
Abstract
Neratinib is a tyrosine kinase inhibitor that is used for the therapy of patients with HER2+ breast tumors. However, despite its clinical benefit, resistance to the drug may arise. Here we have created cellular models of neratinib resistance to investigate the mechanisms underlying such resistance. Chronic neratinib exposure of BT474 human HER2+ breast cancer cells resulted in the selection of several clones resistant to the antiproliferative action of the drug. The clones were characterized biochemically and biologically using a variety of techniques. These clones retained HER2 levels similar to parental cells. Knockdown experiments showed that the neratinib-resistant clones retained oncogenic dependence on HER2. Moreover, the tyrosine phosphorylation status of BT474 and the resistant clones was equally sensitive to neratinib. Transcriptomic and Western analyses showed that peptidylarginine deiminase 3 was overexpressed in the three neratinib-resistant clones studied but was undetectable in BT474 cells. Experiments performed in the neratinib-resistant clones showed that reduction of PADI3 or inhibition of its function restored sensitivity to the antiproliferative action of neratinib. Moreover, overexpression of FLAG-tagged PADI3 in BT474 cells provoked resistance to the antiproliferative action of neratinib. Together, these results uncover a role of PADI3 in the regulation of sensitivity to neratinib in breast cancer cells overexpressing HER2 and open the possibility of using PADI3 inhibitors to fight resistance to neratinib.

Similar content being viewed by others


Introduction
Breast tumors from one out of five patients express elevated levels of the transmembrane tyrosine kinase HER2 [1]. Such characteristics impact the therapeutic options selected to treat those patients, fundamentally based on agents that target HER2 [2]. Two types of agents acting on HER2 have reached the breast cancer clinic [3]. On one hand humanized monoclonal antibodies that recognize the extracellular region of HER2, such as trastuzumab or pertuzumab. This category also includes trastuzumab biosimilars [4] and antibody-drug conjugates such as trastuzumab-emtansine or trastuzumab-deruxtecan, which are modified versions of trastuzumab containing a chemotherapeutic drug linked to the antibody [5,6,7]. The second class of agents that act on HER2 are small molecule tyrosine kinase inhibitors (TKIs) of HER2 [8]. These drugs are membrane-permeable and act on the intracellular region of HER2, inhibiting its kinase activity. Approved drugs of this type include lapatinib, neratinib, pyrotinib, or tucatinib [9].
Neratinib irreversibly binds to cysteines located in the ATP binding pocket of HER2 and the related family members EGFR or HER4, inhibiting their tyrosine kinase activity [8]. Neratinib was approved based on the ExteNET and NALA trials that demonstrated its efficacy to reduce relapses after adjuvant therapy with trastuzumab [10, 11]. Neratinib has shown preclinical activity on models of trastuzumab resistance [12] and increases the antitumoral action of trastuzumab [13]. Moreover, neratinib has shown efficacy inhibiting mutant forms of HER2 in the absence of HER2 gene amplification [14]. The SUMMIT and MutHER trials demonstrated the clinical value of neratinib in combination with hormonal therapies in heavily pretreated patients with HR-positive disease and expressing mutant forms of HER2 [14, 15].
Despite the efficacy of neratinib in HER2+ and HER2 mutant tumors, patients may develop resistance to the drug. Several mechanisms appear to be involved in the resistance to neratinib, since a number of proteins and signaling pathways related to such resistance have been described, including overexpression of Neuromedin U, down-regulation of P-glycoprotein, increased CYP3A4, mutations in HER2, or activation of certain signaling pathways such as the PI3K/AKT/mTOR or YES routes [16,17,18,19,20,21].
To contribute to the understanding of the mechanisms of resistance to neratinib, we generated cellular models of resistance to that drug. We show that the resistant models retain expression of HER2, and the receptor is still sensitive to the kinase inhibitory actions of the drug. Moreover, we show that neratinib-resistant breast cancer cell lines are still dependent on oncogenic HER2. Transcriptomic as well as functional studies led to the identification of the peptidylarginine deiminase PADI3 as a protein that regulated sensitivity to neratinib. Peptidylarginine deiminases (PADs) are a group of calcium-dependent enzymes that catalyze the post-translational conversion of arginine to citrulline, leading to changes in protein structure and function [22,23,24]. Several studies have already demonstrated their involvement in the process of tumorigenesis or tumor progression [25,26,27]. The 5 family members (PADI1, 2, 3, 4, and 6) have distinct substrate affinities and tissue-specific expression patterns [28]. For example, PADI3 is mostly expressed in the epidermis and hair follicles [28], whereas PADI2 and 4 have been associated with the pathogenesis of breast cancer [29,30,31,32].
Although the role of PADI3 in tumorigenesis is still not entirely clear [26], different studies have shown its involvement in several processes that could justify its function as an oncogene [24, 27, 29]. These actions include (i) activation of glycolysis through PKM2 citrullination, favoring cancer cell proliferation, (ii) epigenetic modifications due to citrullination of histones that regulate chromatin status, and (iii) direct regulation of enzymes involved in the transcription of genes related to cell migration, invasion, and survival. Interestingly, it has been observed that PADI3 can also act as a tumor suppressor gene, inhibiting proteins such as SIRT2, Hsp90, CKS1, and positively regulating p21, resulting in cell cycle arrest [24, 26]. Therefore, depending on the molecular context, PADI3 may act both as an oncogene and as a tumor suppressor gene in cancer.
Not only our data uncovers a role of PADI3 as a protein involved in the resistance to neratinib, but they also add evidence to the concept that resistance to neratinib may involve different routes. Therefore, restoration of efficacy of the drug may result difficult considering that several potential escape mechanisms may be present when resistance mechanisms arise.
Materials and methods
Reagents and antibodies
Cell culture media, fetal bovine serum (FBS), antibiotics and trypsin were purchased from Life Technologies (Carlsbad, CA, USA), Immobilon-P membranes from Millipore Corp and Polyethylenimine (PEI) reagent from Polysciences (Hirschber an der Bergstrasse, Germany). Cl-amidine was purchased from Sigma-Aldrich (Sant Louis, Missouri, EEUU). Neratinib, lapatinib and tucatinib were from Selleckchem (Houston, TX, USA). Trastuzumab and T-DM1 were purchased from Farmacia Escudero, Salamanca, Spain and Farmacia Meritxell, Andorra, respectively. Other generic chemicals were from Sigma Chemicals Co, Roche Biochemicals or Merck.
Antibodies against GAPDH (sc-166574), PADI3 (#393622) and pTyr99 (sc-7020) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-calnexin antibody (SPA-860-F) from Stressgen Bioreagents (British Columbia, Canada); anti-HER2 (Ab3, #OP15) used for Western detection of human HER2 was from Calbiochem (La Jolla, CA, USA); anti-pS6 (#2215) and anti-S6 (#2217) were from Cell Signaling Technologies (Beverly, MA, USA). The anti-MGP antibody was from Proteintech (Planegg-Martinsried, Germany). The anti-FLAG antibody (F1804) was from Sigma. The horseradish peroxidase (HRP)-conjugated antibodies against mouse and rabbit IgG were obtained from GE Healthcare Life Sciences (Piscataway, NJ, USA) and Bio-Rad Laboratories (Hercules, CA, USA) and Cy3-conjugated antibodies were from Jackson Immunoresearch. Anti-human-PE was from BD Biosciences.
Cell culture, generation of clones resistant to neratinib, and cell proliferation assays
Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), containing high glucose (4500 mg/L) and antibiotics (penicillin 100 U/ml, streptomycin 100 µg/ml). Cell lines were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. Cell authenticity was analyzed at origin by the ATCC or by STR at the Hematology Service of the University Hospital of Salamanca. Cell lines purchased from the ATCC were expanded from the initially purchased vial to ten 100 mm dishes, and when cultures were confluent, frozen 1 dish:1 vial ratio. Cells were regularly replaced (each 3-6 months) for new ones by thawing a frozen vial.
To generate clones resistant to neratinib, BT474 cells were plated in 150 mm dishes at a density of 20,000 cells per dish. Next day, once attached, cells were treated with neratinib at different doses (1 μM, 500 nM, 100 nM, 50 nM, 25 nM, 10 nM). The medium with the different neratinib concentrations was replaced once a week. After 3 months of exposure to the drug under those culture conditions, 23 clones were isolated resistant to 10 nM neratinib (numbered #1 to #23) and one clone (#24) resistant to 25 nM neratinib.
Cell proliferation was analyzed by conventional MTT uptake or cell counting experiments using a Z1 Coulter Particle Counter. Detailed protocols for these assays have previously been described [33,34,35].
Transfection, lentivirus production and infection
For transfection, PEI reagent was used. It was previously prepared by dissolving 1 mg/ml of the reagent in pre-warmed water. Once dissolved, the pH was adjusted to 7.0, filtered and aliquoted. For the transfection, 500 μl of 150 mM NaCl solution including 10 μg of DNA were mixed with 500 μl of 150 mM NaCl solution containing 40 μl of prepared PEI. After 30 min incubation, complexes were drop-wise added over the monolayers of target cells plated in 10 cm plates 24 h before. 12-24 h later media was replaced by normal culture medium before further analyses. For PADI3 overexpression, the plasmid pCMV-PADI-Myc-DKK was ordered from Origene (Herford, Germany), transfected and neomycin-resistant clones isolated (500 μg/ml, 3 weeks). To obtain a pool of SKBR3 cells overexpressing Flag-tagged PADI3, cells were transfected with the above-mentioned plasmid and selected with G418 (500 μg/ml) for a month. A pool of SKBR3 cells was analyzed for PADI3 expression by western blotting.
For lentivirus production, 4 µg of each of the plasmids pMDLg/RRE, pRSV-Rev and pMD2.G (Addgene, Cambridge, MA, USA), along with 8 µg of the pLKO.1 lentiviral plasmid containing a scramble shRNA (sh-Control) or the shRNAs for HER2 or PADI3 (GE Dharmacon, Lafayette, CO, USA) were co-transfected as described into HEK293T cells. Twenty-four hours later, HEK293T medium was replaced with fresh medium, and 48 h post co-transfection, the medium containing lentiviral particles was collected, filtered, and utilized to infect cells after the addition of 6 µg/ml polybrene (Sigma-Aldrich). Forty-eight hours after the transfection, transduced cells were selected with 3 µg/ml puromycin (Sigma-Aldrich) for another 48 h. Five distinct shRNA sequences targeting HER2 or PADI3 were assessed and used those demonstrating higher levels of knockdown (#1-ATG TAT AGG TAA CCT GTG AT and #2-TAC AAA GCC TGG ATA CTG ACA for shHER2; #36-TGG CAA GGA AGA ACA TGG TTT and #37-CCA GTA TAA GAG GGA CAA GTT for shPADI3).
Protein extraction, immunoprecipitation (IP) and Western blot (WB)
The preparation of cell extracts, IP and WB analyses were performed as described [35, 36]. Blots were visualized by autoradiography or digital capture using a Chemidoc apparatus (Bio-Rad Laboratories, Hercules, CA, USA). Densitometric measurements of the bands were performed using the Image Lab™ Software Version 6.0.1 included with the Chemidoc system.
For cell surface immunoprecipitation, cells were grown at 70-80% confluency, washed twice with Krebs-Ringer-HEPES buffer (128 mM NaCl; 5.0 mM KCl; 5.0 mM MgSO4; 1.3 mM CaCl2; 50 mM Hepes, pH 7.4) and incubated with 10 nM trastuzumab for 2 h at 4 °C in the same buffer. Monolayers were washed twice with PBS and lysed. Cell debris were removed by centrifugation, and supernatants incubated for 30 min with protein A-Sepharose. Immunocomplexes were then washed and loaded in SDS-PAGE gels as described [37].
Immunofluorescence microscopy and cell surface staining
Cells were plated on glass coverslips and 48 h later washed with PBS/Ca2 + /Mg2+ (1 mM CaCl2, 0.5 mM MgCl2 in PBS) and fixed in 2% p-formaldehyde for 30 min at room temperature. Then, cells were washed with PBS/CM, quenched with 50 mM NH4Cl and permeabilized with 0.1% Triton X-100, 0.2% BSA) and then blocked in PBS/CM with 0.2% BSA (blocking solution) for 1 h at room temperature. Monolayers were then incubated with the primary antibody in blocking solution for 2 h at room temperature. After three washes for 10 min each in PBS with 0.2% BSA, the coverslips were incubated with cyanine-3-conjugated secondary antibodies for 30 min, washed three times, 5 min each, in PBS with 0.2% BSA, and mounted. Samples were analyzed by regular epifluorescence microscopy or by confocal immunofluorescence microscopy using a Leica TCS SP5 System (Leica Microsystem CMS, Wetzlar, Germany). Phase contrast images were taken with a Nikon Eclipse Ti-S inverted microscope (Izasa, Barcelona, Spain).
To perform cell surface staining of HER2 for cytometric analyses, cells were trypsinized, collected in culture medium (DMEM + 10% FBS) and resuspended in PBS + 1% BSA. Cells were then incubated with 10 nM trastuzumab for 1 h under rocking agitation at room temperature. Later, cells were washed twice with PBS + 1% BSA and incubated with anti-human-PE (1:100) for 30 min, in agitation at room temperature. After that incubation period, cells were washed twice with PBS + 1% BSA and cytometrically analyzed using a BD AccuriTM apparatus (BD Biosciences). Mean fluorescence intensity of HER2 was quantified using the BD Accuri C6 Software.
Microarray transcriptomic studies
Total RNA extraction was performed with the RNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. RNA concentration was determined with a NanoDropTM 1000 apparatus (Thermo Fisher Scientific). Assessment of the quality of the isolated RNAs was carried out by the Genomics Service of our Institute, using the Bioanalyzer RNA 6000 Nano assay (Agilent). cDNAs were hybridized to Clariom S human arrays (Affymetrix, Santa Clara, USA). Raw CEL files were normalized using the RMA algorithm and the genes that presented differential expression were identified using the Transcriptome Analysis Console (TAC) software version 4.0.2.15 (Affymetrix). The cut-off values taken were fold-change <-2 or >2 and a p-value of ANOVA < 0.01.
HER2 sequencing
Total RNA was isolated as described above and first-strand cDNA was synthesized using M-MLV reverse transcriptase and oligo-dT (Invitrogen, Carlsbad, CA, USA), following the instructions from the manufacturer. The oligonucleotides used to amplify HER2 are described in supplementary fig. 2B. The PCR reactions were as follows: 5 min at 94 °C and 35 cycles of: 30 s at 94 °C, 30 s at 56 °C, 1 min at 72 °C. Next 7 min at 72 °C. After PCR, the amplified fragments were separated on agarose gels and the bands were visualized in a Gel Doc 2000 (Bio-Rad Laboratories, Hercules, CA, USA) apparatus. Bands were excised and amplified DNAs purified using the GFXTM PCR DNA and gel Band purification KIT (GE Healthcare, Buckinghamshire, United Kingdom). DNA concentration was measured in a NanoDropTM 1000 apparatus, and 100 ng used for sequencing. The sequences obtained were compared with the canonical HER2 sequence, available in the databases (Ensembl) (transcript ID ENST00000269571.10), using the Clustal Omega web tool.
Statistics
Data distributions were checked for normality by the Shapiro-Wilk test and homogeneity of variances was checked by the Levene test. The Mann–Whitney test was used to compare continuous variables between two groups and Anova for >2 groups. Statistical analyses were performed using GraphPad Prism 8 (San Diego, CA, USA).
Results
Generation of HER2+ cells resistant to neratinib
To generate cells resistant to neratinib, we followed a classical strategy based on the continuous culture of the target cells with increasing concentrations of the drug against which resistance is desired to be developed [38]. To that end, we used BT474 cells, which constitute a prototypical cell line model of human HER2 positive breast cancer. Those cells were chronically incubated with neratinib (10, 25, 50, 100, 500 or 1000 nM), until clones resistant to the drug were microscopically observed. Three months after the initiation of this treatment, twenty-three clones of BT474 cells resistant to 10 nM neratinib (named BTRN, for BT474 cells Resistant to Neratinib), and one resistant to 25 nM neratinib were isolated (Fig. 1A). Clones BTRN #4, #5, #6, #13, #17, and #24 were initially selected for further studies since they grew better than other clones and their proliferation rate was similar to that of parental BT474. Clones BTRN #4, #5, #6, #13, and #17 were isolated under 10 nM neratinib, while clone BTRN #24 corresponded to the one obtained by continuous treatment with 25 nM of the drug. The different clones were grown in culture media without neratinib, and then tested for the maintenance of resistance. Clones BTRN #5, #6 and #24 retained resistance to neratinib (Fig. 1B), while clones BTRN #4, #13 and #17 showed similar sensitivity to neratinib than the wild type cells (supplementary fig. 1A). Statistical analyses of proliferation values observed in cells treated with 5 nM neratinib showed that clones BTRN #5, #6 and #24 were significantly more resistant than the wild type BT474 cells (Fig. 1C), with IC50 values at least one order of magnitude above that of parental BT474 cells (Fig. 1D). Because clones BTRN #5, #6 and #24 retained resistance to neratinib, grew at similar rates as BT474 (supplementary fig. 1B), and were morphologically similar to the parental BT474 cells (supplementary fig. 1C), they were selected for further analyses.

A Schematic representation of the generation of neratinib resistant cells. BT474 cells were cultured for 3 months in the presence of different concentrations of neratinib. Twenty-three clones were able to grow in the presence of 10 nM neratinib and 1 in the presence of 25 nM neratinib. B Dose-response effect of neratinib on BT474 and neratinib-resistant clones. Cells were plated and 24 h later treated with the indicated doses of neratinib. After 3 days of treatment, cells were counted, and values normalized to untreated controls. Results are shown as mean ± SD of quadruplicates of an experiment that was repeated more than three times. C Plot of the effect of 5 nM neratinib on the indicated cells. **p < 0.05. D IC50 values for the cell lines shown, calculated from data shown in A and using the GraphPad Prism 8 software. E, F Effect of other anti-HER2 therapies on the different cell models generated. Cells were plated and treated with the indicated doses. Proliferation was measured at 3 (E, F) or 5 days (G, H). Results are shown as mean ± SD of quadruplicates of an experiment that was repeated at least twice. I Comparison of the antiproliferative effect of the different anti-HER2 treatments used at 5 nM on the indicated parental or neratinib-resistant cells. **p < 0.05.
Response of the neratinib-resistant clones to other anti-HER2 drugs
With the purpose of evaluating how clones BTRN #5, #6 and #24 responded to other anti-HER2 drugs used in the clinic, dose-effect studies were performed (Fig. 1E-H). The drugs tested included two TKIs (lapatinib and tucatinib) as well as two antibody-based drugs (trastuzumab and T-DM1). In general, the dose–response curves obtained showed that the resistant clones were more resistant to the action of the tested drugs than the parental BT474 cells. In fact, statistical analyses performed on the effect obtained at a concentration of 10 nM of the different drugs, showed that the neratinib-resistant clones had partial resistance to all the tested anti-HER2 compounds (Fig. 1I). These results indicated that chronic treatment with neratinib favored the selection of cells that showed cross-resistance to other anti-HER2 drugs, with independence of whether those drugs were of the same class of neratinib, i.e. small molecule TKIs, or antibodies acting on the extracellular region of HER2.
Neratinib-resistant cells retain HER2 levels and HER2 oncogenic dependency
Loss or decreased expression of HER2 has been reported to result in resistance to anti-HER2 therapies [37]. To explore whether such mechanisms were responsible for the resistance to neratinib or the other anti-HER2 tested drugs, HER2 levels as well as its tyrosine phosphorylation status were analyzed. Western blot analyses showed that the amount of total HER2 was similar among the three resistant clones and with respect to the wild-type BT474 (Fig. 2A and B). Moreover, the tyrosine phosphorylation of the receptor was also similar among the different clones and the parental BT474 cells (Fig. 2A and B). Additional experiments were carried out to explore whether changes in cell surface HER2 could account for the resistance to neratinib and the other anti-HER2 drugs. Cell surface immunoprecipitation followed by Western analysis (Fig. 2C), or flow cytometric studies (Fig. 2D), revealed that the clones and the parental cells expressed similar amounts of surface exposed HER2. Finally, immunofluorescence studies showed that the pattern of expression of HER2 in all the clones was similar to that observed in BT474 cells (Fig. 2E).

A HER2 levels and activation are maintained in neratinib-resistant clones. Fifteen micrograms of cell lysates were used to analyze HER2 levels by Western. In parallel, HER2 tyrosine phosphorylation was determined by immunoprecipitation of 50 μg of protein extracts with the anti-HER2 antibody, followed by Western with an antiphosphotyrosine antibody. Calnexin was used as a loading control. The position of the Mr markers is indicated. B Quantitation of HER2 (top) or pHER2 (bottom) levels shown in A. Results are shown as mean ± SD of 2 independent experiments. C Analysis of cell surface HER2 on neratinib-resistant cells. Cell monolayers were incubated with 10 nM trastuzumab for 2 h at 4 °C, and cell surface HER2 analyzed as described in materials and methods. D Cell-surface HER2 levels in parental and resistant cell lines analyzed by flow cytometry. Cells were incubated with 10 nM trastuzumab (green line) or PBS (black line) for 1 h at RT and analyzed as described in materials and methods. E Detection of HER2 by immunofluorescence. Cells were fixed and stained with trastuzumab to detect HER2 (red) or DAPI to localize DNA (blue). Scale bar: 50 μm.
Molecular alterations of HER2 have been implicated in the resistance to several anti-HER2 drugs, including neratinib [20, 39]. HER2 sequencing with primers designed to cover the entire HER2 sequence (supplementary fig. 2A and B) did not reveal any mutations in the coding sequences derived from the different neratinib-resistant clones (data not shown). In addition to mutations in the HER2 sequence, an alternative molecular alteration that has been linked to resistance to anti-HER2 drugs is a deletion in the extracellular region of HER2, which produces a truncated version of 95 kDa (p95) [40]. Western blotting with an antibody directed to the C-terminus of HER2, failed to detect truncated species of HER2 (supplementary fig. 2C). Together, these results indicate that in our BTRN models, the acquired resistance to neratinib was not caused by loss or molecular alterations of HER2.
To analyze whether neratinib affected tyrosine phosphorylation levels of the resistant clones, cells were treated with increasing concentrations of the drug and tyrosine phosphorylation of HER2 measured by Western. As shown in Fig. 3A, treatment with neratinib reduced tyrosine phosphorylation of HER2 in a dose-dependent manner. Quantitation studies showed that the parental cells and the different clones responded analogously, reaching maximum inhibition of pHER2 at concentrations of 10 nM or above (Fig. 3B). Formerly, it has been reported that cells resistant to neratinib showed activation of the mTORC1 route, and such activation, measured as pS6 levels, was insensitive to the action of the drug [20]. However, in our models, neratinib treatment similarly decreased pS6 levels in parental as well as in the resistant clones (Fig. 3A, B).

A Action of neratinib on HER2, pHER2, S6 and pS6. Cells were treated with the indicated doses of neratinib for 6 h, lysed, and the proteins either directly analyzed by Western blot (HER2, S6 and pS6), or 50 μg of cell lysates immunoprecipitated with anti-HER2 followed by Western against phosphotyrosine. Calnexin was used as a loading control. B Quantitation of pHER2 (top) and pS6 (bottom) levels shown in (A). Each bar shows the mean ± SD of two independent experiments. C=untreated controls. C Knockdown of HER2 in parental and neratinib resistant cells. Cells were infected with viruses containing control vector (pLKO) or two different shRNA sequences targeting HER2 (shRNA #1 and shRNA #2). Cells were lysed and HER2 expression was measured by Western blot (top). Calnexin was also used as a loading control. HER2 knockdown effect on the proliferation rate was analyzed by cell counting after 3 days (bottom). Graph bars represent the mean ± SD of triplicates of an experiment that was repeated twice. **p < 0.01, calculated by the Mann–Whitney U test.
The fact that the isolated clones resulted resistant to neratinib, raised the question of whether the clones had also lost HER2 oncogenic dependence. To explore this possibility, HER2 levels were knocked down by shRNA, and the proliferation of the clones was evaluated. HER2 knockdown drastically affected the proliferation of parental BT474 cells as well as the proliferation of the neratinib-resistant clones (Fig. 3C). In the case of BTRN #24 cells, the effect of HER2 knockdown was similar to that observed in the case of parental BT474 cells. In BTRN clones #5 and #6 the effect of the HER2 knockdown was less pronounced than in parental or BTRN #24 cells. Yet, the inhibition of cell proliferation caused by the knockdown of HER2 in BTRN #5 and #6 was very high. Taken together, the above data indicate that the resistant cell lines continued to be oncogenically dependent on HER2.
Transcriptomic profiling of the neratinib-resistant clones
With the purpose of investigating differences in gene expression that could help in elucidating the mechanism(s) of resistance to neratinib in the resistant clones, a transcriptomic study was carried out. Principal component analyses showed that the four cell lines analyzed (BT474 and the three resistant clones) had different expression patterns (Fig. 4A). Using a fold-change value of ±2, the analysis of the individual clones with respect to parental BT474 cells showed that the number of deregulated genes in the resistant clones with respect to the parental cell line were above 2000, with around half of the transcripts being up-regulated or down-regulated (Fig. 4B and supplementary table 1). Figure 4C illustrates a Venn diagram showing the total number of deregulated genes, 3793 when pooled together, differentially expressed in at least a clone with respect to BT474 cells. This type of plot also indicates that the three studied clones shared 1200 deregulated genes in common when compared to BT474 cells. Of them, 546 were up-regulated, while 654 were down-regulated (Fig. 4D). When compared among them, 1497 genes were differentially expressed between clones #5 and #6; 1421 genes between clones #5 and #24; and 1634 genes between clones #6 and #24 (supplementary table 2). These substantial differences in gene expression were indicative, together with the PCA analysis, that the three clones were different among them.

A Principal component analysis (PCA) of data obtained from RNA microarrays from BT474 and neratinib-resistant clones. All cell lines were grouped according to its intrinsic variability in the 3 axes of the three-dimensional graph (PCA1, 2 or 3) that explains the original variation from among the data set. Replicates for each sample are shown in the same color. B Differentially expressed genes (DEGs) for each resistant clone compared to the parental cell line. C Venn diagram showing both individual and common differentially expressed genes between all resistant clones compared to the parental cell line. D DEGs commonly deregulated among the three neratinib-resistant clones and parental BT474. Cut-off for DEGs was ≥±2-fold change with a p value of 0.01. Genes that meet both criteria are colored green, if downregulated, or red, if upregulated. The y-axis represents the significance as −10 × log of p value. The x-axis represents the base 2 log of the mean differential expression (MDE). E Genes with a higher fold change underexpressed (COLEC12, LMO3, and UPK1A) or overexpressed (MGP, PADI3, and SERPINB5) in the resistant clones compared to the parental cell line BT474. F Analysis of the expression of MGP, PADI3 and SERPINB5 genes comparing normal and tumor tissues of patients with invasive breast carcinoma, using the TNMplot web tool. The p value of the statistical analysis (Mann–Whitney U test) provided by the database is shown.
Of the 1200 genes that were commonly under- or overexpressed, we focused attention on those which showed the highest rate of change. The genes for MGP, PADI3 and SERPINB5 resulted the most overexpressed in the resistant clones, while COLEC12, LMO3 and UPK1A were the three most under expressed genes. The latter genes showed fold changes below those of the overexpressed genes (Fig. 4E). Of the up-regulated genes expressed in the neratinib-resistant clones, RNA-Seq data obtained from breast invasive carcinomas and available from the TNMPlot web tool [41], showed that only PADI3 (Peptidyl Arginine Deiminase 3) was more expressed in tumor tissue compared to healthy tissue (Fig. 4F). We therefore focused on that gene to investigate its relevance as a regulator of the response to neratinib.
Genetic manipulation of PADI3 levels
To validate the transcriptomic data at the protein level, Western blotting studies were performed. These analyses demonstrated that the three neratinib-resistant clones expressed higher amounts of PADI3 when compared to parental BT474 cells (Fig. 5A). Moreover, under these experimental conditions, BT474 cells failed to show detectable levels of the protein. We also analyzed the expression of MGP by western blotting, confirming the up-regulation of the levels of that protein in the neratinib-resistant clones (Fig. 5A).

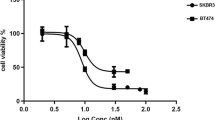
A PADI3 and MGP expression levels in BT474 and the neratinib-resistant clones. PADI3 or MGP levels were analyzed by direct Western blot (top and middle panels) of 50 μg of protein extract with the appropriate antibodies. Loading controls used were calnexin (for PADI3) or GAPDH (for MGP). Quantitative analysis of PADI3 levels (bottom) for each cell line, represented in the graph as mean ± SD of four different experiments. B PADI3 or Flag-PADI3 expression in the different cell lines tested. Protein extracts from the BTRN-resistant clones isolated after transfection of Flag-PADI3 into BT474 cells were prepared and separated by SDS-PAGE. PADI3 levels (top) were determined as in A. The membrane was then stripped and re-probed with an anti-Flag antibody (middle). Calnexin was used as a loading control (bottom). C PADI3 expression levels of Flag-PADI3 clones and BT474 cell line. Data from (B) and another three independent experiments were used for the generation of the graphic, and represent the mean ± SD of the mentioned samples. D PADI3 implication in neratinib resistance. BT474, Flag-PADI3 clones and BTRN cells were plated and treated with 5 nM neratinib. After 3 days of treatment, cells were counted, and the data represented as the mean ± SD of triplicates (normalized to untreated controls) of a representative experiment that was repeated three times. **p < 0.01, calculated by the Mann-Whitney U test.
To explore the possibility that the augmented amount of PADI3 present in the resistant clones could favor resistance to the drug, functional studies were carried out by manipulating the levels of the protein. Gain of function as well as loss of function genetic studies were performed. For the gain of function experiments, BT474 cells were transfected with a plasmid coding for Flag-tagged PADI3, and several clones were isolated. Western blotting with anti-Flag antibodies confirmed the expression of exogenous Flag-tagged PADI3 in several clones derived from naïve BT474 cells (Fig. 5B). As expected, inclusion of the Flag tag resulted in an increase in the Mr of Flag-PADI3 with respect to endogenous PADI3 (Fig. 5B, top Western). Of note, Flag-PADI3 expressing clones did not express detectable levels of endogenous PADI3, behaving in this sense like BT474 cells. Quantitative analysis of the levels of expression of Flag-PADI3 revealed that the transfected clones expressed varying levels of Flag-PADI3 (Fig. 5C). As shown in Fig. 5B, C, clones 7, 11, and 19 expressed high levels of ectopic FLAG-tagged PADI3. Comparatively, these clones had significantly higher levels of Flag-PADI3 than the endogenous PADI3 levels from the originally isolated neratinib-resistant clones (Fig. 5B, C). Of note, the levels of PADI3 in clone 1 were undetectable and in this respect this clone behaved like naïve BT474 cells. Proliferation studies indicated the BT-PADI3 #7, #11 and #19 clones resulted statistically more resistant to the action of neratinib than the parental BT474 cells (Fig. 5D). We used SKBR3 as an alternative cell line to analyze whether overexpression of PADI3 could also lead to resistance to neratinib. After transfection with the PADI3-encoding plasmid and selection, a pool of SKBR3 cells expressing ectopic Flag-tagged PADI3 was obtained (Supplementary Fig. 3A). When compared to naïve SKBR3, the PADI3-overexpressing SKBR3 cells were more resistant to the action of neratinib (Supplementary Fig. 3B).
In addition to the above-described gain of function studies, knockdown experiments were performed to decrease the levels of PADI3 in the neratinib-resistant clones and define whether that action would affect the response of those clones to the drug. Preliminary assays of knockdown efficiency using five different shRNA sequences led to the selection of two of these sequences, #36 and #37, which achieved fair PADI3 knockdown (Fig. 6A). First, the effect of knocking down PADI3 on cell proliferation was analyzed. Infection of resistant clones with lentiviral vectors coding for the two interfering sequences resulted in a decrease in the proliferation of the three neratinib-resistant clones (Fig. 6B). In contrast, infection of parental BT474 cells did not affect its proliferation. To evaluate whether knockdown of PADI3 sensitized to the action of neratinib, resistant cells infected with the PADI3 knockdown sequences or the pLKO control vector were treated with the drug and after three days live cells were counted. As shown in Fig. 6C, knockdown of PADI3 resulted in higher sensitivity to the action of 5 nM neratinib in the three neratinib-resistant clones. In contrast, the same manipulation did not affect the sensitivity of BT474 cells to neratinib.

A Knockdown of PADI3 in parental and neratinib-resistant cells. Cells were infected with lentivirus containing shRNA control (pLKO) or the indicated shRNA sequences targeting PADI3 (shRNA #36 and shRNA #37). After puromycin selection, cells were lysed and gene silencing was evaluated by Western blot by an anti-PADI3 antibody. Calnexin was used as a loading control. B Effect of PADI3 knockdown on cell proliferation rate. The proliferative capacity of transduced cells was evaluated after 3 days by cell counting. The results show the percentage of the mean ± SD of triplicates compared to the shRNA control (pLKO), from a representative experiment that was repeated twice. C Analysis of PADI3 knockdown on the response to neratinib. Transduced cells were treated with 5 nM neratinib for 3 days and counted. Results show the percentage of the mean ± SD of triplicates compared to the pLKO cells of a representative experiment that was repeated three times. **p < 0.01, calculated by the Mann–Whitney U test. D Pharmacological inhibition of PADI3 with Cl-amidine. Dose-response analysis of the effect of Cl-amidine on the proliferation of BT474 cells and its respective neratinib-resistant clones. Cells were counted after 3 days of treatment with the indicated doses and data are represented as the mean ± SD of triplicates. E Cells were plated and treated with 10 μM Cl-amidine, 5 nM neratinib or both, and the re-sensitization capacity of Cl-amidine to neratinib response was evaluated after 3 days by cell counting experiments. Graph bars represent the mean ± SD of triplicates normalized to untreated cells of an experiment that was repeated three times. **p < 0.01, calculated by the Mann–Whitney U test.
Pharmacological inhibition of PADI3 sensitizes to the action of neratinib
To further analyze the role of PADI3 function in the regulation of the antiproliferative action of neratinib, the effect of the PADI3 inhibitor CI-amidine was explored. By itself, CI-amidine did not substantially affect the proliferation of BT474 cells (Fig. 6D). The proliferation of the neratinib-resistant clones was slightly affected, reaching an inhibition of cell proliferation <20% at the highest concentration tested. When the action of CI-amidine was tested in combination with neratinib in BT474 cells, CI-amidine did not increase further the antiproliferative action of neratinib (Fig. 6E). In contrast, in the neratinib-resistant clones, the effect of the combination of CI-amidine and neratinib was higher than the effect of neratinib alone. Moreover, the degree of growth inhibition obtained with the combination of the drugs approached the inhibition caused by neratinib in wild-type BT474 cells.
Discussion
Several clinical trials have demonstrated the benefit of using neratinib for the treatment of HER2+ breast cancer [10, 11]. However, despite its antitumoral efficacy, resistance to the antitumoral action of neratinib can also arise [13]. For that reason, efforts to understand the mechanistic bases of such resistance have raised attention, with the final aim of developing strategies to bypass such resistance. Those studies led to the identification of certain cellular mechanisms used by cancer cells to escape the antitumoral action of neratinib, including adaptation of the drug metabolism machinery or compensatory signaling mechanisms that may bypass the inhibitory effects of the drug [13].
To gain further insights into the mechanisms of resistance to neratinib, we generated HER2+ breast cancer cells resistant to the drug. The strategy used, based on the continuous treatment of BT474 cells with the drug, led to the isolation of several clones significantly resistant to neratinib. When tested for their levels of HER2, the neratinib-resistant clones and naïve BT474 cells showed similar amounts of total, cell surface or tyrosine-phosphorylated HER2. These results are similar to those obtained in BT474 cells made resistant to the action of another HER2 kinase inhibitor, lapatinib, in which no major changes in the level of HER2 were observed [12, 33]. Moreover, sequencing of HER2 did not show molecular alterations in the different neratinib-resistant clones. Therefore, while in other cellular models and clinical settings resistance to anti-HER2 drugs has been reported to depend on the loss, reduction, or molecular alterations of HER2 [42], in the studies herewith reported, such mechanisms do not appear to be the cause of resistance.
The neratinib-resistant clones showed some degree of resistance to the antiproliferative action of other anti-HER2 drugs used in the clinic. That was the case for the reversible HER2 tyrosine kinase inhibitors lapatinib or tucatinib. While neratinib is an irreversible inhibitor, such cross-resistance could be reasonably expected due to similarities in the mechanisms of action of the three TKIs. The resistance also observed in the case of trastuzumab and T-DM1 may have therapeutic implications, as it suggests that neratinib-treated cells may develop generic mechanisms that make them resistant to the available anti-HER2 therapies, with independence of their mechanism of action.
Interestingly, we observed a difference between the dose-response effect of neratinib on the tyrosine phosphorylation of HER2 and its effect on cell proliferation. While the proliferation of neratinib-resistant clones showed significantly decreased sensitivity to neratinib with respect to parental BT474 cells, HER2 tyrosine phosphorylation was equally sensitive to the action of neratinib in the clones resistant to the drug and the parental BT474 cells. In fact, the biochemical analyses of tyrosine phosphorylation of HER2 in the clones resistant to neratinib indicated that at concentrations of 10 nM and above of the drug, tyrosine phosphorylation of HER2 was profoundly compromised, similarly to BT474 cells. Yet, the proliferation of the resistant clones was not inhibited more than 50% at that drug concentration, in contrast to what happens in the parental cells whose proliferation was inhibited >80% by 10 nM neratinib. These results may be interpreted as if resistant cells would have lost HER2 oncogenic dependence. However, the resistant cells are still dependent on the presence of HER2, as indicated by the strong antiproliferative effect obtained in the HER2 knockdown studies. Therefore, it appears that the tyrosine phosphorylation of HER2, which reflects its kinase activity may be partially dispensable for the proliferative signaling allowing neratinib-resistant cells to adapt to such circumstance. However, they are still dependent on HER2 for proliferation. These data give rise to the idea of using drugs such as PROTACS targeting HER2, which can combine kinase inhibitory activity with protein degrading properties, to fight resistance to neratinib.
Transcriptomic profiles of the three neratinib-resistant clones showed a total of 1200 commonly downregulated genes in clones BTRN #5, #6 and #24, compared to BT474 cells. From them, we focused on those genes that were more deregulated by fold-change in the neratinib-resistant clones, and which were differentially expressed in tumor tissue from patients with invasive breast carcinoma, compared to healthy tissue. Among the genes identified, PADI3 led the list of candidates, and for this reason we performed functional analyses to investigate its potential role in neratinib resistance. MGP also resulted up-regulated in the neratinib-resistant clones. However, since the in silico studies indicated that MGP is expressed at higher levels in normal breast tissue when compared to tumoral tissue, we focused our attention on PADI3.
The genetic gain and loss of function experiments performed demonstrated that PADI3 plays an important role in the proliferation of neratinib-resistant BT474 cells and in their response to neratinib. Thus, PADI3 gene silencing in the three neratinib-resistant clones provoked a decrease in their proliferation. In addition, the knockdown of PADI3 resulted in restoration of sensitivity to the antiproliferative action of neratinib. Furthermore, combined treatment of neratinib with Cl-amidine, a PADI enzyme inhibitor [43], resensitized the three resistant clones. Together, these data suggest that PADI3 could be a potential therapeutic target in HER2+ breast cancer with neratinib resistance. On the other hand, gain of function experiments carried out by overexpressing PADI3 in BT474 cells resulted in resistance to the antitumoral action of neratinib, supporting the concept that increased levels of PADI3 may provoke resistance to the drug. Other authors have demonstrated resensitization of NSCLC cells to osimertinib, a TKI against EGFR, after inhibiting PADI1, 2 and 3 with Cl-amidine [27]. However, we are not aware of any studies that have demonstrated the involvement of these enzymes in the acquisition of resistance to any drug used in the therapy of HER2+ breast cancer.
From a translational point of view, the results herewith reported open the possibility of using PADI3 inhibitors to fight neratinib resistance. It will be interesting to analyze in patient samples, whether expression of PADI3 may be used as a biomarker to predict response to neratinib or the development of resistance to the drug. Finally, the finding that neratinib-resistant cells are still oncogenically dependent on HER2 opens the possibility of developing therapeutic strategies aimed at decreasing the levels of HER2 to fight resistance to neratinib.
Data availability
Original data and materials are available from the authors upon reasonable request.
References
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82.
Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303.
Esparis-Ogando A, Montero JC, Arribas J, Ocana A, Pandiella A. Targeting the EGF/HER ligand-receptor system in cancer. Curr Pharm Des. 2016;22:5887–98.
Yang C, Khwaja R, Tang P, Nixon N, King K, Lupichuk S. A review of trastuzumab biosimilars in early breast cancer and real world outcomes of neoadjuvant MYL-1401O versus reference trastuzumab. Curr Oncol. 2022;29:4224–34.
Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, et al. Antibody-drug conjugates: Smart chemotherapy delivery across tumor histologies. CA Cancer J Clin. 2022;72:165–82.
Garcia-Alonso S, Ocana A, Pandiella A. Trastuzumab emtansine: mechanisms of action and resistance, clinical progress, and beyond. Trends Cancer. 2020;6:130–46.
Diaz-Rodriguez E, Gandullo-Sanchez L, Ocana A, Pandiella A. Novel ADCs and strategies to overcome resistance to Anti-HER2 ADCs. Cancers (Basel). 2021;14:154.
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39.
Xia X, Gong C, Zhang Y, Xiong H. The history and development of HER2 inhibitors. Pharmaceuticals (Basel). 2023;16:1450.
Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:367–77.
Saura C, Oliveira M, Feng YH, Dai MS, Chen SW, Hurvitz SA, et al. Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2-positive metastatic breast cancer previously treated with >/= 2 HER2-directed regimens: phase III NALA Trial. J Clin Oncol. 2020;38:3138–49.
Rios-Luci C, Diaz-Rodriguez E, Gandullo-Sanchez L, Diaz-Gil L, Ocana A, Pandiella A. Adaptive resistance to trastuzumab impairs response to neratinib and lapatinib through deregulation of cell death mechanisms. Cancer Lett. 2020;470:161–9.
Guo L, Shao W, Zhou C, Yang H, Yang L, Cai Q, et al. Neratinib for HER2-positive breast cancer with an overlooked option. Mol Med. 2023;29:134.
Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189–94.
Ma CX, Luo J, Freedman RA, Pluard TJ, Nangia JR, Lu J, et al. The phase II MutHER study of neratinib alone and in combination with fulvestrant in HER2-mutated, non-amplified metastatic breast cancer. Clin Cancer Res. 2022;28:1258–67.
Rani S, Corcoran C, Shiels L, Germano S, Breslin S, Madden S, et al. Neuromedin U: a candidate biomarker and therapeutic target to predict and overcome resistance to HER-tyrosine kinase inhibitors. Cancer Res. 2014;74:3821–33.
Zhao XQ, Xie JD, Chen XG, Sim HM, Zhang X, Liang YJ, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharm. 2012;82:47–58.
Collins DM, Conlon NT, Kannan S, Verma CS, Eli LD, Lalani AS, et al. Preclinical characteristics of the irreversible pan-HER kinase inhibitor neratinib compared with lapatinib: implications for the treatment of HER2-positive and HER2-mutated breast cancer. Cancers (Basel). 2019;11:737.
Breslin S, Lowry MC, O’Driscoll L. Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br J Cancer. 2017;116:620–5.
Sudhan DR, Guerrero-Zotano A, Won H, Gonzalez Ericsson P, Servetto A, Huerta-Rosario M, et al. Hyperactivation of TORC1 drives resistance to the pan-HER tyrosine kinase inhibitor neratinib in HER2-mutant cancers. Cancer Cell. 2020;37:183–99.e5.
Takeda T, Yamamoto H, Suzawa K, Tomida S, Miyauchi S, Araki K, et al. YES1 activation induces acquired resistance to neratinib in HER2-amplified breast and lung cancers. Cancer Sci. 2020;111:849–56.
Uysal-Onganer P, D’Alessio S, Mortoglou M, Kraev I, Lange S. Peptidylarginine deiminase inhibitor application, using Cl-amidine, PAD2, PAD3 and PAD4 isozyme-specific inhibitors in pancreatic cancer cells, reveals roles for PAD2 and PAD3 in cancer invasion and modulation of extracellular vesicle signatures. Int J Mol Sci. 2021;22:1396.
Wang Y, Chen R, Gan Y, Ying S. The roles of PAD2- and PAD4-mediated protein citrullination catalysis in cancers. Int J Cancer. 2021;148:267–76.
Zhu C, Liu C, Chai Z. Role of the PADI family in inflammatory autoimmune diseases and cancers: a systematic review. Front Immunol. 2023;14:1115794.
Chai Z, Wang L, Zheng Y, Liang N, Wang X, Zhang Z, et al. PADI3 plays an antitumor role via the Hsp90/CKS1 pathway in colon cancer. Cancer Cell Int. 2019;19:277.
Chang X, Chai Z, Zou J, Wang H, Wang Y, Zheng Y, et al. PADI3 induces cell cycle arrest via the Sirt2/AKT/p21 pathway and acts as a tumor suppressor gene in colon cancer. Cancer Biol Med. 2019;16:729–42.
Li H, Muhetaer G, Xie Y, Yao K, Ma Q, Guan H, et al. Identification of super-enhancer-driven peptidyl arginine deiminases as potential biomarkers and therapeutic targets for osimertinib-resistant non-small cell lung cancer. Front Pharm. 2022;13:1071365.
Malki L, Sarig O, Romano MT, Mechin MC, Peled A, Pavlovsky M, et al. Variant PADI3 in central centrifugal cicatricial alopecia. N. Engl J Med. 2019;380:833–41.
McElwee JL, Mohanan S, Griffith OL, Breuer HC, Anguish LJ, Cherrington BD, et al. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer. 2012;12:500.
Wang Y, Liu C, Zhang N, Song X, Song Y, Cai D, et al. Anti-PADI4 antibody suppresses breast cancer by repressing the citrullinated fibronectin in the tumor microenvironment. Biomed Pharmacother. 2022;153:113289.
Horibata S, Rogers KE, Sadegh D, Anguish LJ, McElwee JL, Shah P, et al. Role of peptidylarginine deiminase 2 (PAD2) in mammary carcinoma cell migration. BMC Cancer. 2017;17:378.
Mohanan S, Cherrington BD, Horibata S, McElwee JL, Thompson PR, Coonrod SA. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem Res Int. 2012;2012:895343.
Diaz-Rodriguez E, Perez-Pena J, Rios-Luci C, Arribas J, Ocana A, Pandiella A. TRAIL receptor activation overcomes resistance to trastuzumab in HER2 positive breast cancer cells. Cancer Lett. 2019;453:34–44.
Almaraz Postigo S, Montero JC. Neuregulin modulates hormone receptor levels in breast cancer through concerted action on multiple signaling pathways. Clin Sci (Lond). 2023;137:1–15.
Esparis-Ogando A, Ocana A, Rodriguez-Barrueco R, Ferreira L, Borges J, Pandiella A. Synergic antitumoral effect of an IGF-IR inhibitor and trastuzumab on HER2-overexpressing breast cancer cells. Ann Oncol. 2008;19:1860–9.
Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002;22:270–85.
Gandullo-Sanchez L, Pandiella A. An anti-EGFR antibody-drug conjugate overcomes resistance to HER2-targeted drugs. Cancer Lett. 2023;554:216024.
Ríos-Luci C, García-Alonso S, Díaz-Rodríguez E, Nadal-Serrano M, Arribas J, Ocaña A, et al. Resistance to the antibody-drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 2017;77:4639–51.
Parra-Palau JL, Morancho B, Peg V, Escorihuela M, Scaltriti M, Vicario R, et al. Effect of p95HER2/611CTF on the response to trastuzumab and chemotherapy. J Natl Cancer Inst. 2014;106:dju291.
Arribas J, Baselga J, Pedersen K, Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011;71:1515–9.
Bartha Á, Győrffy B. TNMplot.com: a web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int J Mol Sci. 2021;22:2622.
Bose R, Ma CX. Breast cancer, HER2 mutations, and overcoming drug resistance. N. Engl J Med. 2021;385:1241–3.
Ahmed D, Puthussery H, Basnett P, Knowles JC, Lange S, Roy I. Controlled delivery of pan-PAD-inhibitor Cl-amidine using poly(3-hydroxybutyrate) microspheres. Int J Mol Sci. 2021;22:12852.
Acknowledgements
The authors thank Ana López for help with some Western blots of Fig. 3.
Funding
AP: Ministry of Economy and Competitiveness of Spain (BFU2015-71371-R and PID2020-115605RB-I00), the Instituto de Salud Carlos III through CIBERONC (groups CB16/12/00275 and CB16/12/00317), Junta de Castilla y León (CSI146P20), ALMOM, ACMUMA, UCCTA, the CRIS Cancer Foundation and the Regional Development Funding Program (FEDER) “A way to make Europe”. IRP was recipient of a predoctoral contract (EDU/556/2019); and later was contracted by the Cancer Research Foundation of the Salamanca University (FICUS). JCM: Instituto de Salud Carlos III through a Miguel Servet program (CPII17/00015) and (PI18/00796).
Author information
Authors and Affiliations
Contributions
IRP, JCM, EDR, LSD: performed the experiments, interpreted the data, prepared figures. AP: directed the project, designed the study, wrote, and edited the paper. The author(s) approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
AP received personal fees from Daiichi-Sankyo and CancerAppy S.L. The rest of the authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Romero-Pérez, I., Díaz-Rodríguez, E., Sánchez-Díaz, L. et al. Peptidylarginine deiminase 3 modulates response to neratinib in HER2 positive breast cancer. Oncogenesis 13, 30 (2024). https://doi.org/10.1038/s41389-024-00531-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-024-00531-4
- Springer Nature Limited