
Abstract
Background
Doxapram is used for the treatment of apnea of prematurity in dosing regimens only based on bodyweight, as pharmacokinetic data are limited. This study describes the pharmacokinetics of doxapram and keto-doxapram in preterm infants.
Methods
Data (302 samples) from 75 neonates were included with a median (range) gestational age (GA) 25.9 (23.9–29.4) weeks, bodyweight 0.95 (0.48–1.61) kg, and postnatal age (PNA) 17 (1–52) days at the start of continuous treatment. A population pharmacokinetic model was developed using non-linear mixed-effects modelling (NONMEM®).
Results
A two-compartment model best described the pharmacokinetics of doxapram and keto-doxapram. PNA and GA affected the formation clearance of keto-doxapram (CLFORMATION KETO-DOXAPRAM) and clearance of doxapram via other routes (CLDOXAPRAM OTHER ROUTES). For a median individual of 0.95 kg, GA 25.6 weeks, and PNA 29 days, CLFORMATION KETO-DOXAPRAM was 0.115 L/h (relative standard error (RSE) 12%) and CLDOXAPRAM OTHER ROUTES was 0.645 L/h (RSE 9%). Oral bioavailability was estimated at 74% (RSE 10%).
Conclusions
Dosing of doxapram only based on bodyweight results in the highest exposure in preterm infants with the lowest PNA and GA. Therefore, dosing may need to be adjusted for GA and PNA to minimize the risk of accumulation and adverse events. For switching to oral therapy, a 33% dose increase is required to maintain exposure.
Impact
-
Current dosing regimens of doxapram in preterm infants only based on bodyweight result in the highest exposure in infants with the lowest PNA and GA.
-
Dosing of doxapram may need to be adjusted for GA and PNA to minimize the risk of accumulation and adverse events.
-
Describing the pharmacokinetics of doxapram and its active metabolite keto-doxapram following intravenous and gastroenteral administration enables to include drug exposure to the evaluation of treatment of AOP.
-
The oral bioavailability of doxapram in preterm neonates is 74%, requiring a 33% higher dose via oral than intravenous administration to maintain exposure.
Similar content being viewed by others

Introduction
A common symptom of neurological and respiratory underdevelopment in preterm born infants is apnea of prematurity (AOP). AOP decreases oxygen saturation and can result in hypoxic episodes that are harmful to the infant’s short- and long-term neurodevelopmental outcomes.1 Endotracheal intubation is a potentially dangerous procedure in patients as small as preterm babies, and invasive ventilation is related to chronic lung disease.2,3
Although caffeine is the first-choice pharmacological agent for the treatment of AOP with a good efficacy and safety profile,4,5,6 in a proportion of patients AOP persists despite adequately dosed caffeine treatment and maximal noninvasive respiratory support. In these patients, the additive use of doxapram in the therapy has been shown to prevent a large proportion of endotracheal intubation and invasive ventilation.7,8 Doxapram has been frequently used for reducing apnea and hypoxic episodes in very preterm infants in countries such as Germany, Belgium, the Netherlands, Austria, Japan, Australia, Canada, France, Denmark, and Italy, although its use in children and neonates is off-label. Evidence about efficacy and safety has been described in a few studies reporting successful control of AOP that was unresponsive to methylxanthines.9,10,11,12,13 Oral administration has also been reported, using a 2-fold higher dosage than the reported intravenous (IV) dosage, despite an unknown bioavailability.14
Doxapram stimulates respiration by inhibition of the K+ channels of the peripheral chemoreceptors located in the carotid bodies.15 Furthermore, doxapram increases minute ventilation and tidal volume via the respiratory neurons in the central nervous system.14,16 Doxapram is primarily metabolized by CYP3A4, and to a lesser extent by CYP3A5, into keto-doxapram, which has a pharmacological activity of ~80% compared to doxapram.17,18,19 Maturation of CYP enzymes is likely to play a significant role in doxapram pharmacokinetics (PKs), which affects the individual drug exposure and therefore the effectiveness and safety. As hardly any data are available on doxapram PKs and pharmacodynamics (PDs) in prematurely born neonates, further investigation is urgently needed.
Population PKs and PD modeling are increasingly applied to data from preterm infants as it can efficiently handle sparse and infrequently collected data.20 Besides quantifying PK parameters, it offers the possibility to quantify inter-individual variability and identify covariates, and can thereby be used to optimize drug dosing.21,22 Reported evidence on a PK–PD relationship of doxapram in preterm infants is limited to two small cohorts: Barbé et al.23 on side effects and Jamali et al.24 on effectiveness; Ogawa et al.18 described the population PKs in 34 preterm Japanese neonates (gestational age (GA) 24–33 weeks)18 and found that the total clearance (CL) of doxapram was affected by postmenstrual age (PMA), bodyweight, and aspartate aminotransferase levels. Volume of distribution was influenced by GA. Despite this maturation of doxapram CL, dosing has yet been based on bodyweight alone, leading to marked differences in exposure between patients. Herewith, over- and undertreatment are caused.
Generating more evidence on the PKs and PDs of doxapram and keto-doxapram is therefore of great importance. In this study, we aimed to investigate the PKs of doxapram and keto-doxapram following IV administration, as well as oral administration using population PK modeling. Covariates were defined and current dosing regimens were evaluated.
Methods
Study and treatment protocol
Data were obtained from the DINO (Drug dosage Improvement in NeOnates) study that prospectively studied nine drugs, including doxapram, in preterm infants (inclusion criteria GA 24–32 weeks) in four Dutch NICUs. Doxapram was used as standard of care and all included infants were born before 32 weeks of gestation. The Erasmus MC ethics review board approved the protocol and written informed consent from parents/legal guardians was obtained prior to study initiation (MEC-2014-067, NL47409.078.14, ClinicalTrials.gov by NCT02421068). Doxapram administration (as hydrochloride, Dopram®, Manage, Belgium) was initiated if the attending physician judged that apnea or bradycardia persisted despite optimal caffeine therapy and maximal noninvasive ventilatory support. The treatment decision was left to the treating physician and was not further defined in the protocol. To reach steady-state blood concentration immediately, doxapram therapy could be started with a loading dose of 2.0–2.5 mg/kg administered in 15 min. The maintenance dose of 0.5–2.0 mg/kg/h was started either by continuous IV infusion or continuous gastroenteral administration of the IV solution via a nasogastric tube. Gastroenteral administration was only considered when enteral feeding was well tolerated. If treatment was effective, doxapram dose could be decreased in a non-standardized stepwise manner, and the route of administration could be switched from IV to gastroenteral, both on the initiative of the attending physician. Doxapram treatment was stopped when the patient required endotracheal intubation for mechanical ventilation or when apnea was absent with a low doxapram dosage ≤0.5 mg/kg/h.
Blood samples
Blood samples of 0.2 mL were collected in EDTA tubes. To minimize the burden for the infants, blood withdrawal was only allowed from an indwelling arterial catheter or in combination with a routinely scheduled blood collection for clinical patient care. This resulted in sparse, opportunistic sampling and small sample volumes. Directly after collection the samples were stored at 2–8 °C, and within 24 h the sample was centrifuged, and plasma was stored at −80 °C until quantification.
Bioanalytical analysis
Doxapram and keto-doxapram plasma concentrations were measured using ultra-performance liquid chromatography-electrospray ionization-tandem mass spectrometry at the Pharmacy Department of the Erasmus Medical Center, Rotterdam, the Netherlands.25 The assay was validated according to Food and Drug Administration (FDA) guidelines, required 50 µL plasma volume, and was linear over a range of 0.05–4.5 mg/L for doxapram, and over 0.05–5.0 mg/L for keto-doxapram. The lower limits of the ranges represent the lower limits of quantification (LLOQs). Concentrations were below the LLOQ for 24 (8.1%) doxapram concentrations with a median of 0.02 mg/L (range 0.002–0.04 mg/L) and for 25 (8.5%) keto-doxapram concentrations with a median of 0.02 mg/L (range 0.001–0.046 mg/L). Six (2.0%) doxapram concentrations were above the upper limit of quantification with a median of 4.8 mg/L (range 4.7–6.3 mg/L). These concentrations were reported by the laboratory and were used in the analysis.
Dataset
From September 2014 to July 2017, 302 plasma samples from 75 newborn infants (28 females, 47 males) were evaluable for analysis (Table 1). In 294 samples, doxapram and keto-doxapram were quantified, and in eight samples only doxapram was measured. In 44 (59%) newborns, doxapram therapy was started with an IV loading dose (median 2.3 mg/kg, interquartile range 2.0–3.0), while the remaining 41% started therapy without a loading dose. Eleven newborns (15%) started with a gastroenteral maintenance dose. The median gastroenteral and IV maintenance dose at the start of therapy was 2.0 mg/kg/h (interquartile range 1.9–2.0 mg/kg/h). In total, 30 newborns (40%) were switched from IV to gastroenteral doxapram therapy for a median duration of 11 days (range 1–41 days). Covariates birthweight, actual bodyweight, GA, postnatal age (PNA), PMA, and gender were retrieved from the Case Record Forms of the DINO study.
Population PK analysis
The analysis was performed using non-linear mixed-effects modelling (NONMEM®, version 7.3, ICON Development Solutions, Ellicott City, MD, USA), supported by Perl-speaks-NONMEM (PsN®) version 3.4.2 and Xpose version 4.3.5.26 Amounts of administered doxapram hydrochloride, and concentrations of doxapram and keto-doxapram in plasma, were expressed in molar equivalents. Model development was performed in four steps: (1) selection of a structural model, (2) selection of an error model, (3) covariate analysis, and (4) model evaluation. A model for doxapram and keto-doxapram was developed by first developing a model for doxapram only (parent model, see Supplemental File), after which the observations of keto-doxapram were included in the analysis of both doxapram and keto-doxapram data (parent and metabolite model). The absorption rate constant (ka) was fixed to a value of 1 h−1, as the sparse opportunistic sampling resulted in very few samples taken after the initial bolus dose, which did not allow for a reliable estimation of ka. A sensitivity analyses on the chosen fixed value between 0.01 and 100 for ka showed no influence on parameter estimates (data not shown). The central volume of distribution of keto-doxapram (V3) could not be estimated and was therefore fixed to a value of 1 L, due to model identifiability. Formation of keto-doxapram from doxapram was assumed to only occur from the central compartment (Fig. 1). The first-order conditional estimation method with interaction was used throughout model development. In case of missing covariate information, the last value observed in the subject was carried forward. For current bodyweight, linear interpolation between available measurements was performed. Parameter uncertainty presented as the relative standard errors (RSE) were calculated with the sampling importance resampling procedure.27

CLDOXAPRAM OTHER ROUTES clearance of doxapram through other routes than metabolization to keto-doxapram, CLFORMATION KETO-DOXAPRAM formation clearance of keto-doxapram from doxapram, CLKETO-DOXAPRAM clearance of keto-doxapram, V1 compartment for oral administration, Q1 intercompartmental clearance doxapram, Q2 intercompartmental clearance keto-doxapram, V2, 3 volume of distribution of central compartment, V4, 5 volume of distribution of peripheral compartment, Ka absorption rate constant of doxapram.
The objective function value (OFV) was used to compare nested models. A drop in OFV of more than 3.84 for one degree of freedom was considered statistically significant (p < 0.05) for structural model selection.
The covariates birthweight, current bodyweight, GA, PNA, PMA, and gender were evaluated using a stepwise covariate modeling procedure.28 A significance level of p ≤ 0.01 was used for the forward inclusion and a significance level of ≤0.005 for the backward elimination. Key models as well as the final model were evaluated using goodness-of-fit plots and normalized prediction distribution errors (npde)29 based on 1000 simulations of the model.
Evaluation and optimization of dosing regimen
Based on the developed model, different dosing regimens were evaluated. As the study population mainly consisted of preterm neonates aged between 24 and 28 weeks of gestation (Table 1), 1000 Monte Carlo simulations were performed for a median individual birthweight of 0.75 kg and GA of 26 weeks and a birthweight on the 25th and 75th percentile (i.e., birthweight 0.6 and 1.0 kg corresponding with a GA of 24 and 28 weeks, respectively) of the population. In order to account for changes in weight during the course of doxapram treatment, the weight of these neonates was assumed to change according to the growth curves published by Anchieta et al.30 for preterm neonates. For the simulations, a continuous infusion of 2 mg/kg/h starting on PNA days 1, 10, 20, and 30 was administered over 10 days, with and without a 2.5 mg/kg bolus doses at start, which was the median bolus and infusion dose in this cohort. Dosing was adjusted to the changing bodyweight of the patient. As no target concentration has yet been defined for doxapram and keto-doxapram in preterm neonates, different dosing regimens were evaluated for their ability to achieve comparable exposure (e.g., steady-state concentrations) across infants with different gestational and PNAs.
Results
We here describe and evaluate a combined population PK model of doxapram and keto-doxapram (parent and metabolite), and refer to the Supplementary Material for the doxapram alone (parent) model.
Population PK analysis of doxapram and keto-doxapram (parent and metabolite model)
Modeling of 302 plasma concentrations of doxapram and 294 keto-doxapram concentrations resulted in a two-compartment model for doxapram with intra-individual variability on CL and volume (Fig. 1). Keto-doxapram concentrations were also best described by a two-compartment model. A combined proportional and additive error yielded the best description of the residual variability. PNA and GA were found as best predictors for changes in the different CL parameters. This applied to the elimination of doxapram through routes other than metabolization to keto-doxapram (CLDOXAPRAM OTHER ROUTES) (p < 0.001 (−17.9 points in OFV)) for PNA, and for GA (p < 0.005 (−8.4 points in OFV)) (Fig. 2a). This was also the case for the formation CL of keto-doxapram from doxapram (CLFORMATION KETO-DOXAPRAM) (p < 0.001 (−21.8 points in OFV for both GA and PNA)) (Fig. 2b). Although the OFV also decreased with −23.1 points with PMA as a covariate on CLFORMATION KETO-DOXAPRAM, we preferred to describe the separate influence of GA and PNA in the final model because this better reflects physiology with intra- and extra-uterine maturation, respectively. No other covariates were found. After introduction of all covariates, inter-occasion variability was identified on CLDOXAPRAM OTHER ROUTES (p < 0.001 (−132.7 points in OFV)). Figure 2 shows how CLDOXAPRAM OTHER ROUTES and CLFORMATION KETO-DOXAPRAM change with PNA for an infant with GA 24, 26, and 28 weeks. Oral bioavailability was estimated at 74% (RSE 13%).

Population clearance expressed in L/h (left) and in L/kg/h (right) of clearance of doxapram through other routes than metabolization to keto-doxapram (CLDOXAPRAM OTHER ROUTES) (a) and formation clearance of keto-doxapram from doxapram (CLFORMATION KETO-DOXAPRAM) (b) for premature infants with a gestational age of 24 (lowest line), 26 (middle line), and 28 (upper line) weeks versus postnatal age [days].
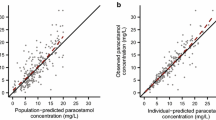
The goodness-of-fit plots of the final model for doxapram and keto-doxapram (Fig. 3) show that the model provides a good description of the data. The inter-individual variability in doxapram CL (CLDOXAPRAM OTHER ROUTES as well as CLFORMATION KETO-DOXAPRAM) and volume of distribution of the final model showed no correlation to parameters relevant for maturation and development. This indicates that the chosen covariates explain most of the variability caused by maturation (Supplementary Fig. S5). There are no remaining trends with respect to time, concentration, GA, or PNA (Supplementary Fig. S6). The npde analysis showed no trends towards a model misspecification for doxapram (Supplementary Fig. S7) and keto-doxapram (Supplementary Fig. S8). The final parameter estimates are displayed in Table 2 as well as their RSEs calculated with the sampling importance resampling procedure.27 Parameter estimates for doxapram in the combined model (parent and metabolite) were comparable to those estimated in the model for the parent compound (Table 2).

a, b, e, f show population predicted versus observed values plotted on linear (left) and log scale (right). c, d, g, h show individual predicted versus observed values plotted on linear (left) and log scale (right).
Evaluation and optimization of dosing regimen
In case of dosing according to bodyweight only, steady-state concentrations predictions varied substantially between preterm neonates of different PNAs and GAs (Fig. 4). In case a continuous infusion (2 mg/kg/h) was started at PNA day 1, more than 2-fold higher doxapram concentrations were observed during the first week of life when compared to infants who started at older PNAs (e.g., 10, 20, or 30 days, Fig. 4a). In addition, infants with a GA of 24 weeks showed higher steady-state plasma concentrations compared to those born after 26 or 28 weeks of gestation. Stable doxapram plasma concentrations were reached after a PNA of ~15 days and were 2.5, 2.0, and 1.7 mg/L for a GA of 24, 26, and 28 weeks, respectively. Starting doxapram treatment with a loading dose of 2.5 mg/kg compared to without a loading dose resulted in a predicted doxapram concentration at 0.5 h post dose of 1.6L versus 0.5 mg/L and 2.0 versus 0.7 mg/L for a GA of 24 and 26 weeks, respectively, at PNA day 1. Figure 4b also illustrates the additive value of a bolus at the start of doxapram therapy.

a A continuous intravenous infusion of 2 mg/kg/h over a period of 10 days without a bolus dose. b A bolus dose of 2.5 mg/kg followed by a continuous infusion of 2 mg/kg/h over a period of 10 days. c compared to a 2.5 mg/kg bolus and 2 mg/kg/h continuous infusion for all patients (see Fig. 5b), continuous infusion was adjusted as follows: PNA: 50% until PNA day 9, 75% from day 10 to 15, and 100% above day 16. GA: 60% for GA 24 and 25 weeks, 80% for 26 and 27 weeks, and 100% for 28 and 29 weeks.
When aiming for a similar steady-state concentration across the entire gestational and PNA range in this study, reducing the continuous dose by 50% up to PNA day 9, and by 25% from PNA day s10 to 15, will reduce the observed high exposure in the early days for all GAs (Fig. 4c in comparison to Fig. 4b). After this adjustment, differences caused by GA can be observed (Fig. 4c), which can be corrected for by (additionally) reducing the dose by 40% for 24 and 25 weeks, and by 20% for 26 and 27 weeks of GA compared to 28 and 29 weeks. Figure 4b illustrates how keto-doxapram concentration time profiles of preterm neonates change with different gestational and PNAs, after a bolus dose of 2.5 mg/kg followed by a doxapram continuous infusion of 2 mg/kg/h over a period of 10 days.
Due to an estimated bioavailability of 74% following gastroenteral administration of doxapram, a 33% dosage increase compared to the IV route would be required in order to achieve comparable exposure.
Discussion
To our best knowledge, we successfully developed the first population PK model to systematically describe the maturation of PKs of doxapram and keto-doxapram in very preterm neonates considering data from both continuous gastroenteral administration and IV infusion. We illustrate that important knowledge may be obtained, despite being limited to observational data from clinical care, and the use of opportunistic, sparse sampled blood to minimize the burden. PKs of doxapram and keto-doxapram were each best described by a two-compartment model. We found that in our data PNA was the most important factor determining the formation CL of keto-doxapram and the CL of doxapram that was eliminated through other pathways. GA also influenced the two CLs, but to a lesser extent. These findings are of relevance for adequate dosing in clinical care, as they reveal the unequal exposure following the current uniform bodyweight-based doxapram dosage regimen. In addition, we report the bioavailability in preterm neonates. Our model-based estimation was 74%.
Our final model suggests that the conversion of doxapram into keto-doxapram and the CL of doxapram other than via this route are subject to intra- and extra-uterine maturation as the parameters were influenced by both GA and PNA, respectively. Namely, intra- and extra-uterine maturation are two separate processes with different rates and drivers. Despite PMA, presenting a combination of GA and PNA, on conversion of doxapram into keto-doxapram led to a comparable OFV reduction as the separate estimation of the two factors, we preferred to describe the separate influence of PNA and GA in the final model. From Table 2 it can be observed that most parameters could be estimated with good reliability. The residual standard error of the peripheral volume of distribution for doxapram in the model for the parent compound showed a rather large residual variability (130%). This is due to the data containing only few samples within 5 h after a loading dose, which complicated the estimation of distribution volumes with the largest impact on the peripheral volume of distribution. Population parameter estimates of the parent model were in line with the model for doxapram and keto-doxapram; total doxapram CL of 0.788 versus 0.760 L/h (sum of CLDOXAPRAM OTHER ROUTES and CLFORMATION OF KETO-DOXAPRAM), central volume of distribution of 2.66 versus 1.54 L, and bioavailability of 74% versus 74%, respectively, indicating a good predictive capacity of the combined model. The comparability of parameter estimates from both models also indicates that the covariates found in the metabolite model were not influenced by the assumptions made.
Previously, Ogawa et al.18 have described the population PKs of intravenously administered doxapram in preterm Japanese infants with a one-compartment model. In line with our findings, Ogawa et al.18 found a CL of 0.698 L/kg/h and volume of distribution of 3.682 L/kg. They also found a correlation between CL and age (postmenstrual age and current bodyweight), albeit without reporting a separate influence of GA and PNA. Although relevant, their model incorporates only IV administration of very low dosages up to 0.4 mg/kg/h in older preterm neonates (median GA 29.1 weeks) and does not describe the PKs of keto-doxapram. Recently, gender and postmenstrual age were used to individualize doxapram dosing and led to a decreased variability in measured concentrations when compared to standard bodyweight dosing.31 In our model, gender was not a significant covariate (Supplementary Fig. S5). We cannot provide an explanation for this.
As in adults, doxapram is primarily metabolized by CYP3A4, and to a lesser extent by CYP3A5 in the liver, which produces the pharmacologically active metabolite keto-doxapram.17,18 An increased CL may be explained by enzyme maturation as well as by an increase in liver blood flow.32,33 This has also been reported for other drugs in preterm born infants who are substrates for CYP3A4, such as midazolam.34,35
The high estimated oral bioavailability of 74% (RSE 10%) is somewhat higher than the reported 60% in healthy adults by Robson et al.36 in 1979. A higher proportion reaching the systemic circulation may be explained by an age-related lower first-pass effect due to low CYP3A activity in the liver and in the gut wall of newborns.37 Our model shows that, for a preterm infant with a median GA (25.6 weeks) and PNA (29 days), conversion of doxapram into keto-doxapram encompasses 15% of doxapram elimination. As both CLDOXAPRAM OTHER ROUTES and CLFORMATION OF KETO-DOXAPRAM are influenced by GA and PNA with different maturation rate, the proportion of the total doxapram CL that follows the conversion into keto-doxapram changes with GA and PNA (Fig. 5).

Three bars for PNA days 1, 5, 15, and 30 represent the population clearance values for GA 24 (left), 26 (middle), and 28 (right) weeks.
Although a target concentration for doxapram has not yet been defined, Jamali et al.24 have speculated that a target concentration of 1.5 mg/L should be aimed for, and Barbe et al.23 suggested an association with high risk of side effects above a concentration of 9 mg/L for doxapram plus keto-doxapram. Yet, validated target concentrations are lacking and the desired concentration might be a moving target and may vary with age due to development of the respiratory center and sensitivity of carotid bodies.38,39 Nevertheless, our study offers the possibility to compare the exposure of different dosing regimen using simulations, which may lead to optimized dosing advices. The performed simulations aimed at achieving comparable steady-state concentrations over all studied gestational and PNAs. Consequently, the dose should be reduced in newborns with low GAs and PNAs. In particular, the continuous dose may be reduced by 50% up to PNA day 9, and by 25% from PNA days 10 to 15. It is important that clinicians are aware of this need for lower dosing because doxapram therapy is increasingly used in these first weeks of life. In addition but less profound, dosing should also be corrected for GA by reducing the dose by 40%, and 20% in newborns with GAs of 24 and 25 weeks and 26 and 27 weeks, respectively, compared to 28 and 29 weeks GA (Fig. 4). Another important finding for clinical practice is the estimation of an oral bioavailability of 74%, which requires a 33% dosage increase compared to the IV route, in order to achieve comparable exposure. Furthermore, our population PK model serves to predict the exposure to doxapram and keto-doxapram in individual patients. By integrating continuous physiological PD data with model-based drug exposure and data on adverse drug reactions (ADRs), we might set one step towards precision medicine in neonatology, as has been illustrated by Poppe et al.40 ADRs are also important to take into account as the use of one bodyweight-based dosage may have led to overexposure to doxapram in a proportion of this vulnerable population in the past. This may have increased the frequency of ADRs that have been reported in preterm infants, including QT interval lengthening41 possibly resulting in an atrioventricular heart block,42 gastrointestinal problems,41,43 tachycardia,23 increased electroencephalographic activity and less sleep–wake cycling,44 irritability and agitation,23,43 and hypokalemia.45,46
Despite the valuable new findings for clinical practice, our model has certain limitations. First, a possible effect of bodyweight on volume of distribution of doxapram may have been missed in our model due to the limitation that the data consisted only of continuous doxapram administration with very few samples taken during the first hours of therapy, which made it difficult to make an accurate estimation of the volume of distribution. Second, although reported by the lab, 27% of the doxapram concentrations were above the upper limit of quantification. This may increase the inaccuracy of these measurements, although Fig. 2 does not indicate inaccurate predictions of high concentrations doxapram and keto-doxapram. Third, as inter-individual variability could not be estimated for the elimination CL of keto-doxapram, the estimate of the inter-individual variability might be higher on the conversion rate than it is in reality and the individual predictions of the keto-doxapram concentrations might be less accurate than those for the parent compound. Fourth, below a PNA of 5 days the model has limited predictive capacity, due to sparse data in this period as doxapram use in these first days is currently retained.
In conclusion, our description of the PKs of doxapram and keto-doxapram in preterm born infants following IV and gastroenteral administration is of great additional value to the recent improvements with regard to treatment of AOP. Steps have been made in the evaluation of the respiratory condition and therewith the effectiveness of treatment.47 The effectiveness of doxapram on arterial oxygenation has been confirmed, as well as some associated side effects. This promising drug urgently needs further investigation. Eventually, an algorithm may be developed to link defined patterns in oxygen saturation profiles to covariates that influence effectiveness of pharmacotherapy, such as changes in doxapram exposure. Herewith, dose adjustments may be proposed tailored to the individual intensively monitored child with a minimal risk of accumulation and consequent adverse events.
References
Poets, C. F. et al. Association between intermittent hypoxemia or bradycardia and late death or disability in extremely preterm infants. JAMA 314, 595–603 (2015).
Sawyer, T. et al. Improving neonatal intubation safety: a journey of a thousand miles. J. Neonatal Perinat. Med. 10, 125–131 (2017).
Sweet, D. G. et al. European Consensus Guidelines on the Management of Respiratory Distress Syndrome—2016 update. Neonatology 111, 107–125 (2017).
Morton, S. U. & Smith, V. C. Treatment options for apnoea of prematurity. Arch. Dis. Child Fetal Neonatal Ed. 101, F352–F356 (2016).
Schmidt, B. et al. Caffeine therapy for apnea of prematurity. N. Engl. J. Med. 354, 2112–2121 (2006).
Schmidt, B. et al. Survival without disability to age 5 years after neonatal caffeine therapy for apnea of prematurity. JAMA 307, 275–282 (2012).
Flint, R. et al. Retrospective study shows that doxapram therapy avoided the need for endotracheal intubation in most premature neonates. Acta Paediatr. 106, 733–739 (2017).
Prins, S. A. et al. Doxapram use for apnoea of prematurity in neonatal intensive care. Int. J. Pediatr. 2013, 251047 (2013).
Alpan, G. et al. Clinical and laboratory observations—doxapram in the treatment of idiopathic apnea of prematurity unresponsive to aminophylline. J. Pediatr. 104, 634–637 (1984).
Eyal, F. et al. Aminophylline versus doxapram in idiopathic apnea of prematurity—a double-blind controlled-study. Pediatrics 75, 709–713 (1985).
Dopram 2 mg/ml-actualisatie-SPC-08-2010. https://www.geneesmiddeleninformatiebank.nl/smpc/h07309_smpc.pdf. Revised 07-03-2012.
Barrington, K. J. et al. Dose–response relationship of doxapram in the therapy for refractory idiopathic apnea of prematurity. Pediatrics 80, 22–27 (1987).
Vliegenthart, R. J., Ten Hove, C. H., Onland, W. & van Kaam, A. H. Doxapram treatment for apnea of prematurity: a systematic review. Neonatology 111, 162–171 (2016).
Poets, C. F., Darraj, S. & Bohnhorst, B. Effect of doxapram on episodes of apnoea, bradycardia and hypoxaemia in preterm infants. Biol. Neonate 76, 207–213 (1999).
Peers, C. Effects of doxapram on ionic currents recorded in isolated type-i cells of the neonatal rat carotid-body. Brain Res. 568, 116–122 (1991).
Barrington, K. J., Finer, N. N., Peters, K. L. & Barton, J. Physiologic effects of doxapram in idiopathic apnea of prematurity. J. Pediatr. 108, 124–129 (1986).
Bairam, A. et al. Doxapram metabolism in human fetal hepatic organ culture. Clin. Pharmacol. Ther. 50, 32–38 (1991).
Ogawa, Y. et al. Population pharmacokinetics of doxapram in low-birth-weight Japanese infants with apnea. Eur. J. Pediatr. 174, 509–518 (2015).
Bairam, A. et al. Pharmacodynamic effects and pharmacokinetic profiles of keto-doxapram and doxapram in newborn lambs. Pediatr. Res. 28, 142–146 (1990).
Krekels, E. H. J. et al. Evidence-based drug treatment for special patient populations through model-based approaches. Eur. J. Pharm. Sci. 109S, S22–S26 (2017).
Brussee, J. M. et al. Children in clinical trials: towards evidence-based pediatric pharmacotherapy using pharmacokinetic-pharmacodynamic modeling. Expert Rev. Clin. Pharmacol. 9, 1235–1244 (2016).
De Cock, R. F. et al. The role of population PK-PD modelling in paediatric clinical research. Eur. J. Clin. Pharmacol. 67(Suppl. 1), 5–16 (2011).
Barbe, F. et al. Severe side effects and drug plasma concentrations in preterm infants treated with doxapram. Ther. Drug Monit. 21, 547–552 (1999).
Jamali, F. et al. Doxapram dosage regimen in apnea of prematurity based on pharmacokinetic data. Dev. Pharm. Ther. 11, 253–257 (1988).
Flint, R. B., Bahmany, S., van der Nagel, B. C. H. & Koch, B. C. P. Simultaneous quantification of fentanyl, sufentanil, cefazolin, doxapram and keto-doxapram in plasma using liquid chromatography—tandem mass spectrometry. Biomed. Chromatogr. 32, e4290 (2018).
Keizer, R. J., Karlsson, M. O. & Hooker, A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacomet. Syst. Pharmacol. 2, e50 (2013).
Dosne, A. G., Bergstrand, M., Harling, K. & Karlsson, M. O. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J. Pharmacokinet. Pharmacodyn. 43, 583–596 (2016).
Lindbom, L., Pihlgren, P. & Jonsson, E. N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput. Methods Prog. Biomed. 79, 241–257 (2005).
Comets, E., Brendel, K. & Mentre, F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput. Methods Prog. Biomed. 90, 154–166 (2008).
Anchieta, L. M., Xavier, C. C., Colosimo, E. A. & Souza, M. F. Weight of preterm newborns during the first twelve weeks of life. Braz. J. Med. Biol. Res. 36, 761–770 (2003).
Greze, E. et al. Doxapram dosing for apnea of prematurity based on postmenstrual age and gender: a Randomized Controlled Trial. Paediatr. Drugs 18, 443–449 (2016).
Gow, P. J. et al. Neonatal hepatic drug elimination. Pharmacol. Toxicol. 88, 3–15 (2001).
Grijalva, J. & Vakili, K. Neonatal liver physiology. Semin. Pediatr. Surg. 22, 185–189 (2013).
Brussee, J. M. et al. Predicting CYP3A-mediated midazolam metabolism in critically ill neonates, infants, children and adults with inflammation and organ failure. Br. J. Clin. Pharmacol. 84, 358–368 (2018).
Voller, S. et al. Recently registered midazolam doses for preterm neonates do not lead to equal exposure: a population pharmacokinetic model. J. Clin. Pharmacol. 59, 1300–1308 (2019).
Robson, R. H. & Prescott, L. F. A pharmacokinetic study of doxapram in patients and volunteers. Br. J. Clin. Pharmacol. 7, 81–87 (1979).
Brussee, J. M. et al. First-pass CYP3A-mediated metabolism of midazolam in the gut wall and liver in preterm neonates. CPT Pharmacomet. Syst. Pharmacol. 7, 374–383 (2018).
Carroll, J. L. & Kim, I. Postnatal development of carotid body glomus cell O2 sensitivity. Respir. Physiol. Neurobiol. 149, 201–215 (2005).
Wong-Riley, M. T., Liu, Q. & Gao, X. P. Peripheral-central chemoreceptor interaction and the significance of a critical period in the development of respiratory control. Respir. Physiol. Neurobiol. 185, 156–169 (2013).
Poppe, J. A. et al. Precision dosing of doxapram in preterm infants using continuous pharmacodynamic data and model-based pharmacokinetics: an illustrative case series. Front. Pharmacol. 11, 665 (2020).
Maillard, C. et al. QT interval lengthening in premature infants treated with doxapram. Clin. Pharm. Ther. 70, 540–545 (2001).
De Villiers, G. S., Walele, A., Van der Merwe, P. L. & Kalis, N. N. Second-degree atrioventricular heart block after doxapram administration. J. Pediatr. 133, 149–150 (1998).
Tay-Uyboco, J. et al. Clinical and physiological responses to prolonged nasogastric administration of doxapram for apnea of prematurity. Biol. Neonate 59, 190–200 (1991).
Czaba-Hnizdo, C. et al. Amplitude-integrated electroencephalography shows that doxapram influences the brain activity of preterm infants. Acta Paediatr. 103, 922–927 (2014).
Fischer, C., Ferdynus, C., Gouyon, J. B. & Semama, D. S. Doxapram and hypokalaemia in very preterm infants. Arch. Dis. Child Fetal Neonatal Ed. 98, F416–F418 (2013).
Shimokaze, T., Toyoshima, K., Shibasaki, J. & Itani, Y. Blood potassium and urine aldosterone after doxapram therapy for preterm infants. J. Perinatol. 38, 702–707 (2018).
Flint, R. B. et al. Big data analyses for continuous evaluation of pharmacotherapy: a proof of principle with doxapram in preterm infants. Curr. Pharm. Des. 23, 5919–5927 (2017).
Acknowledgements
This study was enabled by funding from the Netherlands Organization for Health Research and Development ZonMw (Grant number: 80-83600-98-10190).
Author information
Authors and Affiliations
Consortia
Contributions
R.d.G. initiated the application of the grant that enabled the DINO study. R.B.F., S.H.P.S., R.d.G, D.M.B., and C.A.J.K. designed the study. P.L.J.D., P.A., K.D.L., I.K.M.R., and S.H.P.S. were local coordinating investigators and recruited subjects. B.C.P.K. facilitated the measurement of the plasma concentrations doxapram and keto-doxapram. R.B.F., A.G.J.E., R.t.H., C.A.J.K., and S.V. performed the population PK analyses. All authors have read and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Patient consent
The Erasmus MC ethics review board approved the protocol and written informed consent from parents/legal guardians was obtained prior to study initiation (MEC-2014-067).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Flint, R.B., Simons, S.H.P., Andriessen, P. et al. The bioavailability and maturing clearance of doxapram in preterm infants. Pediatr Res 89, 1268–1277 (2021). https://doi.org/10.1038/s41390-020-1037-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-1037-9
- Springer Nature America, Inc.
This article is cited by
-
Why population pharmacokinetic studies are instrumental to boost clinical research in neonates, and suggestions on how clinicians should assess these papers
Pediatric Research (2024)
-
When will the Glomerular Filtration Rate in Former Preterm Neonates Catch up with Their Term Peers?
Pharmaceutical Research (2024)
-
Doxapram versus placebo in preterm newborns: a study protocol for an international double blinded multicentre randomized controlled trial (DOXA-trial)
Trials (2023)
-
Comment on: "Preterm Physiologically Based Pharmacokinetic Model, Part I and Part II”
Clinical Pharmacokinetics (2021)