
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a well-known lethal and heterogeneous disease. Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like (APOBEC) is an important mutagenic driver that has seldom been investigated in PDAC. Therefore, this study investigated the significance of APOBEC3C in PDAC. First, cytosine deamination-associated mutation signatures were identified in the PDAC cohorts from TCGA and Fudan University Shanghai Cancer Center (FUSCC) datasets, and C > X-enriched kataegis regions were identified in the FUSCC cohort (12 to 27 counts per sample). Patients were stratified according to APOBEC3C expression, and high APOBEC3C expression was found to correlate with a higher motif enrichment score of 5’-CC-3’ and an elevated kataegis count within PCSK5 and NES genes. Second, we compared APOBEC expression in PDAC and normal pancreatic tissues and found that APOBEC3C was substantially upregulated in PDAC. APOBEC3C-overexpressing cell lines were generated to substantiate the effects of APOBEC3C on PDAC genome, including alterations in single-nucleotide variant (SNV) classes (higher proportion of C > T conversions) and the formation of kataegis regions (newly occurring kataegis regions detected in ACHE and MUC6 genes). Three different PDAC cohorts (FUSCC, TCGA, and QCMG) were analysed to evaluate the prognostic value of APOBEC3C, and APOBEC3C overexpression predicted shorter survival. Finally, the APOBEC3C overexpression correalted with the PDAC tumour microenvironment (TME) remodelling, APOBEC3C expression was associated with the invasion of CD4 + T lymphocytes and CD8 + T lymphocytes (cytotoxic T lymphocytes, CTLs), indicating enhanced immune activity and validating the practicality of APOBEC3C for guiding immunotherapy.
Similar content being viewed by others
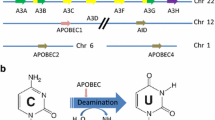


Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a notorious malignancy with a poor prognosis and increasing incidence. PDAC has the lowest 5-year survival rate among all types of cancers [1] and is estimated to become the second leading cause of cancer-related death by 2030. The short survival of patients with PDAC might be largely attributed to systemic therapy resistance [2] and a high recurrence rate; accordingly, studies aiming to determine the mechanisms through which new phenotypes of PDAC develop are urgently needed.
Notably, genomic instability is one of the hallmarks of PDAC [3, 4]. PDAC is characterised by high intratumour heterogeneity and is composed of various subpopulations with diverse somatic mutations [5]. Genomic mutations (such as KRAS and TP53, Fig. S1A) drive the process of tumorigenesis [6] and enhance cancer cell plasticity in response to evolutionary pressures such as treatment [7]. Therefore, elucidating the biological processes that generate mutations has great importance. The underlying mechanisms for generating mutations [8] include exogenous mutagens, DNA replicative infidelity, DNA repair deficiency and enzyme-induced DNA alterations. With the advancement of next-generation sequencing, the genomic landscape of PDAC and the characteristic mutational signatures created by particular mutagenic components [9, 10] have been unveiled. The predominant mutational signatures present in PDAC are associated with age, BRCA mutations, DNA mismatch repair deficiency (dMMR) and apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like (APOBEC) proteins [11].
The APOBEC family comprises activation-induced cytidine deaminase (AICDA), APOBEC1, APOBEC2, APOBEC4 and seven APOBEC3 subfamily homologues [12]. APOBEC deaminates cytosine in single-stranded DNA (ssDNA) or mRNA, resulting in cytosine (C) to uracil (U) conversions [13], and U·G mispairs are generally transformed to C > T single point mutations at replication forks. Moreover, along with the participation of uracyl deglycosylases in base excision repair and mismatch repair (MMR), high levels of C > X mutations and strand breaks lead to chromosome translocation. Thus, APOBEC is capable of causing genomic instability. Physically, APOBEC is involved in biological processes of somatic hypermutation and class switch recombination for B-cell maturation [14], innate immunity to remove foreign retroviruses [15], and epigenetic reprogramming of DNA demethylation [16, 17].
Accumulating evidences suggested APOBEC also promotes tumorigenesis in diverse organs [18,19,20]. APOBEC is implicated not only in haematological malignancies [21,22,23] but also in various solid tumours [24,25,26,27]. APOBEC induces a high tumour mutational burden (TMB), nourishing neoantigens that supplement tumour evolution and adaptation to therapy [28,29,30]. However, research on the role of APOBEC in PDAC is scarce. Here, we performed deep whole-exon sequencing (WES) in 124 PDAC tissues to identify mutational signatures and characteristic kataegis regions and substantiated the effect of APOBEC3C on the PDAC genome by generating APOBEC3C-overexpressing cell lines. Publicly available datasets were used to confirm our conclusions. In addition, the overall survival (OS) of patients with PDAC differed between the subgroups stratified by APOBEC3C expression. Finally, we showed that APOBEC3C expression correlated with PDAC tumour microenvironment (TME) remodelling. This is the first investigation reporting prognostic and therapeutic values of APOBEC3C in PDAC, and our study also contributes to deciphering PDAC genomic features [31].
Methods
Online databases
Open access data (RNA expression and corresponding patient survival times) were sourced from The Cancer Genome Atlas (TCGA) and Queensland Centre for Medical Genomics (QCMG) [32] using Xena browser (https://xenabrowser.net) and cBioPortal (http://www.cbioportal.org/). Comparison of APOBEC expression in TCGA and Genotype-Tissue Expression project (GTEx) databases was performed using GEPIA2 (http://gepia2.cancer-pku.cn) [33], which normalised data by the maximum median expression value across all blocks and then made comparison between PDAC and normal pancreatic tissues. The maf file bcgsc.ca_PAAD.IlluminaHiSeq_DNASeq.1.somatic.maf (n = 147) was downloaded from GDC (https://portal.gdc.cancer.gov).
Cell culture
The human pancreatic cancer cell lines BxPC-3, CFPAC-1, MIA PaCa-2 and SW 1990 were obtained from the National Collection of Authenticated Cell Cultures, Capan-1, Capan-2, and SU.86.86 were obtained from the American Type Culture Collection. The human pancreatic ductal epithelial cell line HPDE6c7 (H6c7) was obtained from Kyushu University. Cell lines were authenticated by STR and tested for mycoplasma contamination.
BxPC-3 and SU.86.86 were cultured in RPMI 1640, Capan-1 and CFPAC-1 were cultured in IMDM, Capan-2 was cultured in McCoy’s 5A modified medium, H6c7 and MIA PaCa-2 were cultured in Dulbecco’s modified Eagle medium (DMEM), and SW1990 was cultured in L15 medium. All cell culture media were supplemented with 10% heat-inactivated foetal bovine serum and 100 U/mL penicillin–streptomycin, and DMEM used for culturing MIA PaCa-2 was additionally supplemented with 2.5% horse serum. All cells were cultured at 37°C with 5% CO2.
Overexpression vector construction and transfection
Wild-type APOBEC3C consensus coding sequence (CCDS) (APOBEC3C-pENTER vector, WZ Biosciences) was amplified by PCR (forward primer: 5’-CCGTCAGATCCGCTAGTAATAC-3’, reverse primer: 5’-CCGGAATTCTGTGGTATGGCTGATTATGATC-3’). Then, BamHI and EcoRI were employed to generate sticky ends. The pCDH-CMV-puro plasmid was digested with the same enzymes, and restriction enzyme-digested products were purified by electrophoresis and Universal DNA Purification Kits (Cat#DP214-03, Tiangen, China). The CCDS fragment and pCDH plasmid were ligated using DNA Ligation Kits (Cat# 6021, Takara, Japan) and then transformed into DH5α cells for further selection and amplification.
EndoFree Mini Plasmid Kit II (Cat# DP118-02, Tiangen) was used to extract pCDH-APOBEC3C (pCDH-A3C). Then, 7.5 µg pCDH-A3C or pCDH backbone plus 5.625 µg psPAX2 and 1.875 µg pMD2.G were incubated with 30 µL Hieff Trans Liposomal Transfection Reagent (Cat# 40802ES03, Yeason, China) in 1 mL DMEM. The mixture was added to 293T cells to generate lentiviruses, which were used to transfect pancreatic cell lines. Puromycin was added to culture mediums (4 µg/mL) for selection. Four months after transfection, total DNA was extracted from A3C-overexpressing cell lines and subjected to Sanger sequencing to verify that A3C sequence in the plasmid did not undergo genetic modifications.
Clonogenic assay
Cells were suspended in PBS, and its density was counted by BECKMAN COULTER (Cat# AW29395, System 7221901). Then, PBS containing 1500 cells was added to 6 mL medium. Cells were resuspended before being transferred into three wells of a 6-well plate and cultured for two weeks. Clones were fixed with 4% paraformaldehyde and stained with crystal violet. Clones were counted, and Welch’s t-test was used to compare the differences.
Patients and specimens
Pancreatic tumour specimens for WES and transcriptomics were obtained from 124 other patients, defined as FUSCC cohort A. Pancreatic tumour specimens for quantitative real-time polymerase chain reaction (qRT–PCR) and survival analyses were obtained from 124 other patients, defined as FUSCC cohort B. Sixty tumour centre tissues and six normal tissues remote from tumour sites for immunohistochemical (IHC) staining were also obtained from FUSCC cohort B, defined as FUSCC cohort B1. All patients were pathologically diagnosed with PDAC after radical surgical resection in the Department of Pancreatic Surgery Shanghai Cancer Center, Fudan University, China.
OS was defined as the length of time from the beginning of diagnosis to death from any cause or the last follow-up. The follow-up period of cohort B ended in November 2019. This study was approved by the Ethics Board of Shanghai Cancer Center, Fudan University, and all patients involved in this study provided written informed consent for use of their specimens and personal data for research purposes.
Mutational signature and SNV distribution analysis
DeconstructSigs R package (version 1.8.0) was used to analyse mutational signatures [34] and compare the results to COSMIC reference signatures (SBS v3.2, March 2021). Ramtools R package (version 2.4.0) was used to obtain flanking sequences centred on each mutation, and APOBEC motif enrichment scores were calculated using the formulas [35]. MeichunCai/KataegisPortal R package (version 1.0.3) [36] was applied to detect and locate kataegis regions and calculate the proportion of C > X mutations in kataegis. Maftools R package (version 2.4.12) [37] was used to detect the differentially altered genes between non-APOBEC-enriched samples and APOBEC-enriched samples in TCGA PDAC cohort.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from cell lines and PDAC tissues using EZ-press RNA Purification Kits (Cat# B0004D-100, EZBioscience, USA) and Tissue RNA Purification Kit PLUS (Cat# EZB-RN001-plus, EZBioscience), respectively. RNA was excluded from further experiments if its 260/280 nm optical density ratio was less than 1.85 or greater than 2.15 or its concentration was less than 100 ng/µL. Reverse transcription was conducted using 4× EZscript Reverse Transcription Mix II (Cat#EZB-RT2GQ, EZBioscience).
qRT–PCR was performed using a QuantStudio™ 7 Flex Real-Time PCR system (Cat#4485701, Applied Biosystems) and 2×Colour SYBR Green qPCR Master Mix (Cat#A0012-R2, EZBioscience). ACTB was used as an internal control, and one PDAC sample was used as an interplate control. All reactions were run in triplicate. All primers for qRT–PCR were designed by Primer Premier. Primer specificity was checked using Basic Local Alignment Search Tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi). All primers produced single-peak melting curves. The primer sequences are listed in Table S1.
Immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) pancreatic specimens were sectioned into 4-micron-thick slices, including 60 tumour tissues and 6 normal tissues. After deparaffinization and rehydration, 3% H2O2 was used to block endogenous peroxidases for 15 min. Then, slices were heated in Tris-EDTA buffer (pH = 9.0) for 10 min to retrieve antigens and blocked with 2.5% goat serum for 1 h. The following primary antibodies were used: polyclonal antibody against APOBEC3C (10591-1-AP, Proteintech, USA), recombinant antibody against CD4 (ab133616, Abcam, England), and polyclonal antibody against CD8 (ab4055, Abcam). The isotype control was ab37415 (polyclonal rabbit IgG, Abcam). Colouration was performed using a GTVisionTM III Detection System/Mo&Rb (Cat#GK500710, Gene Tech, China), and haematoxylin was used for counterstaining. Finally, sections were dehydrated and re-embedded for observation. Expression was considered positive only when positive reaction products were localised in the expected cellular compartment. The counts of immune cells were defined as the mean number of immune cells in three representative high-power fields (20×). Immune cells were counted manually.
Survival analysis
Continuous variables (APOBEC3C mRNA expression level) were dichotomised by optimal cutoff values ascertained by survminer R package (version 0.4.9). Kaplan–Meier method was employed to plot the survival curves. Survival curves of different groups were compared using log-rank (Mantel–Cox) tests, and hazard ratios (HRs) were calculated using Mantel–Haenszel method.
ESTIMATE and CIBERSORT analyses
ESTIMATE method [38] was applied to infer the proportions of immune and stromal components in the TME of TCGA PDAC samples. Single-sample gene set enrichment analysis (ssGSEA) output the immune score, which represents the level of infiltrating immune cells, and the stromal score, which represents the level of stromal cells in tumour tissues; then, the immune score and the stromal score were integrated into the ESTIMATE score, which represents the purity of tumour samples.
CIBERSORT (https://cibersort.stanford.edu/) was used to estimate the fractions of 22 types of immune cells by deconvoluting bulk tumour gene expression profiles based on the characteristic gene signature file “LM22”. The normalised gene expression matrix of TCGA PDAC dataset was uploaded to generate corresponding leucocyte proportion matrix. Spearman’s rank correlation coefficient was calculated to measure the correlation [39].
Results
APOBEC contributes to shaping mutational signatures in PDAC
Mutational signatures in the tumour genome indicate corresponding aetiology. Therefore, we extracted mutational signatures from TCGA PDAC cohort and FUSCC cohort A by referring to the latest COSMIC single-base-substitution signatures (SBS, v3.2, Fig. 1A–D). The two cohorts concurrently reflected that SNVs in the PDAC genome were primarily composed of C > T conversions, consistent with the biological function of APOBEC.

A, B Mutational signatures deconstructed from TCGA PDAC cohort (A) and FUSCC PDAC cohort A (B). The upper x-axis represents the nucleotide in the 5’ end flanking the SNVs, and the lower x-axis represents the nucleotide in the 3’ end flanking the SNVs. C, D Comparison between the deconstructed mutational signatures and validated SBS signatures (v3.2, March 2021). In TCGA cohort (C), the SBS1 weight was 0.115, SBS6 and SBS15 weights were 0.0955 and 0.193, respectively, SBS10b weight was 0.0649, and SBS14 weight was 0.0935. SBS48 correlated with potential artefacts and its weight was 0.0618. In FUSCC cohort A (D), the SBS1 weight was 0.0975, SBS5 and SBS39 weights were 0.142 and 0.068, respectively, SBS6 weight was 0.0685, and SBS87 weight was 0.133. SBS46 and SBS54 correlated with potential artefacts and their weights were 0.111 and 0.234, respectively. E Summary of trinucleotide motifs of all C > X mutations in TCGA cohort and FUSCC cohort A. F, G, H APOBEC-enriched samples referred to those samples with an enrichment score E-TCA > 2. The comparison between non-APOBEC-enriched samples (TCGA PDAC cohort, n = 141) and APOBEC-enriched samples (n = 6) was performed using the “plotApobecDiff” function of the maftools R package, including comparisons of the mutation burden (G), tCw burden (H) and differentially altered genes (I). Detailed information was shown in Table S2. SNV, single-nucleotide variant; PDAC, pancreatic ductal adenocarcinoma; SBS, single-base substitution; tCw, w = A or T.
In TCGA PDAC cohort, SBS1 (associated with spontaneous or enzymatic deamination of 5-methylcytosine) had a weight of 0.115. SBS6 and SBS15 (associated with dMMR) had weights of 0.0955 and 0.193, respectively, SBS10b (associated with polymerase epsilon (POLE) exonuclease domain mutations) had a weight of 0.0649, and SBS14 (associated with concurrent dMMR and POLE mutation) had a weight of 0.0935.
In FUSCC cohort A, SBS1 had a weight of 0.0975, SBS6 had a weight of 0.0685 and SBS87 (associated with thiopurine chemotherapy treatment) had a weight of 0.133. The remaining deconstructed signatures have not yet been correlated with a definite mutagen.
We additionally compared deconstructed mutational signatures of TCGA PDAC genome with classical reference mutational signatures (COSMIC v1.0, August 2013) and matched reference signatures 1, 2, 5, 6, 14 and 18. Note that signature 1 is associated with age, signature 2 is associated with APOBEC and signature 6 is associated with dMMR (Fig. S2E).
Briefly, age, APOBEC, dMMR and POLE mutations established the mutational signature of the PDAC genome.
Single-nucleotide polymorphisms in PDAC match preferred targets of APOBEC3 subfamily
Members of the APOBEC family have a lax sequence preference; thus, we analysed flanking bases of mutated sites to identify the trinucleotide motif of the C > X mutation. The APOBEC3 subfamily has been reported to prefer the 5’-TC-3’ and 5’-CC-3’ sequences [13]. Coincidently, we found that C > X mutations mostly occurred in 5’-TCG-3’ motif in TCGA PDAC cohort. In FUSCC cohort A, however, C > X mutations mainly occurred in 5’-CCG-3’ motif (Fig. 1E).
We selected 25 samples with the highest APOBEC3C expression and 25 samples with the lowest APOBEC3C expression from FUSCC cohort A, calculated motif enrichment scores and made comparisons. The high APOBEC3C expression subgroup had a higher 5’-CC-3’ enrichment score (Table S3, 1.319 versus 1.310, p = 0.0093). However, significant differences in 5’-TC-3’-related motif enrichment scores were not observed.
APOBEC3C is the most abundant member of the APOBEC family in PDAC
We compared the mRNA expression levels of the APOBEC family in PDAC tissues to determine the predominant APOBEC in PDAC and found that APOBEC3C had the highest average mRNA expression level (Fig. 2A, B and Fig. S3A). We were concerned that APOBEC isoforms share similarity in their sequences, thus confounding their transcriptomics quantification results (Table S4). Therefore, we further tested the correlation between the transcripts per million mapped reads (TPM) of APOBEC family members. We found that the APOBEC3C TPM was correlated with the TPM values of AICDA, A1 and other APOBEC3 family members (Fig. 2C, D and Table S5). Whereas, multiple linear regression models showed that only APOBEC3D TPM correlated with APOBEC3C TPM (Table S6, β > 0.8, p < 0.001).

A, B The mRNA expression levels of APOBECs in TCGA (n = 163) and FUSCC cohort A (n = 124). C, D The Pearson correlation coefficients were calculated to examine the correlations between APOBEC expression in TCGA cohort (C) and FUSCC cohort A (D). *p < 0.05, **p < 0.01. Detailed information was shown in Table S5. E The height of the bar represents the median mRNA expression level of a specific APOBEC. GEPIA2 normalised data by the maximum median expression value across all blocks and then compared data between pancreatic tumour tissues (TCGA database, n = 163) and normal tissues (TCGA and GTEx databases, n = 171) using one-way ANOVA, *p-value < 0.01. Corresponding box plots are displayed in Fig. S3. F Expression of APOBEC enzymes in PDAC cell lines. The mRNA expression level was determined by performing qRT–PCR on validated cDNAs in triplicate. G, H Representative images (patient ID.408371, FUSCC cohort B1) of IHC staining for APOBEC3 in PDAC (G) and normal pancreatic tissues (H). Normal pancreatic tissues from six patients with PDAC were stained, and the results are presented in Fig. S4. PDAC, pancreatic ductal adenocarcinoma; TPM: transcripts per million mapped reads.
Afterwards, we compared the median TPM of each APOBEC enzyme in PDAC and normal pancreatic tissues from TCGA and GTEx datasets and obtained consistent results (Fig. 2E and Fig. S3B). Compared to normal pancreatic tissue, almost all APOBEC enzymes were upregulated in PDAC, among which APOBEC3C displayed the highest expression level. APOBEC3C expression in PDAC and normal pancreatic tissues was 20.12 and 1.80 TPM, respectively (p < 0.05).
We then investigated the mRNA expression levels of the APOBEC family in seven PDAC cell lines. APOBEC3C tended to be the main APOBEC enzyme expressed in CFPAC-1, Bxpc-3, and Capan-2 (Fig. 2F and Fig. S3C). Interestingly, the comparatively low expression of APOBEC3C was accompanied by relatively high expression of APOBEC3B and APOBEC3A in Capan-1.
We performed IHC to corroborate the in silico findings by staining for APOBEC3C in PDAC and normal pancreatic tissues and found apparent upregulated expression of APOBEC3C in PDAC (Fig. 2G, H and Fig. S4A).
APOBEC3C is associated with the kataegis distribution pattern in the PDAC genome
Remarkably, APOBECs provides impetus to the formation of kataegis events (Greek for thunderstorms) [40]. Kataegis refers to mutation showers with clustered mutations in small localised genomic regions [41]. Putative kataegis is defined as genomic segments containing six or more consecutive mutations with an average intermutational distance of less than or equal to 1000 bp [11]. Considering the biological function of APOBEC, kataegis with enriched C > X substitutions is regarded as a consequence of APOBEC attack on the genome. Kataegis was observed in normal B lymphocytes [42], haematological malignancies such as B-cell lymphomas [42] and multiple myeloma [43], as well as solid tumours such as breast cancer [40] and osteosarcomas [25]. A recent pancancer analysis also reported the presence of kataegis in PDAC [31].
We performed WES (>200×) in FUSCC cohort A to sensitively and accurately detect SNVs and analyse the presence of kataegis. A deep sequencing depth was essential because tumour-derived DNA was mixed with normal DNA from tumour adjacent tissues, and mutations with a low variant allele frequency (VAF) are only detectable using deep sequencing [44, 45]. The total kataegis count was 125 to 173 per FUSCC cohort A sample (Fig. 3A and Table S7) and did not show an association with the APOBEC3C TPM (Fig. 3B, r = 0.0996, p = 0.2709). We further discerned the kataegis with more than five C > X conversions per 1000 bp to more clearly reflect the possible effect of APOBEC. The count of kataegis with enriched C > X conversions ranged from 12 to 27 per sample and was marginally correlated with the APOBEC3C TPM (r = 0.1803, p = 0.0451). Kataegis with enriched C > X conversions were primarily located on chromosomes 1, 7, 9, 11, and 17 within MAP2K3, GPRIN2, MUC3A, and BMS1P20 genes (Fig. 3C, D; Tables S8 and S9). We then used a stricter filtering condition to examine whether TpC to TpX mutations formed kataegis in the PDAC genome. Kataegis completely consisting of TpC to TpX mutations were observed in MIR4273 gene and etc., accounting for 0 to 2 counts per sample (Fig. S2F and Table S10).

The MeiChuncai/KataegisPortal R package was used to detect kataegis in FUSCC cohort A (n = 124) and calculate the C > X mutation proportion in each kataegis region. The Ramtools R package was used to obtain trinucleotide motifs of mutations. Each PDAC sample harboured 125 to 173 kataegis regions. Detailed information about kataegis in FUSCC cohort A is recorded in Tables S7-10. A Representative rainfall plot showing a PDAC sample (Patient ID: 442043) harbouring 159 kataegis regions. The x-axis represents the SNV position, and the y-axis represents the intermutational distance. As the density of the mutation distribution may influence the observation of kataegis, we separately display kataegis regions on chromosomes 1, 7, 9, 11, and 17 to show the kataegis regions with enriched C > X conversions. Kataegis were pointed out by arrows, and kataegis with enriched C > X mutations were labelled by solid arrows. B Nonparametric Spearman correlation test to determine whether the count of kataegis regions with enriched C > X substitutions or the count of total kataegis regions correlated with APOBEC3C expression. C Heatmap showing the chromosomal distribution of kataegis regions with enriched C > X substitutions (more than 5 C > X substitutions per 1000 bp). The colour ladder was based on the counts of kataegis regions. The y-axis represents the hospital ID of each sample in FUSCC. D Genes with enriched C > X kataegis regions. Genes with frequencies lower than 20% were omitted from this figure. The complete list is recorded in Table S9. SNV, single-nucleotide variant; PDAC, pancreatic ductal adenocarcinoma; chr, chromosome; r, correlation. E, F, G Comparison of total kataegis count (E), C > X mutation-enriched kataegis count (F) and proportion of C > X mutation-enriched kataegis in total kataegis (G) between APOBEC3C highest expressed samples (n = 25) and APOBEC3C lowest expressed samples (n = 25) from FUSCC cohort A.
We then compared the kataegis distribution of the 25 samples with the highest APOBEC3C expression and the 25 samples with the lowest APOBEC3C expression from FUSCC cohort A. Higher kataegis counts were observed within CMYA5, ZNF49, and ACSF3 genes in the A3C high subgroup. The counts of kataegis with enriched C > X conversions were higher in PCSK5, UGT1A5, NES, OR52L1, MROH5 genes in A3C high subgroup, the proportion of kataegis with enriched C > X conversions among total kataegis increased within PCSK5, NES, TRMT112, KRTAP10-2, and TRG-AS1 genes (Fig. 3E, F, G). Therefore, we speculated that APOBEC3C may influence the formation of kataegis within PCSK5, NES genes and etc.
APOBEC3C overexpression results in newly occruing SNVs and kataegis in pancreatic cell lines
A normal human pancreatic ductal epithelial cell line (H6c7) and two PDAC cell lines (SU.86.86 and Capan-1) with relatively low expression of APOBEC3C were used to construct APOBEC3C-overexpressing cell lines and simulate the effect of APOBEC3C on the genome. We excluded MIA PaCa-2 because they have a hypotriploid karyotype with a few absent normal chromosomes and thus lack representativeness. Ectopic APOBEC3C overexpression was validated by qRT–PCR and western blotting (Fig. 4A and Fig. S4B). Cell lines with APOBEC3C overexpression tended to form more and larger clones in the colony formation assay (Fig. 4B, C, increases in Capan-1 A3C and SU.86.86-A3C, and a tendency to increase in H6c7 A3C).

A APOBEC3C-overexpressing cell lines were generated by transfecting a pCDH plasmid loaded with the APOBEC3C CCDS, and the control group was transfected with the pCDH backbone plasmid. Multiple APOBEC3C primer pairs were applied to perform qRT–PCR in triplicate at different time points after transfection to validate that the cell lines transfected with the pCDH-A3C plasmid had a steadily elevated APOBEC3C mRNA expression level compared to the corresponding control cell lines. The corresponding western blotting results are shown in Fig. S4. B Representative images of the clonogenic assay. The assay was repeated in triplicate. C Summary of clone counts for H6c7-pCDH (mean = 272.7, SD = 39.5), H6c7-A3C (mean = 322.3, SD = 42.91), Capan-1-pCDH (mean = 56.33, SD = 23.54), Capan-1-A3C (mean = 132.3, SD = 13.8), SU.86.86-pCDH (mean = 280.3, SD = 8.505) and SU.86.86-A3C (mean = 280.3, SD = 8.505) cells. Welch’s t-test was applied to perform statistical analyses and obtain a two-tailed p-value. D, E, F Classification of newly occurring variants in APOBEC3C-overexpressing cell lines. Missense mutation was the major variant type (D), SNV was more prevalent than INS and DEL (E), and C > T mutation was the primary SNV type (F). G Mutation spectrum of newly occurring SNVs in APOBEC3C-overexpressing cell lines compared to corresponding control cell lines. H Mutational signatures of newly occurring SNVs in H6c7, SU.86.86 and Capan-1 cells. The upper x-axis represents the nucleotide at the 5’ end flanking the mutated sites, and the lower x-axis represents the nucleotide at the 3’ end flanking the mutated sites. I Rainfall plots showing that some of the newly occurring SNVs in APOBEC3C-overexpressing cell lines had short intermutational distances and formed characteristic kataegis regions on chromosomes 1, 2, 3, 7, 11, 12, and 13. The x-axis represents the SNV position, and the y-axis represents the intermutational distance. The complete list of newly formed kataegis regions is recorded in Table S12. CCDS, consensus coding sequence; SNV/SNP, single-nucleotide variant/polymorphism; INS, insertion; DEL, deletion.
Cell lines were cultured for another four months to accumulate mutations before WES. We extracted newly occurring SNVs in APOBEC3C-overexpressing cell lines compared to the corresponding control cell line (Table S11). In diploid cell lines (H6c7 and SU.86.86), the newly occurring SNVs were primarily composed of C > T conversions, while in Capan-1, an even distribution of the mutational spectrum of newly occurring SNVs was observed (Fig. 4D–H). In addition, the mutational signature reflected that most newly occurring C > X substitutions tended to be located in TpCpX trinucleotides in APOBEC3C-overexpressing cell lines, consistent with the preferred APOBEC3C target sequence. All three APOBEC3C-overexpressing cell lines had newly occurring kataegis regions in ACHE and MUC6 genes (Fig. 4I and Table S12). Additionally, APOBEC3C-overexpressing cell lines had newly occurring kataegis regions in HRNR (H6c7 and SU.86.86), PABPC3 (Capan-1) and TNS1 (H6c7) genes. Kataegis oriented within HRNR, PABPC3, and TNS1 genes were also observed in FUSCC cohort A. Therefore, we speculated that APOBEC3C overexpression was capable of inducing C > X substitution in pancreatic cell lines and generating characteristic kataegis.
A high level of APOBEC3C expression predicts shorter overall survival for patients with PDAC
Given the importance of A3C in causing PDAC genomic instability, we further investigated the correlation between APOBEC3C expression and the survival of patients with PDAC. We quantified the APOBEC3C mRNA expression level in FUSCC cohort B by performing qRT–PCR with APOBEC3C primer 1 (targeting A3C specific sequence to avoid other APOBECs’ confounding) and obtained its correlation with patient prognosis. The APOBEC3C mRNA expression level measured by qRT–PCR was cross-validated at protein level by performing IHC in 60 FFPE tumour tissues (FUSCC cohort B1, Fig. 5A, B).

The Mantel–Cox test was used to compare Kaplan–Meier survival curves. A, B Representative IHC images showing relatively high expression (A patient ID: 470506) and relatively low expression (B patient ID: 372428) of APOBEC3C in PDAC tissues. Sixty PDAC specimens (FUSCC cohort B1) underwent IHC staining with an APOBEC3C antibody to corroborate the qRT–PCR results. Positive APOBEC3C staining was located in the nucleus and cytoplasm of pancreatic tumour cells. C A cutoff value of 0.011802837 (qRT–PCR result) stratified the patients in FUSCC cohort B into the high APOBEC3C expression subgroup (n = 109) and low APOBEC3C expression subgroup (n = 15) with a difference in OS. The mRNA expression level was determined by performing qRT–PCR with cDNAs in triplicate, and the normalised mean value was used for analysis. D A cutoff value of 353.34 TPM stratified TCGA cohort into the high APOBEC3C expression subgroup (n = 69) and the low APOBEC3C expression subgroup (n = 94) with a trend towards a difference in OS. E A cutoff value of 372.77 TPM stratified the QCMG cohort into the high APOBEC3C expression subgroup (n = 62) and the low APOBEC3C expression subgroup (n = 8) with a difference in OS. PDAC, pancreatic ductal adenocarcinoma; A3C, APOBEC3C; HR, hazard ratio.
We calculated the optimal cutoff values and then stratified the three cohorts accordingly. FUSCC cohort B was divided into high and low APOBEC3C expression groups (n = 109 and n = 15, respectively), and patients without ectopic APOBEC3C expression experienced a longer OS (Fig. 5C, mean days of survival = 947.1 days versus 856.7, HR = 2.690, p = 0.0274). TCGA PDAC cohort was also stratified into high and low APOBEC3C expression subgroups (n = 69 and n = 94, respectively), and patients with comparatively low APOBEC3C expression tended to experience longer OS (Fig. 5D, mean days of survival = 634.0 days versus 518.0 days, HR = 1.325, p = 0.1892). QCMG PDAC dataset includes 70 patients with PDAC who have complete survival data and APOBEC3C expression data, and they were stratified into high and low APOBEC3C expression groups (n = 62 and n = 8, respectively). The latter group had a longer OS (Fig. 5E, mean days of survival were 1190.8 days versus 645.5 days, HR = 2.982, p = 0.0022). Stratification of FUSCC and QCMG datasets were in accordance with the result that most PDAC samples overexpressed APOBEC3C and few PDAC samples did not (Fig. 2A, B, E and Fig. S3A, B).
As APOBEC3D expression correlated with APOBEC3C expression in transcriptomics (Tables S5 and 6), we further performed survival analysis based on APOBEC3D expression to assess possible confounding effects. Patients with comparatively low APOBEC3D expression in TCGA cohort and QCMG cohort experienced longer survival (Fig. S5A, TCGA cohort, mean days of survival = 614.0 days versus 517.0 days, HR = 1.408, p = 0.1776; B, QCMG cohort, mean days of survival = 912.0 days versus 429.0 days, HR = 2.106, p = 0.0148). However, multivariable Cox analysis showed that neither APOBEC3C nor APOBEC3D expression correlated with survival when analysing transcriptimics data (Table S13).
APOBEC3C remodels the PDAC immune microenvironment
ESTIMATE and CIBERSORT analyses revealed that APOBEC3C was correlated with PDAC stromal modification and immune cell distribution in TCGA dataset (Fig. 6A, r = 0.364 and 0.404, respectively, ESTIMATE score r = 0.415, p < 0.001). Activated CD4 + T cells and CTLs were marginally positively correlated with APOBEC3C expression (relevance = 0.397 and 0.230, respectively, p < 0.05), while natural killer cells and M0 macrophages were marginally negatively correlated with APOBEC3C expression (relevance = –0.309 and –0.251, respectively, p < 0.05).

A ssGSEA was used to analyse the stroma of TCGA PDAC samples (n = 162) and output the immune score and the stromal score. The immune score and the stromal score were integrated into the ESTIMATE score. The CIBERSORT analytical tool was used to estimate the fractions of 22 types of immune cells in the matrix of TCGA PDAC dataset (based on the characteristic gene signature file “LM22”). Spearman’s rank correlation coefficient was calculated to measure the correlation between APOBEC3C expression and leucocyte fractions in the PDAC TME. B, C Representative images showing CD4 IHC staining (B) and CD8 IHC staining (C) in FUSCC cohort B1 (n = 60). For each sample, immune cells were manually counted in three representative high-power fields. D, E Nonparametric Spearman’s correlation test to determine whether the count of CD4 T cells (D) or the count of CD8 T cells (E) correlated with APOBEC3C expression in FUSCC cohort B1 (n = 60). The APOBEC3C expression level was quantified using qRT–PCR. The immune cells were counted manually, and taken the mean values of three high-power fields (20×). PDAC, pancreatic ductal adenocarcinoma; ssGSEA, single-sample gene set enrichment analysis; NK cells, natural killer cells; Tfh, follicular helper T cells; Tregs, regulatory T cells; r, correlation.
We then counted the numbers of infiltrated CD4 + T cells and CD8 + T cells in PDAC stroma and correlated them with APOBEC3C expression in FUSCC cohort B1 (n = 60, Fig. 6B–E). APOBEC3C expression marginally correlated with CD4 + T cell counts (r = 0.2566, p = 0.0478) and CD8 + T cell counts (r = 0.2973, p = 0.0211).
Discussion
The dismal prognosis of PDAC has not improved for decades, which might largely be attributed to therapeutic resistance and ubiquitous recurrence [46]. Notably, PDAC features high intratumor heterogeneity [5, 47], which accounts for its phenotypes development and adaptation to evolutionary pressure [48]. Patients have an increased mutational burden after first-line treatment [49], and the heterogeneous polyclonality of PDAC contributes to metastatic progression [50]. Accordingly, investigations of the factors resulting in PDAC genomic instability and promoting PDAC heterogeneity are urgently needed.
Remarkably, APOBEC3, along with MMR, promotes focal hypermutation in the tumour genome [51]. We analysed TCGA and FUSCC cohort A, and mutational signatures of the two cohorts coincidently showed a predominance of C > T mutations. Moreover, C > T mutations more frequently occurred in APOBEC3 preferred motifs, indicating that APOBEC3 participates in modelling mutational features in the PDAC genome. We located characteristic kataegis regions and correlated the count of C > X mutation-enriched kataegis regions to APOBEC3C mRNA expression level. We compared the high APOBEC3C expression subgroup and the low APOBEC3C expression subgroup and speculated that kataegis located within PCSK5 and NES genes were fuelled by APOBEC3C. Vector transfected APOBEC3C overexpression in cell lines also resulted in altered SNV distribution and new kataegis occurrence. The alteration of SNV distributions in diploid cell lines met our expectation, whereas it did not in the hypotriploid cell line (Capan-1). The seemingly paradoxical results may be attributed to the comprehensive effects of multiple mutagens, such as ADAR, POLE and POLH (Fig. S5C–H).
Notably, ssDNA is more vulnerable to DNA-modifying enzymes that cause clustered DNA damage, thus forming kataegis regions [52]; hence, the kataegis location reflects the ssDNA region, which is more likely to be translated or modified. Kataegis is not only observable evidence of hypermutagenic factors but is also associated with structural or functional alterations in the genome. Previous studies have reported that kataegis stabilises the expression of neighbouring genes [41] and colocalizes with genomic rearrangements [20] or is accompanied by chromothripsis [53].
Furthermore, this research revealed that APOBEC3C is the predominant APOBEC enzyme expressed in PDAC and that universal ectopic expression of APOBEC3C occurs in PDAC. We established survival curves showing that universally elevated APOBEC3C expression was related to shorter OS of PDAC patients. High APOBEC3C expression contributes to tumour plasticity; thus, tumours are able to adapt more easily to evolutionary pressure, such as chemotherapy [54], and are more prone to developing new phenotypes for recurrence or metastasis, which account for the worse prognosis.
Notably, factors involved in inducing genomic instability are also promising actionable targets for precise therapy in PDAC, such as BRCA mutations [55, 56] and dMMR [57, 58]. A real-world study revealed that patients with PDAC receiving matched precise therapy (including PARP inhibitors and PD-1 inhibitors) experienced prolonged survival [59], which reinforces the importance of identifying precise therapeutic targets to overcome the low survival rate of patients with PDAC. Regarding APOBEC, its increased TMB also suggests the potential for immune therapy [60,61,62]. APOBEC-induced genomic instability nourishes neoantigens, which are promising targets for immune cells to recognise and attack tumour cells. Our study revealed a positive correlation between APOBEC3C expression and the invasion of activated CD4 + T cells and CTLs, which are major effector lymphocytes involved in antitumour immune processes. Therefore, APOBEC3C expression denotes enhanced immune activity in the PDAC TME and predicts immunotherapy responses of PDAC. A previous pancancer analysis also revealed that APOBEC and kataegis are associated with programmed death-ligand 1/2 [63], and the potential of APOBEC for guiding immunotherapy has been affirmed in other tumours [64, 65]. In addition, ongoing clinical trials are investigating the practicality of APOBEC in guiding precise tumour therapy [66], such as NCT02576444 (olaparib, phase II) and NCT03989089 (pembrolizumab, phase II). Further studies on the application of APOBEC in PDAC immunotherapy are anticipated.
Given the importance of APOBEC for guiding immunotherapy, the potential to inhibit APOBEC family members for the purpose of limiting tumour adaptation also exists [19]. However, APOBEC has numerous isoforms, and each single isoform should be evaluated thoroughly to ensure the effect of inhibitors. The APOBEC expression burden may also be applied as an indicator for the risk of developing cancerous disease in the future. Moreover, APOBECs are promising base editing tools and probably have therapeutic implications in gene therapy [67].
In conclusion, APOBEC3C, a mutagenic driver, plays a dual role in PDAC treatment. On the one hand, its induction of genomic instability supplements tumour heterogeneity and evolution; on the other hand, it is a promising target for precise therapy.
Data availability
The datasets used and analysed during this study available from the corresponding author on reasonable request.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Grossberg AJ, Chu LC, Deig CR, Fishman EK, Hwang WL, Maitra A, et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J Clin. 2020;70:375–403.
Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–13.
Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–74.
Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–38.
Balmain A. The critical roles of somatic mutations and environmental tumor-promoting agents in cancer risk. Nat Genet. 2020;52:1139–43.
Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 2017;542:362–6.
Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Abe T, Blackford AL, Tamura K, Ford M, McCormick P, Chuidian M, et al. Deleterious germline mutations are a risk factor for neoplastic progression among high-risk individuals undergoing pancreatic surveillance. J Clin Oncol. 2019;37:1070–80.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Siriwardena SU, Chen K, Bhagwat AS. Functions and malfunctions of mammalian DNA-cytosine deaminases. Chem Rev. 2016;116:12688–710.
Salter JD, Bennett RP, Smith HC. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci. 2016;41:578–94.
Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–6.
Chiu YL, Greene WC. Multifaceted antiviral actions of APOBEC3 cytidine deaminases. Trends Immunol. 2006;27:291–7.
Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J, et al. DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep. 2015;12:2086–98.
Kumar R, DiMenna L, Schrode N, Liu TC, Franck P, Munoz-Descalzo S, et al. AID stabilizes stem cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 2013;500:89–92.
Nowarski R, Kotler M. APOBEC3 cytidine deaminases in double-strand DNA break repair and cancer promotion. Cancer Res. 2013;73:3494–8.
Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC Enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5:704–12.
Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019;176:1282–1294.e20.
Talluri S, Samur MK, Buon L, Kumar S, Potluri LB, Shi J, et al. Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma. Blood Cancer J. 2021;11:166.
Li Z, Abraham BJ, Berezovskaya A, Farah N, Liu Y, Leon T, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T-ALL. Leukemia. 2017;31:2057–64.
Ren W, Ye X, Su H, Li W, Liu D, Pirmoradian M, et al. Genetic landscape of hepatitis B virus–associated diffuse large B-cell lymphoma. Blood. 2018;131:2670–81.
Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer Michael. Nature. 2013;494:366–70.
Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–1271.e11.
Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell. 2018;34:996–1011.e8.
Nowarski R, Wilner OI, Cheshin O, Shahar OD, Kenig E, Baraz L, et al. APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair. Blood. 2012;120:366–75.
Brady SW, Gout AM, Zhang J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2022;38:194–208.
Venkatesan S, Rosenthal R, Kanu N, McGranahan N, Bartek J, Quezada SA, et al. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann Oncol. 2018;29:563–72.
Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–60.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31.
Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47:1067–72.
Yin X, Bi R, Ma P, Zhang S, Zhang Y, Sun Y, et al. Multiregion whole-genome sequencing depicts intratumour heterogeneity and punctuated evolution in ovarian clear cell carcinoma. J Med Genet. 2020;57:605–609.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Yoshihara K, Shahmoradgoli M, Martínez E, Martinez E, Vegesna R, Kim H, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–59.
Nik-zainal S, Alexandrov LB, Wedge DC, Loo PV, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93.
Antonio MD, Tamayo P, Jill P, Frazer KA. Kataegis expression signature in breast cancer is associated with late onset, better prognosis, and higher HER2 levels. Cell Rep. 2016;16:672–83.
Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Noia JMD. Mutations, kataegis, and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16:164–76.
Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997.
Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods. 2009;6:291–5.
Dai P, Wu LR, Chen SX, Wang MX, Cheng LY, Zhang JX, et al. Calibration-free NGS quantitation of mutations below 0.01% VAF. Nat Commun. 2021;12:6123.
Jain T, Dudeja V. The war against pancreatic cancer in 2020 — advances on all fronts. Nat Rev Gastroenterol Hepatol. 2021;18:99–100.
Bernard V, Semaan A, Huang J, Lucas FAS, Mulu FC, Stephens BM, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res. 2019;25:2194–205.
Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553–65.
Sakamoto H, Attiyeh MA, Gerold JM, Makohon-Moore AP, Hayashi A, Hong J, et al. The evolutionary origins of recurrent pancreatic cancer. Cancer Discov. 2020;10:792–805.
Maddipati R, Stanger BZ. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 2015;5:1086–97.
Mas-Ponte D, Supek F. DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nat Genet. 2020;52:958–68.
Chan K, Gordenin DA. Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet. 2015;49:243–67.
Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet. 2020;52:884–90.
Yoo SH, Yun J, Keam B, Hong S, Ock C, Koh J, et al. Discovery of acquired molecular signature on immune checkpoint inhibitors in paired tumor tissues. Cancer Immunol Immunother. 2021;70:1755–69.
Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q, et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol Cancer. 2020;19:49.
Golan T, Hammel P, Reni M, Cutsem EV, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl J Med. 2019;381:317–27.
Marabelle A, Le DT, Ascierto PA, Giacomo AMD, Jesus-Acosta AD, Delord J, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38:1–10.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl J Med. 2017;377:2500–1.
Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020;21:508–18.
Chapuy B, Stewart C, Dunford AJ, Kim J, Wienand K, Kamburov A, et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood. 2019;134:2369–82.
Ladányi A, Tímár J. Immunologic and immunogenomic aspects of tumor progression. Semin Cancer Biol. 2020;60:249–61.
Boichard A, Pham TV, Yeerna H, Goodman A, Tamayo P, Lippman S, et al. APOBEC-related mutagenesis and neo-peptide hydrophobicity: implications for response to immunotherapy. Oncoimmunology. 2018;8:1550341.
Boichard A, Tsigelny IF, Kurzrock R. High expression of PD-1 ligands is associated with kataegis mutational signature and APOBEC3 alterations. Oncoimmunology. 2017;6:e1284719.
Luo C, Wang S, Liao W, Zhang S, Xu N, Xie W, et al. Upregulation of the apobec3 family is associated with a poor prognosis and influences treatment response to raf inhibitors in low grade glioma. Int J Mol Sci. 2021;22:10390.
Kim MH, Kim JH, Lee JM, Choi JW, Jung D, Cho H, et al. Molecular subtypes of oropharyngeal cancer show distinct immune microenvironment related with immune checkpoint blockade response. Br J Cancer. 2020;122:1649–60.
Chumsri S, Sokol ES, Soyano-Muller AE, Parrondo RD, Reynolds GA, Nassar A, et al. Durable complete response with immune checkpoint inhibitor in breast cancer with high tumor mutational burden and APOBEC signature. J Natl Compr Canc Netw. 2020;18:517–21.
Xu W, Biswas J, Singer RH, Rosbash M. Targeted RNA editing: novel tools to study post-transcriptional regulation. Mol Cell. 2022;82:389–403.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 82072693, 81902417, 82172884), the Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), Clinical and Scientific Innovation Project of Shanghai Hospital Development Center (SHDC12018109), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1006A), and National Key Research and Development Program of China (2020YFA0803202).
Author information
Authors and Affiliations
Contributions
Conceptualisation and funding acquisition: XY and CL. Project administration and supervision: XW. Specimens acquirement and validation: HC and GL. Data collection: ZD and YC. Cell culture and plasmid construction: YQ. RNA extraction and PCR assay: YQ and XL. Western blotting: RW and YT. Immunohistochemical evaluation: YL. Rstudio analysis and visualisation: XZ. Writing—original draft: YQ and YG. Writing—review editing: XW.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Ethics Board of Shanghai Cancer Center, Fudan University. All patients involved in this study provided informed consent for the use of their specimens and personal data for research purposes.
Consent for publication
Written informed consent for publication was obtained from all participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qian, Y., Gong, Y., Zou, X. et al. Aberrant APOBEC3C expression induces characteristic genomic instability in pancreatic ductal adenocarcinoma. Oncogenesis 11, 35 (2022). https://doi.org/10.1038/s41389-022-00411-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-022-00411-9
- Springer Nature Limited