
Abstract
Background
Optic pathway gliomas (OPGs) are classified by anatomic location and the association with neurofibromatosis type 1 (NF1). Children with OPGs face sequelae related to tumor location and treatment modalities. We assessed the prevalence of endocrine dysfunction in children with OPGs and compared outcomes between those with and without NF1.
Methods
We performed a retrospective medical record review of medical history, and clinical and laboratory data, of children diagnosed with OPGs (n = 59, 61% with NF1) during 1990–2020, followed at a tertiary endocrine clinic. Growth and puberty parameters and occurrence of endocrine dysfunction were evaluated.
Results
Isolated optic nerve involvement was higher among patients with than without NF1. Patients without NF1 were younger at OPG diagnosis and more often treated with debulking surgery or chemotherapy. At the last endocrine evaluation, patients without NF1 had comparable height SDS, higher BMI SDS, and a higher rate of endocrine complications (78.3% vs. 41.7%, p = 0.006). Younger age at diagnosis, older age at last evaluation, and certain OPG locations were associated with increased endocrine disorder incidence.
Conclusions
Endocrine dysfunction was more common in patients without NF1; this may be related to younger age at presentation, tumor locations, a greater progressive rate, and more aggressive treatments.
Impact
-
The literature is sparse regarding sporadic OPGs, and the mean duration of follow-up is shorter than at our study.
-
Our data show a higher rate of endocrine dysfunction in patients with OPGs than previously described.
-
We also found a higher prevalence of endocrine dysfunctions among patients without compared to those with NF-1.
-
A better understanding of the true prevalence of endocrine disabilities that may evolve along time can help in guiding physicians in the surveillance needed in patients with OPG.
Similar content being viewed by others


Introduction
Optic pathway gliomas (OPGs) are low-grade astrocytic tumors that account for 3–5% of all pediatric brain tumors.1 OPGs occur in the optic nerve, chiasm, or both and can also involve the posterior part of the pathway. Often asymptomatic, OPGs sometimes demonstrate rapid growth and, depending on location and rate of progression, may cause considerable visual dysfunction, neurologic deficits, and endocrine disturbances.
OPGs are common in individuals with neurofibromatosis type 1 (NF1), up to 20% of whom develop an OPG at a mean age of 4.5–5 years.2 An anatomical classification for OPGs, proposed by Dodge et al.,3 defines tumors as involving the optic nerves alone (Stage 1), the chiasm with or without nerve involvement (Stage 2), or the hypothalamus or other adjacent structures (Stage 3). The original Dodge criteria (DC) were modified to provide a more detailed description of tumor number, location, and size. Modified DC1 includes a single/bilateral/cisternal segment of the optic nerve. Modified DC2 includes central/asymmetric chiasmatic involvement. Modified DC3 includes optic/asymmetric tract involvement. Modified DC4 refers to posterior/asymmetric tract involvement. H+ signs define hypothalamic involvement.4 In NF1, OPGs are more commonly classified as MDC1, while tumors in non-NF1 are more frequently classified as MDC2/3/4.
The primary presenting symptoms of OPGs tend to differ in patients with and without NF1. This may be due to differences in surveillance, as monitoring for OPG is recommended in young children with NF1, whereas routine surveillance is not conducted for sporadic cases. Sporadic OPGs are more likely to present with signs of increased intracranial pressure and hydrocephalus. NF1-associated OPGs are most commonly detected in children younger than 6 years, during ophthalmologic surveillance, even before any vision complaint.5
Endocrine dysfunction due to hypothalamic extension is present in 10–20% of individuals with OPG.1 The most frequent endocrine dysfunction is central precocious puberty (CPP) due to involvement of the hypothalamic–pituitary–gonadal axis. This occurs in up to 39% of children with NF1 and chiasmal OPG and is associated with transient rapid growth and reduced final height.6 Overall, individuals with NF1 are shorter than the general population. However, growth hormone deficiency (GHD) has been reported in individuals with NF1, without suprasellar abnormalities.7 This suggests that the mechanism of short stature may be related to NF1 itself.
Children with OPG face sequelae related also to tumor location and treatment modalities. The optimal management of OPG is controversial. Treatment decisions should take into account the patient’s age, presence or absence of NF1, and the location of the tumor.8,9,10,11 Current guidance for the management of OPG relies on whether the tumors affect or threaten vision.12 Observation without specific treatment is appropriate for tumors graded as MDC1, until evidence arises of either extension into the optic canal or progressive visual compromise.13,14
Chemotherapy is commonly selected as first treatment when a tumor progresses, particularly in children younger than 5 years, to avoid adverse effects on cognitive function from radiation or other therapies.15 If progression is associated with proptosis, and vision is poor/absent in that eye, debulking surgery should be considered; however, surgery invariably results in complete loss of vision in the involved eye.16,17 Radiation therapy is generally avoided in children due to an increased risk of secondary malignancy and other sequelae,18 and especially in those with NF1 due to genetic predisposition to tumor formation.19 Successful outcomes have been reported following treatment of advanced tumors with pathway inhibitors. These are medications whose interactions belong to the RAS MAPK pathway and include both BRAF and MEK1/MEK2 inhibitors20,21 This is important in light of the high proportion of optic gliomas with pilocytic astrocytoma histology that have been reported to involve BRAF alterations.
A number of studies have evaluated endocrine disorders related to OPG, especially in patients with NF1.22,23,24,25 The literature is sparse regarding sporadic OPG.
The aim of this study was to assess the prevalence of endocrine dysfunction in children with OPG, in the setting of an endocrine clinic in a tertiary referral center, and to compare outcomes between those with and without NF1.
Materials and methods
Study setting and patients
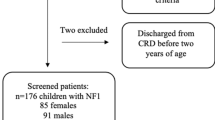
In this retrospective study, the computerized clinical database of Schneider Children’s Medical Center of Israel, a major tertiary university-affiliated hospital, was searched for patients who were diagnosed with OPG between January 1990 and January 2020. Inclusion criteria were age <18 years at diagnosis and <35 years at the time of data collection; at least 1 year of follow-up at the hospital’s institutes of oncology, neurology, and endocrinology; and ≥2 visits at the hospital’s Institute of Endocrinology and Diabetes (Fig. 1). All the data described in the current study are limited to this specific population of patients with OPG who were followed at a tertiary care center.

Study cohort flow chart.
Surveillance of patients with NF1 and with sporadic OPG in our center
After a clinical or molecular diagnosis of NF1, our surveillance includes ophthalmologist examination every 6 months, annual optical coherence tomography examination beginning at age 4 years, and annual vision field examination beginning at age 8 years. Magnetic resonance imaging (MRI) is performed following a pathological finding in an eye examination or presentation of neurological symptoms or signs. When OPG is diagnosed by MRI, the follow-up includes repeated MRI imaging once or twice annually, depending on whether the imaging is stable or shows dynamic changes.
Data collection
From the medical files of eligible patients, demographic parameters (sex, ethnicity), documentation of the presence of NF1, OPG-related data, growth and endocrine data, and parental data were extracted. The OPG-related data included the age at diagnosis, tumor location, and mode of therapy. Tumor location was classified according to the modified DC: stage I, optic nerve alone (MDC1); stage II, optic chiasm with or without optic nerve involvement (MDC2/MDC3/MDC4); and stage III, involvement of the hypothalamus or other adjacent structures (H+). The mode of therapy was categorized as surgical resection, chemotherapy, biological therapy (BRAF/MEK inhibitor therapy), cranial radiation, and combined treatment. Growth and endocrine data included pubertal stage and age at onset of puberty; growth parameters during the endocrine follow-up (height, weight, body mass index (BMI)); and occurrence, characteristics, and age at onset of endocrine disorders. Endocrine dysfunction was determined by documentations of clinical manifestations and laboratory findings. Parental data included anthropometric measurements and history of onset of puberty.
Evaluation of endocrine dysfunction
The following parameters regarding endocrine disorders were retrieved from the medical files: short stature, GHD, GH excess, CPP, hypogonadism, central hypothyroidism, central hypocortisolism, central diabetes insipidus (DI), and obesity.
Patients at our clinic undergo thorough physical examinations, measurement of height (using a Harpenden–Holtain stadiometer) and weight, and determination of pubertal stage according to the criteria of Marshall and Tanner.26,27 Height was expressed as the standard deviation score (Ht SDS) for age and sex.28 Short stature was defined as height <−2SDS. Adult height was defined as the attainment of bone age ≥15 years for females or ≥17 years for males, or when the annual growth rate slowed to <2 cm at age ≥18 years. Adult Ht SDS was calculated for an age set at 21 years.
BMI was calculated as weight (in kilograms) divided by height (in meters) squared and expressed as the standard deviation score BMI-SDS.28 Obesity was defined as BMI ≥1.645 SDS (≥95th percentile for age and sex) and overweight as 1.036 <BMI SDS <1.644 (≥85th and <95th percentile for age and sex).
Precocious puberty was defined as the onset of puberty (breast budding in females, testicular volume >4 ml in males) before age 8 years in girls and 9 years in boys, coincident with elevated plasma concentrations of gonadotropin-releasing hormone (GnRH)-stimulated gonadotropic hormones and plasma levels of sex hormones. Hypogonadism was defined as failure of spontaneous puberty: the absence of testicular enlargement in boys at age ≥14 years, accompanied by the absence of phallic development29; and the absence of breast development in girls at age ≥13 years,30 with estrogen or testosterone levels in the pre-pubertal range.
Laboratory assessments included determinations of fasting blood glucose and lipid profile [triglycerides, total cholesterol, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C)] performed at >10 years, or at a younger age if there was a family history of dyslipidemia or of early cardiovascular disease. Dyslipidemia was defined in children as age- and sex-specific levels of triglycerides >the 95th percentile, LDL-C >the 95th percentile, or HDL-C <the 10th percentile;31 and in adults, as LDL-C >130 mg/dL, HDL-C <40 mg/dL, or triglycerides >150 mg/dL.32
Thyroid-stimulating hormone (TSH) and free thyroxine (FT4) were measured annually. Hypothyroidism was defined as free T4 < 10.5 pmol/L (normal, 10.5–25) with TSH > 10.0 mIU/L (normal, 0.5–4.5). Central hypothyroidism was defined as a subnormal FT4 level (<10.5 pmol/mL) with a low, normal, or mildly raised TSH level.
GH stimulation tests were performed in patients with short stature (height ≤−2SD) or with growth deceleration. Sex hormone priming was used in prepubertal females aged >11.5 years and in prepubertal males aged >13 years. The priming was with estradiol pills, 4 mg/m2 for 2 days before stimulation testing in females; and with intramuscular injection of Testoviron depot 100 mg, 10 days before stimulation testing in males. GHD was defined as peak GH < 10 ng/mL until 2011. Thereafter (when the measurement method was changed), the definition was peak GH < 7.5 ng/mL in response to stimulation with clonidine HCl (0.15 mg/m2, maximal dose 0.15 mg), glucagon (0.03 mg/kg, maximal dose 1 mg), or arginine (0.5 g/kg, maximal dose 30 g). GHD was diagnosed according to growth velocity/growth data and low levels of GH on two stimulation tests or low levels of GH on one stimulation test in patients previously treated with cranial radiation. In patients suspected for GH excess, insulin-like growth factor-1 levels and GH were assessed in an oral glucose suppression test.
Basal follicle-stimulating hormone (FSH), luteinizing hormone (LH), estradiol levels, and testosterone levels were measured in patients for whom precocious puberty was suspected, as per patient sex. GnRH stimulation test (50 mg/m2) was performed in patients with failure of spontaneous puberty, pubertal arrest, or precocious puberty. Hypogonadism secondary to gonadal dysfunction was defined as either basal FSH > 15 IU/L or basal LH > 10 IU/L, together with decreased levels of estradiol in girls and testosterone in boys (interpreted according to pubertal stage). Hypogonadotropic hypogonadism was defined as the lack of an increase in serum LH level >0.8 IU/L after exogenous GnRH stimulation at the age of >13 years in girls and >14 years in boys, concurrent with sex hormone levels in the prepubertal range. The latter was defined as estradiol <20 pmol/L in females and testosterone <0.7 nmol/L in males, or decreased age- and pubertal-stage-corrected levels of estradiol in adult females and of testosterone in adult males.
Morning cortisol level was measured and a cortisol level of <270 nmol/L precipitated re-evaluation following adrenocorticotropic hormone (ACTH) stimulation. Cortisol deficiency was defined as peak cortisol following ACTH stimulation test <500 nmol/L.
Central DI was defined as the presence of polyuria and polydipsia with serum osmolality >300 mOsm/kg and urine osmolality <300 mOsm/kg.
Statistical analysis
Statistical analysis was performed using the SPSS software, version 27 (SPSS, Inc., Chicago, IL). Data were expressed as mean ± standard deviation (SD) for normally distributed variables and median (interquartile range) for skewed variables. Variables were compared between those with and without NF1) using the independent t test (for variables with normal distribution), the Mann–Whitney test (for skewed variables), or the Pearson’s chi-square test (for categorical variables). A backward logistic regression model was performed to identify predictors for the occurrence of endocrine disorders. Significance was set at p ≤ 0.05. Linear mixed model analyses were performed to evaluate the between group (OPG with NF1 and OPG without NF1) differences in anthropometric parameters (height SDS and BMI SDS) during 8 years of follow-up.
Results
Clinical and anthropometric data of the study cohort are presented in Table 1. Of the 59 patients (29 males) fulfilling the study criteria, 36 (61%) had NF1. Of these, 18 (50%) were diagnosed based on clinical criteria only, and 18 (50%) also had genetic confirmation of NF1. Isolated optic nerve involvement presented in 30 and 0% of patients with and without NF1, respectively. The hypothalamus or adjacent structures were involved in only 22% of those with NF1 compared to 52% of those without NF1 (p < 0.01).
Patients without NF1 compared to those with NF1 were significantly younger at diagnosis of OPGs (median age: 1.5 vs. 4.2 years, p = 0.04), older at the last endocrine evaluation (14.7 + 6.3 vs. 11.5 ± 5.0 years, p = 0.03), and more often treated with debulking surgery (65 vs. 8%, p < 0.001) and chemotherapy (83 vs. 31%, p < 0.001).
At the last endocrine evaluation, for patients without NF1 compared to those with NF1, the mean height SDS was comparable (−0.97 ± 1.39 vs. −0.72 ± 1.13, p = 0.45), and the mean BMI SDS was higher (1.08 ± 1.26 vs. 0.37 + 1.02, p = 0.020). No significant difference was found between the groups in the age at pubertal onset.
One patient in the NF1 group had an aggressive tumor that was non-responsive to conventional treatments, including chemotherapy, radiation, and BRAF/MEK inhibitor therapy. NF1 was diagnosed according to clinical criteria, without molecular confirmation. His OPG diagnosis was confirmed on biopsy. This patient developed obstructive hydrocephalus and underwent recurrent surgeries of drainage and insertion of a ventriculoperitoneal shunt as a palliative treatment. He died shortly after he was diagnosed.
Among patients without compared to those with NF1, the proportion with endocrine complications was higher (78.3 vs. 41.7%, p = 0.006) (Table 2). Specifically, proportions were higher for central hypothyroidism (47.8 vs. 2.8%, p < 0.001), hypocortisolism (26 vs. 2.8%, p = 0.011), central DI (26 vs. 2.8%, p = 0.011), and obesity (43.5 vs. 8.3%, p = 0.001). The proportions with GHD, CPP, and hypogonadotropic hypogonadism did not differ significantly between the groups.
In a logistic regression analysis, adjusted to the type of treatment (surgery vs. chemotherapy/radiation/BRAF/MEK inhibitor therapy), significant differences were not found in the rate of endocrinopathies between individuals with and without NF1 (Table 2). However, debulking surgery was found to be a risk factor for central hypothyroidism, odds ratio (OR) = 52.15 (4.62, 589.41) p = 0.001; central DI, OR = 13.12 (1.23, 139.85) p = 0.033; obesity, OR = 6.82 (1.38, 33.65) p = 0.018; and GHD, OR = 136.39 (4.16, 4469.5), p = 0.006.
Three patients were treated with recombinant growth hormone due to GHD (of them, two had NF1), ten were treated with GnRH analogs due to precocious puberty (of them seven had NF1), and one of them was treated with aromatase inhibitor as well. Five patients received hormone-replacement therapy due to hypogonadism. Of them, only one had NF1.
Mixed model analysis showed no significant change in boys’ height SDS during the 8 years of follow-up, with no difference between patients with and without NF1 (Fig. 2a and Table 3). In all females, a significant reduction in height SDS occurred during follow-up (ptime = 0.002), with no difference between those with and without NF1 (Fig. 2b and Table 3).

a Height SDS boys. b Height SDS girls. c BMI-SDS boys. d BMI-SDS girls.
Mixed model analysis for the change in BMI SDS during the 8 years of follow-up showed no significant changes over time in boys or in girls (non-significant time effect). BMI SDS was significantly lower in boys with than without NF1 (pgroup = 0.032) (Fig. 2c and Table 3). BMI SDS showed a trend of lower values in girls with than without NF1 (pgroup = 0.102), and a difference in the interaction between group and time in girls (p = 0.042) (Fig. 2d and Table 3). This difference is attributed to the change in BMI SDS during the first year following diagnosis, as seen in Fig. 2d.
Only a small number of our patients reached adult final height at the time of their last visit at our endocrine clinic. Three males (16.7%) with NF1 reached a mean adult final height of 166 ± 0.04 cm, while five males without NF1 (45.4%) reached a mean adult final height of 164 ± 0.14 cm (p = 0.84). Among females, the mean adult final height was 158 ± 0.58 cm and 157 ± 0.03 cm (p = 0.83) in 4 (22.2%) with NF1 and 4 (33.3%) without NF1, respectively.
Table 4 presents the backward logistic regression model for prediction of the occurrence of endocrine abnormality risk factors. Older age at diagnosis was associated with a lower risk of developing an endocrine disorder later in life (OR = 0.7, confidence interval (CI) = 0.55–0.89, p = 0.004). Age at the last endocrine evaluation was significantly associated with increased incidence of endocrine disorders (OR = 1.29, CI = 1.09–1.52, p = 0.002).
Tumor location that included the hypothalamus or adjacent structures, compared to the optic nerve and chiasm only, was associated with a higher risk of developing an endocrine disorder later in life (OR = 5.25, CI = 1.1–25.0, p = 0.038). Sex, the presence of NF-1, and surgical treatment did not enter the final model.
Discussion
Our data show a higher rate of endocrine dysfunction in patients with OPGs than previously described.24,33 This may be due to the older age of our patients at data collection. We report higher prevalence of endocrine dysfunctions among patients without compared to with NF-1.
Santoro et al.24 reported that, in patients with NF1 with OPG, tumor location was the most important predictor of endocrine disorders, particularly hypothalamic involvement. In our cohort, tumor location differed significantly between patients with and without NF1. Involvement of the optic nerve alone (MDC1) was found only among patients with NF1. Hypothalamic involvement (MDC, H+) was much more common in the non-NF1 group. The chiasm (MDC2) was involved in almost half the patients in both groups, similar to previous reports.33 In addition, a higher proportion of patients without NF1 than with NF1 were treated with debulking surgery, chemotherapy, or cranial radiation. Such differences in anatomic characteristics and therapy modality may explain in part the higher rates of endocrine complications that we found in the non-NF1 group.
Other cross-sectional studies showed associations of tumor location, radiation, and debulking surgery, with the development of endocrinopathies. However, they were not able to distinguish whether the tumor itself, or the treatment for it, was the cause.33,34,35,36,37 In contrast, in a multivariate longitudinal analysis, Gan et al.38 demonstrated the impact of both tumor location and treatment modality on long-term neuroendocrine morbidity. Tumor location was found to be the predominant factor that influences the rate of endocrinopathy; while radiation was strongly associated with the nature of the endocrine complications, particularly with GHD. Other endocrine complications were predicted more by hypothalamic involvement than by the treatment.
In our cohort, both among patients with and without NF1, treatment modality was not found to correlate with the incidence of endocrinopathies. However, debulking surgery was found to be a risk factor for central hypothyroidism, central DI, obesity, and GHD. This may be explained by the insult to the hypothalamic–pituitary axis. A recent study with a long-term follow-up reported that 58% of 90 children diagnosed with OPG before age 3 years had some degree of endocrine complications.39 Both NF-1 and anterior tumor position were more common in those without endocrinopathies (42.2%). Endocrine dysfunction of the anterior pituitary was greatly increased by hypothalamic involvement, radiotherapy, and surgery.
The most common endocrine complication we found in the non-NF1 group was central hypothyroidism (47.8%), which occurred in only 1 patient with NF1 (2.8%). All of those with central hypothyroidism had chiasmatic involvement; two-thirds of them had involvement of hypothalamic or adjacent structures as well. All but one underwent debulking surgery, and the one who did not (from the non-NF1 group) was treated with chemotherapy at a very young age (9 months). Terashima et al.40 reported a similar prevalence of central hypothyroidism (42%) in a population of children aged 0–4 years with low-grade glioma (without differentiating between NF1 and non-NF1). They found hypothyroidism to be more common in younger patients (diagnosed before age 5 years). Among those aged ≥5 years, the prevalence was 6%, and the overall prevalence of this endocrinopathy was 27% in the entire cohort. In our cohort, the median age at diagnosis of central hypothyroidism was 5.36 ± 4.31 in the non-NF1 group, and 5.5 years in the NF1 patient who developed hypothyroidism. Gan et al.38 revealed that hypothalamic involvement was more predictive of most endocrine deficits, including hypothyroidism, than treatment-related factors such as radiotherapy exposure. This observation can explain the higher prevalence of hypothyroidism that we found in the non-NF1 group, which had a higher rate of hypothalamic involvement.
In our cohort, hypogonadotropic hypogonadism developed in 7.7% of the NF1 group and among 33.3% of the non-NF1 group. All of them had chiasmatic involvement, and one had hypothalamic involvement as well. All but the single patient with NF1 were treated with debulking surgery, and four of five of them were treated with chemotherapy as well. One patient without NF1 had CPP as a child, which evolved to gonadal failure as an adolescent. He was diagnosed as an infant, had hypothalamic involvement, and was treated with debulking surgery, chemotherapy, and radiation. Gan et al.38 reported a 20.4% prevalence rate of gonadal failure among 166 children with OPGs. Hypothalamic involvement was more predictive of gonadal failure than treatment-related factors. Another interesting observation, which was also seen in one of our patients, is the progression from CPP to hypogonadotropic hypogonadism. Sani et al.22 also described a girl who was previously treated with GnRH analog for CPP and later developed gonadal failure. Terashima et al.40 reported a lower prevalence of this endocrinopathy: 16% among those diagnosed with OPG before age 5 years, and zero among those diagnosed after age 5 years. In our cohort, 4 of the 6 patients (67%) who developed hypogonadotropic hypogonadism were younger than age 5 years at OPG diagnosis. The other two who did not have NF1 were treated with debulking surgery and one of them with chemotherapy as well.
In our cohort, hypocortisolism developed in 1 (2.8%) patient with NF1 and in 6 (26%) without NF1. All of them had chiasmal or hypothalamic involvement, and they were all treated with debulking surgery. All except one were treated with chemotherapy as well. Sani et al.22 reported a 5.5% prevalence rate of hypocortisolism among children with NF1 and OPG, with chiasmatic or hypothalamic involvement. One of them was treated with debulking surgery and both of them were treated with chemotherapy. Shalitin et al.41 found that 9 of the 114 long-term survivors of various childhood brain tumors (including optic glioma) developed ACTH deficiency. This endocrinopathy was associated with cranial radiation and suprasellar tumor location. Interestingly, in the present cohort, only one of the seven patients who developed hypocortisolism was treated with radiation. Therefore, our relatively high prevalence rate of ACTH deficiency could be attributed to the hypothalamic involvement found in five of the seven patients who developed hypocortisolism.
Central DI developed in 1 (2.8%) patient with NF1 and in 6 (26%) without NF1. All seven had chiasmal and/or hypothalamic involvement. All but one underwent debulking surgery, and the remaining patient was treated with chemotherapy and radiation. Listernick et al.42 reported a 15.8% prevalence rate of DI among patients without NF1, compared to none among those with NF1. All of them had involvement of the chiasm, and they were all treated with debulking surgery, chemotherapy, or radiation. However, the authors did not mention the exact treatment received by each of the children who developed DI. Gan et al.38 and Picariello et al.39 reported central DI prevalence rates of 4.2 and 8.9%, respectively. A risk factor for this endocrinopathy was treatment with debulking surgery. Therefore, we can assume that central DI is associated with hypothalamic involvement and surgical manipulation.
We found that obesity was more common in the non-NF1 group. The etiology for obesity in the setting of central nervous system tumors is multifactorial.43 Gan et al.38 demonstrated a high prevalence of obesity in optic pathway, hypothalamic, and suprasellar gliomas, which may be increased by radiotherapy. They found that any form of hypothalamic damage was a risk factor for an abnormal increase in BMI. In addition, younger age at diagnosis was found to be a risk factor for developing obesity. Indeed, in our cohort, patients without NF1 compared to those with NF1 were significantly younger at diagnosis of OPG. Moreover, 14% of our patients without NF1 were treated with radiotherapy vs. only 3% in the NF1 group. Hypothalamic involvement in our patients was also more common in the non-NF1 group. Therefore, these factors together may explain the difference in obesity prevalence between the groups.
Our single patient with oversecretion of GH did not have NF1. Josefson et al.44,45 described seven children with OPG and GH excess, five of them had NF1 and two had sporadic OPG. GH excess resolved in five. Of these, only three were treated for GH excess for a sustained period. Thus, it is unclear if treatment altered outcomes or prevented tumor progression. The authors suggested that GH excess in children with OPG, both sporadic and NF1-associated, is a transient phenomenon.
In children with NF1 and OPG, GH excess is mainly associated with central precocious puberty. Among 64 children with NF1 and OPG,46 7 (10.9%) showed GH excess, all of whom had a tumor involving the chiasma. Their study demonstrated that GH excess should be considered as a relatively frequent endocrine manifestation in patients with NF1, similar to CPP. The presence of OPG has been hypothesized to inhibit the somatostatin tone, thus leading to unregulated release of GH.47
We found prevalence of 8.3 and 13% of GHD in the NF1 and non-NF1 groups, respectively. None of them were treated with radiation. All but one underwent debulking surgery, and two of the three patients without NF1 were treated with chemotherapy. This prevalence of GHD is lower than 40.3, 41.1, and 36.1% reported by Gan et al.,38 Picariello et al.,39 and Sani et al.,22 respectively. This lower prevalence of GHD in our cohort may be explained by the fact that in our cohort GH stimulation test was performed only in patients with short stature (height ≤−2SD) or with growth deceleration, and some of the patients with hypothalamic obesity may have normal growth in the setting of GHD although we found that short stature frequency was higher than GHD. The short stature may be explained by higher frequency of skeletal anomalies, the impact of a germline NF1 gene mutation on bone, and somatic growth.
GHD is probably more common in the NF1 population, even in the absence of OPG. Vassilopoulou-Sellin et al.7 described children with NF1 as having short stature and GHD, without suprasellar tumors or medical treatment. These findings imply that GHD may be relatively common in children with NF-1 without suprasellar abnormalities, thus suggesting an association with NF-1 independent of pituitary damage.
We found a higher rate of CPP in the NF1 compared to the non-NF1 group: 22.2 vs. 13%, although the median age was significantly younger in the latter. Only three patients with NF1 had isolated optic nerve involvement and received no treatment for their OPG. Picariello et al.39 found that CPP was the earliest endocrine dysfunction that affected 26.7% of their young cohort with OPG.
Listernick et al.42 demonstrated CPP exclusively among patients with NF1, at age 4.8–5.9 years. In another study, Listernick et al.48 found that in 30% of all children with NF1 and OPG involving the chiasm, the presenting sign was CPP. Among 219 children with NF1,7 CPP was found exclusively in those who had OPGs involving the optic chiasm. CPP occurred in 39% of the children with NF1 who had tumors of the optic chiasm; however, the true incidence may be even higher, because many of the children were still very young at the time of the study and could develop CPP later.
The relationship of precocious puberty to the exact location of the tumor is unknown. However, we assume that lesions located near the hypothalamus interfere with tonic central nervous system inhibition of the hypothalamic–pituitary–gonadal axis, resulting in premature onset of puberty.49 This can explain why in our cohort patients without NF1, with chiasm, or hypothalamic involvement developed CPP.
The NF1 population as a whole is significantly shorter than the general population.50,51 In our cohort, probably due to the relatively small sample, we did not find a significant difference between height SDS among patients with and without NF1. Also, no significant change was found in boys’ height SDS during the 8-year follow-up period; but all our female patients demonstrated a significant reduction in height SDS. Boys with NF1 were not significantly shorter than boys without NF1.
BMI SDS did not change significantly during the 8-year follow-up. However, BMI SDS was significantly higher in boys without than with NF1, and a trend of higher values in girls without than with NF1 was observed. The latter can be attributed to higher prevalence of hypothalamic involvement in this population, which is associated with obesity, as discussed above.
A multivariate regression analysis identified younger age at diagnosis and tumor location that included the hypothalamus as risk factors for endocrine disorders. Older age at the last endocrine evaluation was associated with increased incidence of endocrine disorders. This could be explained by earlier presentation of tumors causing endocrinopathies than of asymptomatic tumors, and by the older age at the last follow-up, of patients who received active endocrine follow-up.
The main strength of our study is the longitudinal long-term follow-up evaluated in a single tertiary center. This compares with most previous studies of OPG, with and without NF1, which were cross-sectional or with a short follow-up. The limitations of our study include its retrospective design, which raises the potential for confounding. In addition, the absence of data relating to height and weight of the patients at some of the visits during the follow-up period might have influenced the true impact of height and BMI SDS over the years. We therefore applied a mixed model analysis to estimate the missing numbers. Moreover, as some of the patients included in the study had a relatively short period of follow-up after the diagnosis of OPG, the true incidence of endocrine complications may be higher than presented here.
Notably, our results are limited to the specific population of patients with OPG who were followed at a tertiary care center. In addition, some of the 128 patients whose records were initially reviewed for the current study did not continue follow-up at our institution, and we had no access to their medical files. Therefore, the true incidence of endocrinopathies in the entire population of patients with OPG might be different than described.
The overall survival of children with low-grade OPG is generally excellent, regardless of tumor location, patient demographics, histopathologic subtype, or initial treatment.40,52,53,54 The progressive nature of endocrine deficits over prolonged follow-up suggests that lifelong repeated endocrine assessment is mandatory, both in patients with and without NF1.
Conclusions
Among our patients with OPGs, endocrine dysfunction presented at a younger age in those without NF1 than in those with NF1. This may be associated with tumor location and more aggressive treatments. The differences between OPG with and without NF1should dictate separate concerns in the follow-up of children with OPG. All children with OPG, and especially those without NF1, should have close follow-up for signs of endocrine disorders. Nevertheless, we note that, in our hospital, the management of OPG changed in the past decade, and smaller proportions of patients currently undergo major debulking surgeries and radiation. Therefore, we can expect that, in the upcoming years, the rate of endocrine complications may decrease.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to patients’ confidentiality but are available from the corresponding author on reasonable request.
References
Hill, C. S. et al. Neurosurgical experience of managing optic pathway gliomas. Childs Nerv. Syst. 37, 1917–1929 (2021).
Helfferich, J. et al. Neurofibromatosis type 1 associated low-grade gliomas: a comparison with sporadic low-grade gliomas. Crit. Rev. Oncol. Hematol. 104, 30–41 (2016).
Dodge, H. W. et al. Glioma of the optic nerves. Arch. Neurol. Psych. 79, 607–621 (1958).
Taylor, T. et al. Radiological classification of optic pathway gliomas: experience of a modified functional classification system. Br. J. Radiol. 81, 761 (2008).
Miller, D. T. et al. Health supervision for children with neurofibromatosis type 1. Pediatrics 143, 1–16 (2019).
Habiby, R., Silverman, B., Listernick, R. & Charrow, J. Precocious puberty in children with neurofibromatosis type 1. J. Pediatr. 126, 364 (1995).
Vassilopoulou-Sellin, R., Klein, M. J. & Slopis, J. K. Growth hormone deficiency in children with neurofibromatosis type 1 without suprasellar lesions. Pediatr. Neurol. 22, 355–358 (2000).
Grill, J. et al. When do children with optic pathway tumors need treatment? An oncological perspective in 106 patients treated in a single center. Eur. J. Pediatr. 159, 692–696 (2000).
Kortmann, R. D. et al. Current and future strategies in radiotherapy of childhood low-grade glioma of the brain. Part I: treatment modalities of radiation therapy. Strahlenther. Onko. 179, 509–520 (2003).
Thiagalingam, S., Flaherty, M., Billson, F. & North, K. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology 111, 568–577 (2004).
Listernick, R., Louis, D. N., Packer, R. J. & Gutmann, D. H. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann. Neurol. 41, 143–149 (1997).
Farazdaghia, M. K., Katowitza, W. R. & Averya, R. A. Current treatment of optic nerve gliomas. Curr. Opin. Ophthalmol. 30, 356–363 (2019).
Medlock, M. D. et al. Optic chiasm astrocytoma’s of childhood 1. Long-term follow-up. Pediatr. Neurosurg. 27, 121–128 (1997).
Binning, M. J., Liu, J. K., Kestle, J. R., Brockmeyer, D. L. & Walker, M. L. Optic pathway gliomas: a review. Neurosurg. Focus 23, 1–8 (2007).
Massimino, M. et al. High response rate to cisplatin/etoposide regimen in childhood low-grade glioma. J. Clin. Oncol. 20, 4209–4216 (2002).
Astrup, J. Natural history and clinical management of optic pathway glioma. Br. J. Neurosurg. 17, 327–335 (2003).
Tow, S. L., Chandela, S., Miller, N. R. & Avellino, A. M. Long-term outcome in children with gliomas of the anterior visual pathway. Pediatr. Neurol. 28, 262–270 (2003).
Rodriguez, L. A., Edwards, M. S. & Levin, V. A. Management of hypothalamic gliomas in children. An analysis of 33 cases. Neurosurgery 26, 242–247 (1990).
Attina, G. et al. Management of children with optic gliomas and neurofibromatosis type 1. Biomed. Pharmacol. J. 13, 1601–1606 (2020).
Miller, C., Guillaume, D., Dusenbery, K., Clark, H. B. & Moertel, C. Report of effective trametinib therapy in 2 children with progressive hypothalamic optic pathway pilocytic astrocytoma: documentation of volumetric response. J. Neurosurg. Pediatr. 19, 319–324 (2017).
Upadhyaya, S. A. et al. Marked functional recovery and imaging response of refractory optic pathway glioma to BRAFV600E inhibitor therapy: a report of two cases. Childs Nerv. Syst. 34, 605–610 (2018).
Sani, I. & Albanese, A. Endocrine long-term follow-up of children with neurofibromatosis type 1 and optic pathway glioma. Horm. Res. Paediatr. 87, 179–188 (2017).
Cambiaso, P. et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 173, 2353–2358 (2017).
Santoro, C. et al. Pretreatment endocrine disorders due to optic pathway gliomas in pediatric neurofibromatosis type 1: multicenter study. J. Clin. Endocrinol. Metab. 105, 2214–2221 (2020).
Martínez, R., Honegger, J., Fahlbusch, R. & Buchfelder, M. Endocrine findings in patients with optico-hypothalamic gliomas. Exp. Clin. Endocrinol. Diabetes 111, 162–167 (2003).
Marshall, W. A. & Tanner, J. M. Variations in the pattern of pubertal changes in boys. Arch. Dis. Child. 45, 12–23 (1970).
Marshall, W. A. & Tanner, J. M. Variations in the pattern of pubertal changes in girls. Arch. Dis. Child. 44, 291–303 (1969).
Kuczmarski, R. J., Ogden, C. L. & Grummer-Strawn, L. M. CDC growth charts: United States. Adv. Data 8, 1–27 (2000).
Styne, D. M. in Pediatric Endocrinology 2nd edn (ed. Sperling, M. A.) 599 (Elsevier Science, 2002).
Rosenfield, R. in Pediatric Endocrinology 2nd edn (ed. Sperling, M. A.) 494 (Elsevier Science, 2002).
Daniels, S. R. & Greer, F. R. Committee on Nutrition: lipid screening and cardiovascular health in childhood. Pediatrics 122, 198–208 (2008).
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106, 3143–3421 (2002).
Parkhurst, E. & Abboy, S. Optic glioma in neurofibromatosis type 1. J. Pediatr. Ophthalmol. Strabismus 53, 334–338 (2016).
Jeuken, J. W. & Wesseling, P. MAPK pathway activation through BRAF gene fusion in pilocytic astrocytomas; a novel oncogenic fusion gene with diagnostic, prognostic, and therapeutic potential. J. Pathol. 222, 324–328 (2010).
Fouladi, M. et al. Survival and functional outcome of children with hypothalamic/chiasmatic tumors. Cancer 97, 1084–1092 (2003).
Collet-Solberg, P. F. et al. Endocrine outcome in long-term survivors of low-grade hypothalamic/chiasmatic glioma. Clin. Endocrinol. 47, 79–85 (1997).
Merchant, T. E., Conklin, H. M., Wu, S., Lustig, R. H. & Xiong, X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J. Clin. Oncol. 27, 3691–3697 (2009).
Gan, H. W. et al. Neuroendocrine morbidity after pediatric optic gliomas: A longitudinal analysis of 166 children over 30 years. J. Clin. Endocrinol. Metab. 100, 3787–3799 (2015).
Picariello, S. et al. A 40-year cohort study of evolving hypothalamic dysfunction in infants and young children (<3 years) with optic pathway gliomas. Cancers 14, 1–16 (2022).
Terashima, K. et al. Long term outcomes of centrally located low grade glioma in children. Cancer 119, 2630–2638 (2013).
Shalitin, S. et al. Endocrine outcome in long term survivors of childhood brain tumors. Horm. Res. Pediatr. 76, 113–122 (2011).
Listernick, R., Darling, C., Greenwald, M., Strauss, L. & Charrow, J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J. Pediatr. 127, 718 (1995).
Lustig, R. H. et al. Risk factors for the development of obesity in children surviving brain tumors. J. Clin. Endocrinol. Metab. 88, 611–616 (2003).
Josefson, R. L., Listernick, B., Charrow, J. & Habiby, R. L. Growth hormone excess in children with optic pathway tumors is a transient phenomenon. Horm. Res. Pediatr. 86, 35–38 (2016).
Josefson, J., Listernick, R., Fangusaro, J. R., Charrow, J. & Habiby, R. Growth hormone excess in children with neurofibromatosis type 1-associated and sporadic optic pathway tumors. J. Pediatr. 158, 433–436 (2011).
Cambiaso, P. et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. 173A, 2353–2358 (2017).
Manski, T. J., Haworth, C. S., Duval-Arnould, B. J. & Rushing, E. J. Optic pathway glioma infiltrating into somatostatinergic pathways in a young boy with gigantism. Case report. J. Neurosurg. 81, 595–600 (1994).
Listernick, R., Charrow, J., Greenwald, M. J. & Esterly, N. B. Optic gliomas in children with neurofibromatosis type 1. J. Pediatr. 114, 788–792 (1989).
Stein, D. T. Southwestern Internal Medicine Conference. New developments in the diagnosis and treatment of sexual precocity. Am. J. Med. Sci. 303, 53–71 (1992).
Clementi, M. et al. Neurofibromatosis type 1 growth charts. Am. J. Med. Genet. 87, 317–323 (1999).
Szudek, J., Birch, P. & Friedman, J. M. Growth in North American white children with neurofibromatosis 1 (NF1). J. Med. Genet. 37, 933–938 (2000).
Gajjar, A. et al. Low-grade astrocytoma: a decade of experience at St. Jude Children’s Research Hospital. J. Clin. Oncol. 15, 2792–2799 (1997).
Fisher, P. G. et al. Outcome analysis of childhood low-grade astrocytomas. Pediatr. Blood Cancer 51, 245–250 (2008).
Stokland, T. et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro. Oncol. 12, 1257–1268 (2010).
Funding
No financial assistance was received in support of the study.
Author information
Authors and Affiliations
Contributions
M.G.M.—substantial contributions to acquisition of data, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content, and final approval of the version to be published. M.Y.-G.—substantial contributions to analysis and interpretation of data, revising the article critically for important intellectual content, and final approval of the version to be published. H.T., A.T., R.C., and M.P.—substantial contributions to acquisition of data, revising the article critically for important intellectual content, and final approval of the version to be published. S.S.—substantial contributions to conception and design, acquisition of data, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content, and final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the local Institutional Review Board. Patient consent was not required.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gil Margolis, M., Yackobovitz-Gavan, M., Toledano, H. et al. Optic pathway glioma and endocrine disorders in patients with and without NF1. Pediatr Res 93, 233–241 (2023). https://doi.org/10.1038/s41390-022-02098-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-02098-5
- Springer Nature America, Inc.