor-inhibiting effects of SH003 and kaempferol appear to be a combination of G9a inhibition and endoplasmic reticulum (ER) stress, which involves the IRE1/JNK/CHOP and PERK/ATF4/CHOP signaling pathways.217,218
S-adenosyl-L-methionine (SAM)-competitive inhibitors is a major class of EZH2 inhibitors, such as EPZ005687, EI1, GSK126, UNC1999 and GSK343. In pancreatic cancer, GSK343 suppresses cell proliferation and induces apoptosis and autophagy via AKT/mTOR signaling pathway.219 In colorectal cancer, GSK343 and UNC1999 transcriptionally upregulated autophagy to promote cell death, but it is in an EZH2-independent manner.220 A comparative assessment of antitumor effects of drugs in epithelioid sarcoma indicated that autophagy induction is a possible survival mechanism in residual tumor cells following EZH2 inhibitor EPZ-011989 treatment, in which HMGA2 is a main player in this process.67 In contrast, in the progress of aortic dissection, UNC1999 employment to inhibit EZH2 activity appears to be detrimental for the function of vascular muscle cells (VSMCs), which is due to autophagic cell death exacerbation via MEK-ERK1/2 signaling pathway, highlighting from point to surface that the heterogeneity of outcomes of EZH2-induced autophagy in context-dependent diseases.64
Drugs targeting phosphorylation modification and autophagy
Since phosphorylation is a fairly significant process in autophagic signaling transduction pathway, efforts have been made to seek for some chemical agents that could modulate the phosphorylation modification of critical autophagy-related components to specifically and efficiently activate or suppress autophagy (Table 3).
A cell-based screen has found that a potent and highly selective ULK1 kinase inhibitor, SBI-0206965, suppresses ULK1-mediated phosphorylation events by decreasing Beclin1 and VPS34 phosphorylation, upregulating autophagy level and inducing tumor cell apoptosis.221 PIKfyve kinase inhibitors, such as WX8, can disrupt lysosome function in autophagy and selectively kill certain cancer cells. Analysis of biochemical changes caused by PIKfyve kinase inhibitors reveals that resistant cancer cells contain higher level of p38MAPK protein and phosphorylation, which subsequently results in phosphorylation of lysosomal-associated membrane protein 2 (LAMP2) and compensatory autophagy function. Combined use of PIKfyve and p38MAPK inhibitors, WX8 and SB202190, synergistically blocks autophagy and markedly reduces the viability of multiple cancer cell types.222 Under nutrient-replete conditions, the vertebrate nonreceptor tyrosine kinase SRMS phosphorylates FKBP51, disrupts the FKBP51-PHLPP complex. This prevents PHLPP-mediated dephosphorylation of AKT, causing sustained AKT activation that inhibits autophagy and promotes tumor growth. SRMS kinase inhibition activates autophagy and inhibits tumor growth, and can be accomplished by using FDA-approved tyrosine kinase inhibitor Ibrutinib.223 In gastric cancer, Salidroside decreases the phosphorylation of PI3K/AKT/mTOR and induces apoptosis and protective autophagy. Treatment with the autophagy inhibitor chloroquine enhanced Salidroside-induced apoptosis.224 TGF-β-mediated super-activation of urethra fibroblasts contributes to the progression of traumatic urethral stricture. However, this kind of super-activation can be reversed by the use of a Rho-associated kinase inhibitor Fasudil and autophagy inhibitors. During this process, Fasudil is able to hamper TGF-β-induced super-activation by hampering autophagy in an AKT/mTOR pathway-dependent manner.225 It has reported that PREP suppresses autophagy via PP2A-DAPK1-Beclin1 signaling pathway in the progression of neurodegenerative disorders. Further, a small-molecule inhibitor KYP2407 has been screened out to stimulate autophagy and reduces the accumulation of α-SYN aggregates.226 Exendin-4 is the GLP1R agonist and stimulates autophagic flux in a setting of chronic nutrient excess, such as in type 2 diabetes. Mechanistically, RAPGEF4/EPAC2 and downstream calcium signaling contributes dominantly to activated autophagic flux by exendin-4. Instead of being dependent on AMPK and mTOR signaling, this pathway regulates PPP3/calcineurin and then dephosphorylates its downstream effector TFEB to upregulate transcription of autophagy-related genes.227 Analogously, the natural compound trehalose promotes autophagy and ameliorates neurodegenerative phenotype by inducing rapid and transient lysosomal enlargement and membrane permeabilization. This effect correlated with the calcium-dependent phosphatase PPP3/calcineurin activation, TFEB dephosphorylation and nuclear translocation228 (Table 4).
Conclusion
Autophagy can be aberrantly activated or inhibited during disease occurrence and progression, displaying cytoprotective or cell-damage, oncogenic or tumor-inhibiting functions. Although the multifactor and heterogeneity of disease-relevant autophagy requires further elucidation, systematic experimental studies across diverse disease types have revealed a wealth of regulatory mechanisms to help understand the reasons for aberrant alteration of autophagy level. This review focuses on epigenetic and post-translational modifications, including classic DNA methylation, histone methylation and acetylation, microRNAs and non-histone acetylation, ubiquitination, phosphorylation to integrate the multiplicity of chemically modified enzymes, related modified targets, and their modification sites, providing potential disease-specific autophagic biomarkers and predicting the development of novel clinical therapeutic targets. Importantly, this review also investigates the effects of autophagy, accompanied by the application of a series of epigenetic drugs and some phosphorylase inhibitors. The distinct consequences of autophagy induction or inhibition may potentiate an optimal combination of autophagy inducers or inhibitors with drug therapies. However, there are still various constrains remain to be further investigated and solved.
At present, epigenetic and post-translational regulation of autophagy may not be comprehensive. Research on many other emerging modifications, such as those modified on mRNA, including N6-methyladenosine (m6A) and N1-methyladenosine (m1A), histone or non-histone phosphorylation, glycosylation, and sumoylation are still have not been elucidated clearly. It indicates that these unknown modifications in autophagy regulation deserve an investigation, which will facilitate to further understand regulatory mechanisms and explore new molecular targets. Moreover, autophagy-relevant detection in humans remains difficult and vacant in clinical tests. Due to the scarcity of certain molecules that can specifically represent autophagy activation or inhibition, it highlights the importance of seeking for highly specific autophagic indicators and the development of new detection approaches. Additionally, although many experimental studies have demonstrated the efficacy of combined treatment of autophagy activators or inhibitors with chemotherapy, RT, and immunotherapy, very few studies have investigated the outcomes of treatment with autophagy activators or inhibitors combined with epigenetic therapies. Simultaneously, we should also pay attention to the specificity of autophagy activators and inhibitors that would possibly result in unexpected treatment effects and unsensitive response. Exploring and identifying small-molecule modulators targeting specific autophagic proteins could be a promising approach to solve sensitivity and effectiveness of autophagy-related drugs.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Kim, K. H. & Lee, M. S. Autophagy–a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 10, 322–337 (2014).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Amaravadi, R., Kimmelman, A. C. & White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 30, 1913–1930 (2016).
Garcia-Martinez, L., Zhang, Y., Nakata, Y., Chan, H. L. & Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 12, 1786 (2021).
Ling, C. & Ronn, T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 29, 1028–1044 (2019).
Mizushima, N. & Levine, B. Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576 (2020).
Zhu, Z. et al. Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 20, 728 (2019).
Sun, Y., Cai, Y. & Zang, Q. S. Cardiac autophagy in sepsis. Cells 8, 141 (2019).
Lorzadeh, S., Kohan, L., Ghavami, S. & Azarpira, N. Autophagy and the Wnt signaling pathway: a focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118926 (2021).
Senft, D. & Ronai, Z. A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148 (2015).
Kimmelman, A. C. & White, E. Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043 (2017).
Jiang, G. M. et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 18, 17 (2019).
Sahni, S., Merlot, A. M., Krishan, S., Jansson, P. J. & Richardson, D. R. Gene of the month: BECN1. J. Clin. Pathol. 67, 656–660 (2014).
Gong, Y. et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 46, 588–594 (2014).
Zhao, Y. et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675 (2010).
Poillet-Perez, L. & White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 33, 610–619 (2019).
White, E., Mehnert, J. M. & Chan, C. S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 21, 5037–5046 (2015).
Wang, L. et al. Aberrant SIRT6 expression contributes to melanoma growth: Role of the autophagy paradox and IGF-AKT signaling. Autophagy 14, 518–533 (2017).
Rohn, T. T. et al. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 43, 68–78 (2011).
Pickford, F. et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199 (2008).
Tian, Y., Chang, J. C., Fan, E. Y., Flajolet, M. & Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl Acad. Sci. USA 110, 17071–17076 (2013).
Coffey, E. E., Beckel, J. M., Laties, A. M. & Mitchell, C. H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246e mutation can be reversed with camp. Neuroscience 263, 111–124 (2014).
Kim, S. et al. NDP52 associates with phosphorylated tau in brains of an Alzheimer disease mouse model. Biochem. Biophys. Res. Commun. 454, 196–201 (2014).
Usenovic, M., Tresse, E., Mazzulli, J. R., Taylor, J. P. & Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246 (2012).
Zhang, M. et al. Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis. Theranostics 11, 8624–8639 (2021).
Dewanjee, S. et al. Autophagy in the diabetic heart: a potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res. Rev. 68, 101338 (2021).
Bates, S. E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 383, 650–663 (2020).
Hogg, S. J., Beavis, P. A., Dawson, M. A. & Johnstone, R. W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 19, 776–800 (2020).
Muhammad, J. S. et al. Autophagy impairment by Helicobacter pylori-induced methylation silencing of MAP1LC3Av1 promotes gastric carcinogenesis. Int. J. Cancer 140, 2272–2283 (2017).
Liao, Y. P. et al. Hypomethylation signature of tumor-initiating cells predicts poor prognosis of ovarian cancer patients. Hum. Mol. Genet. 23, 1894–1906 (2014).
Dunwell, T. et al. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol. Cancer 9, 44 (2010).
Wang, J. et al. The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J. Hematol. Oncol. 12, 81 (2019).
Hervouet, E. et al. The autophagy GABARAPL1 gene is epigenetically regulated in breast cancer models. BMC Cancer 15, 729 (2015).
Zhang, X. et al. Aberrant methylation of ATG2B, ATG4D, ATG9A and ATG9B CpG island promoter is associated with decreased mRNA expression in sporadic breast carcinoma. Gene 590, 285–292 (2016).
Fu, R. et al. TET1 exerts its tumour suppressor function by regulating autophagy in glioma cells. Biosci. Rep. 37, BSR20160523 (2017).
Fu, R. et al. Ten-eleven translocation 1 regulates methylation of autophagy-related genes in human glioma. Neuroreport 29, 731–738 (2018).
Xue, H. et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy 12, 1129–1152 (2016).
Yan, J. et al. AEG-1 is involved in hypoxia-induced autophagy and decreases chemosensitivity in T-cell lymphoma. Mol. Med. 24, 35 (2018).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 162, 454 (2015).
Huang, S., Qi, P., Zhang, T., Li, F. & He, X. The HIF1alpha/miR2243p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxiainduced protective autophagy in glioblastoma and astrocytoma. Oncol. Rep. 41, 1759–1768 (2019).
Bellot, G. et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581 (2009).
Ma, Z. et al. BNIP3 induces apoptosis and protective autophagy under hypoxia in esophageal squamous cell carcinoma cell lines: BNIP3 regulates cell death. Dis. Esophagus 30, 1–8 (2017).
Cruzeiro, G. A. V. et al. HIF1A is Overexpressed in Medulloblastoma and its Inhibition Reduces Proliferation and Increases EPAS1 and ATG16L1 Methylation. Curr. Cancer Drug Targets 18, 287–294 (2018).
Gordevicius, J. et al. Epigenetic inactivation of the autophagy-lysosomal system in appendix in Parkinson’s disease. Nat. Commun. 12, 5134 (2021).
Bayer, C. et al. DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes. Int. J. Mol. Sci. 21, 5420 (2020).
Gonzalez-Rodriguez, P. et al. The DNA methyltransferase DNMT3A contributes to autophagy long-term memory. Autophagy 17, 1259–1277 (2021).
Gonzalez-Rodriguez, P., Cheray, M., Keane, L., Engskog-Vlachos, P. & Joseph, B. ULK3-dependent activation of GLI1 promotes DNMT3A expression upon autophagy induction. Autophagy 18, 2769–2780 (2022).
Ahmad, F., Dixit, D., Joshi, S. D. & Sen, E. G9a inhibition induced PKM2 regulates autophagic responses. Int. J. Biochem. Cell Biol. 78, 87–95 (2016).
Yin, C. et al. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 33, 14036–14050 (2019).
Li, K. C. et al. Inhibition of G9a induces DUSP4-dependent autophagic cell death in head and neck squamous cell carcinoma. Mol. Cancer 13, 172 (2014).
Artal-Martinez de Narvajas, A. et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell. Biol. 33, 3983–3993 (2013).
Park, S. E. et al. Inhibition of EHMT2/G9a epigenetically increases the transcription of Beclin-1 via an increase in ROS and activation of NF-kappaB. Oncotarget 7, 39796–39808 (2016).
Yuan, Y. et al. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 4, e690 (2013).
Kim, H., Choi, S. Y., Lim, J., Lindroth, A. M. & Park, Y. J. EHMT2 Inhibition induces cell death in human non-small cell lung cancer by altering the cholesterol biosynthesis pathway. Int. J. Mol. Sci. 21, 1002 (2020).
Seo, Y. K. et al. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 13, 367–375 (2011).
Chen, T. Q. et al. EHMT2/G9a inhibits aortic smooth muscle cell death by suppressing autophagy activation. Int. J. Biol. Sci. 16, 1252–1263 (2020).
Sengupta, S. et al. Mycobacterium tuberculosis phosphoribosyltransferase promotes bacterial survival in macrophages by inducing histone hypermethylation in autophagy-related genes. Front. Cell Infect. Microbiol. 11, 676456 (2021).
Shin, H. J. et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534, 553–557 (2016).
Wei, X. et al. SKP2 promotes hepatocellular carcinoma progression through nuclear AMPK-SKP2-CARM1 signaling transcriptionally regulating nutrient-deprived autophagy induction. Cell. Physiol. Biochem. 47, 2484–2497 (2018).
Zhou, K. et al. TFE3, a potential therapeutic target for Spinal Cord Injury via augmenting autophagy flux and alleviating ER stress. Theranostics 10, 9280–9302 (2020).
Liu, Y. et al. A C9orf72-CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 32, 1380–1397 (2018).
Yao, Y. et al. Downregulation of enhancer of zeste homolog 2 (EZH2) is essential for the induction of autophagy and apoptosis in colorectal cancer cells. Genes (Basel) 7, 83 (2016).
Yang, Y. et al. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. Onco Targets Ther. 11, 8455–8463 (2018).
Li, R. et al. EZH2 inhibits autophagic cell death of aortic vascular smooth muscle cells to affect aortic dissection. Cell Death Dis. 9, 180 (2018).
Yang, A. et al. Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis. 9, 169 (2018).
Wei, F. Z. et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 11, 2309–2322 (2015).
Stacchiotti, S. et al. Comparative assessment of antitumor effects and autophagy induction as a resistance mechanism by cytotoxics and EZH2 inhibition in INI1-negative epithelioid sarcoma patient-derived xenograft. Cancers (Basel) 11, 1015 (2019).
Yang, K. et al. Knockdown of HMGA2 regulates the level of autophagy via interactions between MSI2 and Beclin1 to inhibit NF1-associated malignant peripheral nerve sheath tumour growth. J. Exp. Clin. Cancer Res. 38, 185 (2019).
Liu, F. et al. miR92b promotes autophagy and suppresses viability and invasion in breast cancer by targeting EZH2. Int. J. Oncol. 53, 1505–1515 (2018).
Zhang, H., Liang, H., Wu, S., Zhang, Y. & Yu, Z. MicroRNA-638 induces apoptosis and autophagy in human liver cancer cells by targeting enhancer of zeste homolog 2 (EZH2). Environ. Toxicol. Pharmacol. 82, 103559 (2021).
Hou, Z., Chen, J., Yang, H., Hu, X. & Yang, F. PIAS1 alleviates diabetic peripheral neuropathy through SUMOlation of PPAR-gamma and miR-124-induced downregulation of EZH2/STAT3. Cell Death Discov. 7, 372 (2021).
Ma, J. et al. MiR-124 induces autophagy-related cell death in cholangiocarcinoma cells through direct targeting of the EZH2-STAT3 signaling axis. Exp. Cell Res. 366, 103–113 (2018).
Xu, L. et al. MicroRNA-101 inhibits human hepatocellular carcinoma progression through EZH2 downregulation and increased cytostatic drug sensitivity. J. Hepatol. 60, 590–598 (2014).
Chen, L., Jia, J., Zang, Y., Li, J. & Wan, B. MicroRNA-101 regulates autophagy, proliferation and apoptosis via targeting EZH2 in laryngeal squamous cell carcinoma. Neoplasma 66, 507–515 (2019).
Kuntimaddi, A. et al. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 Di- and tri-methylation on target genes and transformation potential. Cell Rep. 11, 808–820 (2015).
Bernt, K. M. et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78 (2011).
Wang, W. T. et al. Activation of the lysosome-associated membrane protein LAMP5 by DOT1L serves as a bodyguard for MLL fusion oncoproteins to evade degradation in leukemia. Clin. Cancer Res. 25, 2795–2808 (2019).
Tan, J. et al. JMJD2B-induced amino acid alterations enhance the survival of colorectal cancer cells under glucose-deprivation via autophagy. Theranostics 10, 5763–5777 (2020).
Sha, J. et al. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J. Cell. Physiol. 235, 2129–2138 (2020).
Ambrosio, S. et al. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 36, 6701–6711 (2017).
Zhuo, X. et al. Knockdown of LSD1 meliorates Ox-LDL-stimulated NLRP3 activation and inflammation by promoting autophagy via SESN2-mesiated PI3K/Akt/mTOR signaling pathway. Life Sci. 233, 116696 (2019).
Scher, H. I. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 367, 1187–1197 (2012).
Pasha, M., Eid, A. H., Eid, A. A., Gorin, Y. & Munusamy, S. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid. Med. Cell. Longev. 2017, 3296294 (2017).
Wolfson, R. L. et al. METABOLISM Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48 (2016).
Zhang, F. et al. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 5, 5775 (2014).
Amente, S. et al. Lysine-specific demethylase (LSD1/KDM1A) and MYCN cooperatively repress tumor suppressor genes in neuroblastoma. Oncotarget 6, 14572–14583 (2015).
He, M. et al. LSD1 contributes to programmed oocyte death by regulating the transcription of autophagy adaptor SQSTM1/p62. Aging Cell 19, e13102 (2020).
Eckschlager, T., Plch, J., Stiborova, M. & Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 18, 1414 (2017).
Del Bufalo, D. et al. Histone deacetylase inhibition synergistically enhances pemetrexed cytotoxicity through induction of apoptosis and autophagy in non-small cell lung cancer. Mol. Cancer 13, 230 (2014).
Annunziata, I. et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat. Commun. 10, 3623 (2019).
Li, X. et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell 66, 684–697 (2017).
Gabay, M., Li, Y. L. & Felsher, D. W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4, a014241 (2014).
Wang, W. et al. Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy 13, 941–954 (2017).
Ma, X. & Wang, Q. Short-chain fatty acids attenuate renal fibrosis and enhance autophagy of renal tubular cells in diabetic mice through the HDAC2/ULK1 axis. Endocrinol. Metab. (Seoul.) 37, 432–443 (2022).
Chen, L. et al. The microRNA-155 mediates hepatitis B virus replication by reinforcing SOCS1 signalling-induced autophagy. Cell Biochem. Funct. 38, 436–442 (2020).
Liang, L., Zhou, Q. & Feng, L. Decreased microRNA-155 in Behcet’s disease leads to defective control of autophagy thereby stimulating excessive proinflammatory cytokine production. Arthritis Res. Ther. 23, 135 (2021).
Wang, J. et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 9, e1003697 (2013).
Chen, W. et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. 11, 198 (2020).
Fu, X. T. et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 412, 108–117 (2018).
Xu, R., Liu, S., Chen, H. & Lao, L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol. Rep. 35, 1757–1763 (2016).
Li, B. B., Chen, Y. L. & Pang, F. MicroRNA-30a Targets ATG5 and Attenuates Airway Fibrosis in Asthma by Suppressing Autophagy. Inflammation 43, 44–53 (2020).
Shi, D. et al. Myocardin/microRNA-30a/Beclin1 signaling controls the phenotypic modulation of vascular smooth muscle cells by regulating autophagy. Cell Death Dis. 13, 121 (2022).
Zhang, K. et al. PU.1/microRNA-142-3p targets ATG5/ATG16L1 to inactivate autophagy and sensitize hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 9, 312 (2018).
Qu, Y. et al. MicroRNA-142-3p inhibits autophagy and promotes intracellular survival of Mycobacterium tuberculosis by targeting ATG16L1 and ATG4c. Int. Immunopharmacol. 101, 108202 (2021).
Lin, X. T. et al. MicroRNA-143 Targets ATG2B to Inhibit Autophagy and Increase Inflammatory Responses in Crohn’s Disease. Inflamm. Bowel Dis. 24, 781–791 (2018).
Zhang, H. et al. MicroRNA-143 sensitizes acute myeloid leukemia cells to cytarabine via targeting ATG7- and ATG2B-dependent autophagy. Aging (Albany, N. Y.) 12, 20111–20126 (2020).
Du, F., Feng, Y., Fang, J. & Yang, M. MicroRNA-143 enhances chemosensitivity of Quercetin through autophagy inhibition via target GABARAPL1 in gastric cancer cells. Biomed. Pharmacother. 74, 169–177 (2015).
Ma, W. et al. By Targeting Atg7 MicroRNA-143 mediates oxidative stress-induced autophagy of c-Kit(+) mouse cardiac progenitor cells. EBioMedicine 32, 182–191 (2018).
Liu, J., Qian, C. & Cao, X. Post-translational modification control of innate. Immun. Immun. 45, 15–30 (2016).
Pecuchet, N., Cluzeau, T., Thibault, C., Mounier, N. & Vignot, S. [Histone deacetylase inhibitors: highlight on epigenetic regulation]. Bull. Cancer 97, 917–935 (2010).
Matsuzaki, H. et al. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl Acad. Sci. USA 102, 11278–11283 (2005).
Zhang, J. et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 14, 1043–1059 (2018).
Wang, Y. et al. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep. 21, e48335 (2020).
Naidu, S. R., Lakhter, A. J. & Androphy, E. J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle 11, 2717–2728 (2012).
Jiang, X. & Wang, J. Knockdown of TFAM in tumor cells retarded autophagic flux through regulating p53 acetylation and PISD expression. Cancers (Basel) 12, 493 (2020).
Xu, X. et al. Autophagic feedback-mediated degradation of IKKalpha requires CHK1- and p300/CBP-dependent acetylation of p53. J. Cell Sci. 133, jcs246868 (2020).
Sun, M. et al. p53 deacetylation alleviates sepsis-induced acute kidney injury by promoting autophagy. Front. Immunol. 12, 685523 (2021).
Zhang, J. et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 11, 629–642 (2015).
Xiao, Q. et al. Histone deacetylase inhibitors promote epithelial-mesenchymal transition in Hepatocellular Carcinoma via AMPK-FOXO1-ULK1 signaling axis-mediated autophagy. Theranostics 10, 10245–10261 (2020).
Zhao, Y. et al. Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy 6, 988–990 (2010).
Yang, D. et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc. Res. 114, 1016–1028 (2018).
Banreti, A., Sass, M. & Graba, Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 9, 819–829 (2013).
Xu, Y. & Wan, W. Acetylation in the regulation of autophagy. Autophagy 1–9 (2022) https://doi.org/10.1080/15548627.2022.2062112. [Online ahead of print].
Sun, T. et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 6, 7215 (2015).
Sun, T., Ming, L., Yan, Y., Zhang, Y. & Xue, H. Beclin 1 acetylation impairs the anticancer effect of aspirin in colorectal cancer cells. Oncotarget 8, 74781–74790 (2017).
Deng, Z. et al. SIRT1 attenuates sepsis-induced acute kidney injury via Beclin1 deacetylation-mediated autophagy activation. Cell Death Dis. 12, 217 (2021).
Pi, Q. Z. et al. Melatonin alleviates cardiac dysfunction via increasing Sirt1-mediated Beclin-1 deacetylation and autophagy during sepsis. Inflammation 44, 1184–1193 (2021).
Han, L. L., Jia, L., Wu, F. & Huang, C. Sirtuin6 (SIRT6) promotes the EMT of hepatocellular carcinoma by stimulating autophagic degradation of E-cadherin. Mol. Cancer Res. 17, 2267–2280 (2019).
Sun, T., Jiao, L., Wang, Y., Yu, Y. & Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 9, 136 (2018).
Liu, J. et al. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 6, e1827 (2015).
Tao, Z. et al. Sirt1 coordinates with ERalpha to regulate autophagy and adiposity. Cell Death Disco. 7, 53 (2021).
Wei, Q. & Dong, Z. HDAC4 blocks autophagy to trigger podocyte injury: non-epigenetic action in diabetic nephropathy. Kidney Int. 86, 666–668 (2014).
Lee, J. Y. et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 (2010).
Wang, R. et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 218, 267–284 (2019).
Zheng, Z. et al. Histone deacetylase 6 inhibition restores autophagic flux to promote functional recovery after spinal cord injury. Exp. Neurol. 324, 113138 (2020).
Jiang, X. et al. Metastatic prostate cancer-associated P62 inhibits autophagy flux and promotes epithelial to mesenchymal transition by sustaining the level of HDAC6. Prostate 78, 426–434 (2018).
Majora, M. et al. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci. Transl. Med. 10, eaam7510 (2018).
Esteves, A. R. et al. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim Biophys. Acta Mol. Basis Dis. 1865, 2008–2023 (2019).
Melino, G., Cecconi, F., Pelicci, P. G., Mak, T. W. & Bernassola, F. Emerging roles of HECT-type E3 ubiquitin ligases in autophagy regulation. Mol. Oncol. 13, 2033–2048 (2019).
Han, S. H. et al. GCA links TRAF6-ULK1-dependent autophagy activation in resistant chronic myeloid leukemia. Autophagy 15, 2076–2090 (2019).
Raimondi, M. et al. USP1 (ubiquitin specific peptidase 1) targets ULK1 and regulates its cellular compartmentalization and autophagy. Autophagy 15, 613–630 (2019).
Liu, C. C. et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol. Cell 61, 84–97 (2016).
Shi, C. S. & Kehrl, J. H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal 3, ra42 (2010).
Zhan, Z. Z. et al. Autophagy facilitates TLR4-and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 10, 257–268 (2014).
Min, Y., Kim, M. J., Lee, S., Chun, E. & Lee, K. Y. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy 14, 1347–1358 (2018).
Kim, M. J. et al. p62 is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells 9, 1142 (2020).
Han, T. et al. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy 14, 2035–2048 (2018).
Chen, Y. H. et al. VPS34 K29/K48 branched ubiquitination governed by UBE3C and TRABID regulates autophagy, proteostasis and liver metabolism. Nat. Commun. 12, 1322 (2021).
Wu, H. et al. TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/beta-catenin degradation and is targeted for GSK3B/GSK3beta-mediated phosphorylation and degradation. Autophagy 15, 1506–1522 (2019).
Peng, H. et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 27, 657–674 (2017).
Lee, Y. et al. Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 19, 188–202 (2017).
Shi, Q. et al. SPOP mutations promote p62/SQSTM1-dependent autophagy and Nrf2 activation in prostate cancer. Cell Death Differ. 29, 1228–1239 (2022).
Peng, H. et al. The ubiquitin-specific protease USP8 directly deubiquitinates SQSTM1/p62 to suppress its autophagic activity. Autophagy 16, 698–708 (2020).
He, Y. et al. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 217, 315–328 (2018).
Lu, H. et al. TOPK inhibits autophagy by phosphorylating ULK1 and promotes glioma resistance to TMZ. Cell Death Dis. 10, 583 (2019).
Ryu, H. Y. et al. GSK3B induces autophagy by phosphorylating ULK1. Exp. Mol. Med. 53, 369–383 (2021).
Deng, Z. et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 16, 917–931 (2020).
Wold, M. S., Lim, J., Lachance, V., Deng, Z. & Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 11, 76 (2016).
Keil, E. et al. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 20, 321–332 (2013).
Feng, X. et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 17, 723–742 (2021).
Richter, B. et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl Acad. Sci. USA 113, 4039–4044 (2016).
Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233 (2011).
Gay, C. M., Balaji, K. & Byers, L. A. Giving AXL the axe: targeting AXL in human malignancy. Br. J. Cancer 116, 415–423 (2017).
Lozneanu, L. et al. Computational and immunohistochemical analyses highlight AXL as a potential prognostic marker for ovarian cancer patients. Anticancer Res. 36, 4155–4163 (2016).
Rao, L. et al. p85beta regulates autophagic degradation of AXL to activate oncogenic signaling. Nat. Commun. 11, 2291 (2020).
Zhang, N. et al. HSF1 upregulates ATG4B expression and enhances epirubicin-induced protective autophagy in hepatocellular carcinoma cells. Cancer Lett. 409, 81–90 (2017).
Liu, P. F. et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy 10, 1454–1465 (2014).
Thompson, B. J. & Sahai, E. MST kinases in development and disease. J. Cell Biol. 210, 871–882 (2015).
Huang, T. et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 32, 840–855 (2017).
Ni, Z. et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy 14, 685–701 (2018).
Cheong, J. K. et al. Casein kinase 1alpha-dependent feedback loop controls autophagy in RAS-driven cancers. J. Clin. Invest. 125, 1401–1418 (2015).
Cai, J. et al. CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat. Cell Biol. 20, 465–478 (2018).
Zuo, Q. et al. Targeting PP2A with lomitapide suppresses colorectal tumorigenesis through the activation of AMPK/Beclin1-mediated autophagy. Cancer Lett. 521, 281–293 (2021).
Svarcbahs, R. et al. Prolyl oligopeptidase inhibition activates autophagy via protein phosphatase 2A. Pharmacol. Res. 151, 104558 (2020).
Magnaudeix, A. et al. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol. Aging 34, 770–790 (2013).
Ma, H. et al. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 101, 315–330 (2022).
Zubiete-Franco, I. et al. Methionine and S-adenosylmethionine levels are critical regulators of PP2A activity modulating lipophagy during steatosis. J. Hepatol. 64, 409–418 (2016).
Lorenzi, P. L., Claerhout, S., Mills, G. B. & Weinstein, J. N. A curated census of autophagy-modulating proteins and small molecules: candidate targets for cancer therapy. Autophagy 10, 1316–1326 (2014).
Feng, X. et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 15, 1130–1149 (2019).
Cheng, X. & Sun, Q. RUBCNL/Pacer and RUBCN/Rubicon in regulation of autolysosome formation and lipid metabolism. Autophagy 15, 1120–1121 (2019).
Dong, X. et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca(2+)-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell Signal 53, 68–79 (2019).
Li, X. et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat. Commun. 8, 1159 (2017).
Wu, J. C. et al. Se-Allylselenocysteine induces autophagy by modulating the AMPK/mTOR signaling pathway and epigenetic regulation of PCDH17 in human colorectal adenocarcinoma cells. Mol. Nutr. Food Res. 59, 2511–2522 (2015).
Schnekenburger, M. et al. Sustained exposure to the DNA demethylating agent, 2’-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 81, 364–378 (2011).
Li, L. et al. Decitabine downregulates TIGAR to induce apoptosis and autophagy in myeloid leukemia cells. Oxid. Med. Cell. Longev. 2021, 8877460 (2021).
Romano, A. et al. ProteoMic analysis reveals autophagy as pro-survival pathway elicited by long-term exposure with 5-azacitidine in high-risk myelodysplasia. Front. Pharmacol. 8, 204 (2017).
Dubois, A. et al. LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia 33, 1501–1513 (2019).
Carew, J. S. et al. Disruption of autophagic degradation with ROC-325 antagonizes renal cell carcinoma pathogenesis. Clin. Cancer Res. 23, 2869–2879 (2017).
Mrakovcic, M. & Frohlich, L. F. Molecular determinants of cancer therapy resistance to HDAC inhibitor-induced autophagy. Cancers (Basel) 12, 109 (2019).
Foggetti, G. et al. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 39, BSR20181345 (2019).
Lee, J. Y. et al. Inhibition of HDAC3- and HDAC6-promoted survivin expression plays an important role in SAHA-induced autophagy and viability reduction in breast cancer cells. Front. Pharmacol. 7, 81 (2016).
Bai, Y. et al. Trichostatin A activates FOXO1 and induces autophagy in osteosarcoma. Arch. Med. Sci. 15, 204–213 (2019).
Gao, L. et al. Histone deacetylase inhibitor trichostatin A and autophagy inhibitor chloroquine synergistically exert anti-tumor activity in H-ras transformed breast epithelial cells. Mol. Med. Rep. 17, 4345–4350 (2018).
Cao, D. J. et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl Acad. Sci. USA 108, 4123–4128 (2011).
Mu, N. et al. Inhibition of SIRT1/2 upregulates HSPA5 acetylation and induces pro-survival autophagy via ATF4-DDIT4-mTORC1 axis in human lung cancer cells. Apoptosis 24, 798–811 (2019).
Sun, J. et al. Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. FEBS J. 287, 2118–2133 (2020).
Dong, L. H. et al. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J. Hematol. Oncol. 6, 53 (2013).
Shi, X., Liu, Y., Zhang, D. & Xiao, D. Valproic acid attenuates sepsis-induced myocardial dysfunction in rats by accelerating autophagy through the PTEN/AKT/mTOR pathway. Life Sci. 232, 116613 (2019).
Pietrocola, F. et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 22, 509–516 (2015).
Yang, F. et al. Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells. Life Sci. 213, 149–157 (2018).
Chiu, H. W. et al. A new histone deacetylase inhibitor enhances radiation sensitivity through the induction of misfolded protein aggregation and autophagy in triple-negative breast cancer. Cancers (Basel) 11, 1703 (2019).
De, U. et al. A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. Int. J. Mol. Sci. 19, 2743 (2018).
Liu, J. et al. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 9, 322 (2018).
Cui, S. et al. Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis. Int. Immunopharmacol. 77, 105973 (2019).
Sakamaki, J. I. et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell 66, 517–532 e519 (2017).
Wen, X. & Klionsky, D. J. BRD4 is a newly characterized transcriptional regulator that represses autophagy and lysosomal function. Autophagy 13, 1801–1803 (2017).
Li, Y. et al. Inhibition of Brd4 by JQ1 promotes functional recovery from spinal cord injury by activating autophagy. Front. Cell. Neurosci. 14, 555591 (2020).
Mu, J., Zhang, D., Tian, Y., Xie, Z. & Zou, M. H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 149, 1–14 (2020).
Sui, S. et al. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 10, 331 (2019).
Pan, Z. et al. Discovery of thieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as bromodomain-containing protein 4/histone deacetylase dual inhibitors induce autophagic cell death in colorectal carcinoma cells. J. Med. Chem. 63, 3678–3700 (2020).
Jang, J. E. et al. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 13, 761–762 (2017).
Luan, W. et al. Akt/mTOR-mediated autophagy confers resistance to BET Inhibitor JQ1 In Ovarian Cancer. Onco Targets Ther. 12, 8063–8074 (2019).
Kim, Y. et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 9, 2126–2139 (2013).
Ke, X. X., Zhang, R., Zhong, X., Zhang, L. & Cui, H. Deficiency of G9a Inhibits Cell Proliferation and Activates Autophagy via Transcriptionally Regulating c-Myc Expression in Glioblastoma. Front Cell Dev. Biol. 8, 593964 (2020).
De Smedt, E. et al. G9a/GLP targeting in MM promotes autophagy-associated apoptosis and boosts proteasome inhibitor-mediated cell death. Blood Adv. 5, 2325–2338 (2021).
Tu, W. B. et al. MYC interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell 34, 579–595 (2018).
Kim, T. W., Cheon, C. & Ko, S. G. SH003 activates autophagic cell death by activating ATF4 and inhibiting G9a under hypoxia in gastric cancer cells. Cell Death Dis. 11, 717 (2020).
Kim, T. W., Lee, S. Y., Kim, M., Cheon, C. & Ko, S. G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 9, 875 (2018).
Xu, H. et al. GSK343 induces autophagy and downregulates the AKT/mTOR signaling pathway in pancreatic cancer cells. Exp. Ther. Med. 18, 2608–2616 (2019).
Hsieh, Y. Y., Lo, H. L. & Yang, P. M. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am. J. Cancer Res. 6, 1661–1680 (2016).
Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59, 285–297 (2015).
O’Connell, C. E. & Vassilev, A. Combined inhibition of p38MAPK and PIKfyve synergistically disrupts autophagy to selectively target cancer cells. Cancer Res. 81, 2903–2917 (2021).
Park, J. M. et al. The nonreceptor tyrosine kinase SRMS inhibits autophagy and promotes tumor growth by phosphorylating the scaffolding protein FKBP51. PLoS Biol. 19, e3001281 (2021).
Rong, L. et al. Salidroside induces apoptosis and protective autophagy in human gastric cancer AGS cells through the PI3K/Akt/mTOR pathway. Biomed. Pharmacother. 122, 109726 (2020).
Feng, H. et al. A Rho kinase inhibitor (Fasudil) suppresses TGF-beta mediated autophagy in urethra fibroblasts to attenuate traumatic urethral stricture (TUS) through re-activating Akt/mTOR pathway: An in vitro study. Life Sci. 267, 118960 (2021).
Rostami, J. et al. Prolyl oligopeptidase inhibition by KYP-2407 increases alpha-synuclein fibril degradation in neuron-like cells. Biomed. Pharmacother. 131, 110788 (2020).
Zummo, F. P. et al. Exendin-4 stimulates autophagy in pancreatic beta-cells via the RAPGEF/EPAC-Ca(2 + )-PPP3/calcineurin-TFEB axis. Autophagy 18, 799–815 (2022).
Rusmini, P. et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82272388, 82072365, 82273566), Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011666), The Youth Innovation Project of the University of Science and Technology of China (WK9110000203), the Natural Science Foundation of Anhui Province (2008085MH241).
Author information
Authors and Affiliations
Contributions
G.-M.J. designed the outline of this manuscript. F.S. and H.X. were responsible for writing the initial manuscript and depicting all the figures and tables. Q.-N.L., X.-S.R., Z.-G.L., and B.-W.H. reviewed the manuscript and amended the reference. G.-M.J., H.W., H.-S.W. revised the manuscript. F.S. and H.X. contributed equally to this review. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shu, F., Xiao, H., Li, QN. et al. Epigenetic and post-translational modifications in autophagy: biological functions and therapeutic targets. Sig Transduct Target Ther 8, 32 (2023). https://doi.org/10.1038/s41392-022-01300-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01300-8
- Springer Nature Limited
This article is cited by
-
m1A regulator-mediated methylation modification patterns correlated with autophagy to predict the prognosis of hepatocellular carcinoma
BMC Cancer (2024)
-
Epigenetic regulation of diverse cell death modalities in cancer: a focus on pyroptosis, ferroptosis, cuproptosis, and disulfidptosis
Journal of Hematology & Oncology (2024)
-
GSK3β and UCHL3 govern RIPK4 homeostasis via deubiquitination to enhance tumor metastasis in ovarian cancer
Oncogene (2024)
-
Revealing the role of epigenetic and post-translational modulations of autophagy proteins in the regulation of autophagy and cancer: a therapeutic approach
Molecular Biology Reports (2024)
-
The emerging role of CARM1 in cancer
Cellular Oncology (2024)