
Abstract
Arginine methylation is an important posttranslational modification catalyzed by protein arginine methyltransferases (PRMTs). However, the role of PRMTs in colorectal cancer (CRC) progression is not well understood. Here we report that non-POU domain-containing octamer-binding protein (NONO) is overexpressed in CRC tissue and is a potential marker for poor prognosis in CRC patients. NONO silencing resulted in decreased proliferation, migration, and invasion of CRC cells, whereas overexpression had the opposite effect. In a xenograft model, tumors derived from NONO-deficient CRC cells were smaller than those derived from wild-type (WT) cells, and PRMT1 inhibition blocked CRC xenograft progression. A mass spectrometry analysis indicated that NONO is a substrate of PRMT1. R251 of NONO was asymmetrically dimethylated by PRMT1 in vitro and in vivo. Compared to NONO WT cells, NONO R251K mutant-expressing CRC cells showed reduced proliferation, migration, and invasion, and PRMT1 knockdown or pharmacological inhibition abrogated the malignant phenotype associated with NONO asymmetric dimethylation in both KRAS WT and mutant CRC cells. Compared to adjacent normal tissue, PRMT1 was highly expressed in the CRC zone in clinical specimens, which was correlated with poor overall survival in patients with locally advanced CRC. These results demonstrate that PRMT1-mediated methylation of NONO at R251 promotes CRC growth and metastasis, and suggest that PRMT1 inhibition may be an effective therapeutic strategy for CRC treatment regardless of KRAS mutation status.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the second most deadly cancer worldwide [1]. Neoadjuvant chemoradiotherapy and total mesorectal excision surgery have improved the outcome of locally advanced rectal cancer [2], and adding adjuvant chemotherapy decreases the rate of relapse in patients with locally advanced colon cancer [3]. However, the efficacy of the epidermal growth factor receptor (EGFR) inhibitor cetuximab is limited to patients with wild-type (WT) KRAS [4,5,6]. Moreover, distant metastasis is the leading cause of mortality in CRC; the 5-year survival of patients with metastasis is markedly lower than that of patients with nonmetastatic disease (90.0% vs 14.0%) [7]. Identifying novel therapeutic targets can provide a basis for the development of drugs that improve the outcome of metastatic CRC.
Non-POU domain-containing octamer-binding protein (NONO), also known as 54-kDa nuclear RNA- and DNA-binding protein (p54nrb), belongs to the Drosophila behavior/human splicing family and functions in a variety of physiologic processes including mRNA splicing [8], transcriptional regulation [9], DNA repair [10], and nuclear retention of defective RNA [11]. NONO also plays an important role in the regulation of malignant phenotypes in cancer cells [12]. In breast cancer, NONO increased nuclear sterol regulatory element-binding protein (SREBP)−1a protein stability and stimulated SREBP-1a–mediated transcription of lipogenic genes and lipid production, thus promoting breast cancer growth [13]. As a component of the cyclic (c)AMP signaling pathway, NONO was shown to interact with the long noncoding (lnc)RNAs LINC00473 and MetaLnc9 to facilitate transcription through cAMP-responsive element-binding protein (CREB) and CREB-regulated transcription coactivator to promote lung cancer growth and metastasis [14, 15]. However, the mechanism by which NONO regulates distant metastasis of cancer cells is not known.
NONO is posttranslationally modified in several ways including by phosphorylation, ubiquitination, and arginine methylation. As a cell cycle regulator, NONO is phosphorylated by cyclin-dependent kinase (CDK)1 at T412, T430, and T452 during mitosis for Pin1 binding [16]. In response to ultraviolet-induced DNA damage, NONO is ubiquitinated by RING finger protein (RNF)8 for degradation [17]. Polyubiquitination of NONO by F-box and WD repeat domain-containing (FBW)7a is triggered by glycogen synthase kinase (GSK)3β-mediated phosphorylation, which is involved in chromosomal rearrangement in cancer cells [18]. Moreover, coactivator-associated arginine methyltransferase (CARM)1-mediated NONO methylation regulates the nuclear retention of mRNAs containing inverted-repeat Alu elements under poly(I:C) treatment [19]. Additionally, NONO posttranscriptionally promotes the expression of S-phase-associated kinase (SKP)2 and E2F transcription factor (E2F)8 by directly binding to their mRNAs, leading to increased breast cancer cell proliferation [20].
Arginine methylation is a common posttranslational modification and it is catalyzed by protein arginine methyltransferases (PRMTs) that mainly occurs in nuclear proteins of eukaryotic cells. PRMTs—which are broadly classified as type I, II, or III—transfer methyl groups from S-adenosylmethionine (SAM) to the guanidine nitrogen of specific arginine residues of target proteins. Type I and II PRMTs catalyze the formation of ω-NG-monomethylarginine (MMA) as an intermediate; the former (including PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8) stimulate the production of ω-NG, NG-asymmetric dimethylarginine (aDMA), while the latter (including PRMT5 and PRMT9) catalyze the formation of ω-NG, N,G-symmetric dimethylarginine (sDMA) [21]. In contrast, type III PRMT (PRMT7) only generates MMA [22]. Arginine methylation has been implicated in tumorigenesis and metastasis [23, 24]. For example, CARM1-mediated arginine methylation of BRG1-associated factor (BAF)155 was shown to control the expression of genes in the c-Myc pathway and regulate cell migration and metastasis in breast cancer [25]. We previously reported that PRMT1-mediated methylation of EGFR maintains its activation and promotes cell proliferation; increased EGFR methylation was correlated with worse survival in CRC patients who received cetuximab treatment [26]. Although PRMT1 has been reported to asymmetrically dimethylate splicing factor proline- and glutamine-rich (SFPQ)/PTB-associated splicing factor (PSF) to increase its association with mRNA without affecting complex formation with NONO in mammalian cells [27], it is not known whether NONO is a substrate of PRMT1.
In this study we demonstrate that NONO is overexpressed in CRC tissue and promotes CRC progression. We found that asymmetric dimethylation at R251 by PRMT1 was required for NONO-induced CRC cell proliferation, migration, and invasion. Inhibition of PRMT1 by gene silencing or pharmacological treatment abrogated NONO methylation-associated increases in cell proliferation, migration, and invasion, regardless of KRAS mutation status. These results were confirmed in a CRC xenograft model treated with PRMT1 inhibitor. Thus, therapeutic strategies targeting PRMT1-mediated asymmetric arginine dimethylation of NONO are a promising treatment for CRC, irrespective of KRAS mutation status.
Results
NONO is overexpressed in CRC tissue and promotes cell proliferation, migration, and invasion
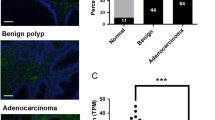
To investigate the role of NONO in CRC progression, we examined NONO expression levels in clinical specimens and found that it was overexpressed in CRC compared to adjacent normal tissue at both the protein (Fig. 1a and Supplementary Fig. S1a) and mRNA (Fig. 1b) levels. Consistent with these observations, NONO transcript level was increased in CRC samples of The Cancer Genome Atlas (TCGA) (Fig. 1b). We analyzed the correlation between NONO expression in 93 CRC tissue specimens and patients’ clinicopathologic features (Fig. 1c) and found that elevated expression of NONO was correlated with poor differentiation of intestinal tissue (P = 0.002; Supplementary Table S1). Kaplan–Meier survival analysis revealed that NONO overexpression was correlated with shorter overall survival (P = 0.012; Fig. 1d).

a NONO protein is highly expressed in CRC tissue. NONO protein level was analyzed in 28 paired colorectal tumor (T) and adjacent normal (N) tissue samples by western blotting. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. b NONO mRNA level is higher in T than in N. NONO mRNA level was examined by qPCR in 29 paired tissue samples. U6 was used as an internal control (right panel). NONO mRNA expression level in TCGA N (n = 41) and T (n = 286) CRC samples (left panel). c, d Higher NONO protein level is correlated with shorter overall survival in CRC patients. The 93 patients were divided into NONO low (n = 48) and high (n = 45) subgroups (c) and subjected to Kaplan–Meier survival analysis (d). Scale bar, 1200 μm. e NONO overexpression promotes CRC cell growth. KM12 (1 × 104) and HCT8 (2 × 104) CRC cells were seeded on day 0 and counted on days 2 and 4. f NONO KO leads to a decrease in tumor size. KM12 WT or NONO KO cells (1 × 106) were injected into nude mice (n = 5) and tumors were weighed on day 21. g–j NONO overexpression enhances CRC cell migration and invasion. The wound-healing assay was performed using KM12 (8 × 104) or HCT8 (1 × 105) cells (g, i). The transwell assay was performed using KM12 (4 × 104) or HCT8 (8 × 104) cells for 24 h (h, j). Scale bar, 400 μm. EV empty vector (pCDH-CMV)-transfected cells, NONO-OE NONO-overexpressing cells (transfected with pCDH-CMV-Flag-NONO). *P < 0.05, **P < 0.01, ***P < 0.001.
NONO was highly expressed in different CRC cell lines irrespective of KRAS mutation status (Supplementary Fig. S1b). We used KM12 and HCT8 cells—which harbor WT and mutant KRAS, respectively—to investigate the function of NONO in CRC. NONO knockout (KO) in both cell lines significantly inhibited proliferation, whereas NONO overexpression had the opposite effect (Fig. 1e and Supplementary Fig. S1c, d). Consistent with these results, KM12 and HCT8 NONO KO cell-derived tumors in an in vivo xenograft model were smaller than those derived from NONO WT cells (Fig. 1f and Supplementary Fig. S1e). Furthermore, the wound-healing and transwell assays showed that NONO deficiency suppressed CRC cell migration and invasion (Fig. 1g, h), while NONO overexpression enhanced these behaviors in KM12 and HCT8 cells (Fig. 1i, j).
PRMT1 mediates NONO arginine methylation in CRC cells
To investigate the mechanism by which NONO enhances CRC progression, Flag-NONO recombinant protein was immunoprecipitated from KM12 cells and subjected to liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis (Supplementary Fig. S2a). Several arginine residues in NONO were found to be methylated (Supplementary Table S2). To identify the PRMT responsible for arginine methylation of NONO in CRC cells, we examined PRMT1, PRMT3, PRMT4, PRMT5, PRMT6, and PRMT8 mRNA and protein levels in paired tumor and adjacent normal tissues. PRMT1 was overexpressed in CRC tissue compared to normal tissue (Fig. 2a, b). In contrast, PRMT3, PRMT4, and PRMT5 levels were comparable between groups, whereas PRMT6 and PRMT8 were undetectable in CRC tissue specimens (Fig. 2a, b). Immunofluorescence analysis and the Duolink proximity ligation assay (PLA) showed that NONO was asymmetrically dimethylated in KM12 and HCT8 CRC cells (Fig. 2c and Supplementary Fig. S2b). To determine whether asymmetric dimethylation of NONO is mediated by PRMT1, endogenous NONO was immunoprecipitated from control or PRMT1-silenced CRC cells, followed by immunoblotting with pan-ADMA and -MMA antibodies. Compared to control cells, NONO aDMA level was significantly decreased in PRMT1-deficient cells, while NONO and whole-cell lysate MMA levels were increased (Fig. 2d and Supplementary Fig. S2c). This finding was confirmed with the Duolink PLA in which NONO aDMA level was reduced in cells lacking PRMT1 (Fig. 2e). Notably, PRMT1 silencing had no effect on PRMT3, PRMT4, and PRMT5 mRNA levels in KM12 and HCT8 cells (Supplementary Fig. S2d). These results indicate that NONO is a substrate of PRMT1 in CRC cells.

a Compared to adjacent normal (N) tissue, PRMT1 mRNA level was higher in CRC tissue (T), as determined by qPCR (n = 29 paired tissues). There were no differences in PRMT3, PRMT4, and PRMT5 mRNA levels between groups. b PRMT1 protein is highly expressed in CRC tissue. PRMT5 protein expression was similar between T and N tissues, as determined by western blotting (n = 28 paired tissues; left panel). Protein band intensity was quantified using ImageJ software (right panel). c NONO and the aDMA modification colocalized in the nucleus. Immunofluorescence analysis was performed using anti-NONO and -ADMA antibodies (right panel) in KM12 and HCT8 cells, and colocalization was analyzed using ZEN v3.0 software (Zeiss, Oberkochen, Germany) (left panel). Scale bar, 10 μm. d, e PRMT1 silencing reduces NONO aDMA level. Endogenous NONO was immunoprecipitated (IP) from control and PRMT1-deficient KM12 and HCT8 cells and analyzed by western blotting (d). NONO methylation was detected using an anti-ADMA antibody. For Duolink PLA (e), NONO aDMA level was detected using anti-NONO and -ADMA antibodies (left panel). At least 100 nuclei were counted in each group (right panel). **P < 0.01, ***P < 0.001; ns no significance.
NONO interacts with PRMT1 in CRC cells
To confirm whether NONO interacts with PRMT1, we performed coimmunoprecipitation (coIP) assays. PRMT1 was detected in Myc-NONO complexes and NONO was present in Flag-PRMT1 complexes immunoprecipitated from HEK 293T cells (Fig. 3a). The coIP assay confirmed the interaction between NONO and PRMT1 in KM12 and HCT8 cells (Fig. 3b). Immunofluorescence analysis and the Duolink PLA showed that NONO and PRMT1 colocalized and interacted in the nucleus (Fig. 3c, d). A coIP experiment with NONO and various truncation mutants of PRMT1 showed that the catalytic domain of PRMT1 mediates its interaction with NONO (Fig. 3e).

a Myc-NONO and Flag-PRMT1 interact in HEK 293T cells. pCDH-CMV-Myc-NONO and pCDH-CMV-Flag-PRMT1 were cotransfected into HEK 293T cells for 24 h before coIP analysis. b NONO interacts with PRMT1 in situ. CoIP was performed in KM12 and HCT8 cells using anti-NONO and -PRMT1 antibodies. Arrows indicate NONO or PRMT1, and circles indicate the heavy chain. c NONO and PRMT1 colocalize in the nucleus. Immunofluorescence analysis was performed using anti-NONO and -PRMT1 antibodies in KM12 and HCT8 cells (right panel). NONO and PRMT1 colocalization was analyzed using ZEN v3.0 software (left panel). Scale bar, 10 μm. d NONO interacts with PRMT1. Duolink PLA was performed with indicated antibodies. Blank, no primary antibody added in Duolink PLA. Scale bar, 10 μm. e NONO binds to the catalytic domain of PRMT1. NONO truncations are illustrated schematically at the top. HEK 293T cells were cotransfected with Myc-NONO and Flag-PRMT1 truncations for 24 h before CoIP analysis. aa amino acid, CD catalytic domain, POD post domain, PRD pre domain.
PRMT1-mediated methylation at R251 is required for the oncogenic function of NONO
To identify the arginine residues of NONO that are methylated by PRMT1, Flag-NONO was immunoprecipitated from control and PRMT1-silenced KM12 cells and subjected to LC–MS/MS analysis (Fig. 4a). Compared to control cells, Flag-NONO protein from PRMT1-silenced cells showed decreased methylation at R251, R287, R364, and R383, although only R251 showed a statistically significant difference in methylation level (P < 0.05; Fig. 4b, Supplementary Fig. S3a, and Supplementary Table S2). To confirm these findings, a series of R-to-K NONO mutants and PRMT1 were coexpressed in HEK 293T cells, followed by analysis of aDMA status. aDMA was not detected in R251K mutants but was observed in R287K, R364K, and R383K mutants (Fig. 4c). After in vitro incubation with PRMT1, purified NONO protein showed an increase in asymmetric arginine dimethylation, whereas the R251K mutant had a lower aDMA level (Fig. 4d).

a Flag-NONO protein were immunoprecipitated from control or PRMT1-silenced KM12 cells, separated by SDS–PAGE, and subjected to silver staining. b LC–MS/MS analysis of the NONO R251 methylation site. The fragmentation pattern of the typical NONO peptide EREQPPR is shown. Fragment ions are shown as b and y ions; ++ represents the loss of doubly charged ions. c R251K mutation abolishes PRMT1-mediated aDMA modification of NONO. NONO R-to-K mutants are illustrated schematically at the top. WT or R-to-K mutant NONO protein was immunoprecipitated with Flag beads and then immunoblotted with anti-ADMA antibody. d NONO is methylated by PRMT1 at R251, as determined with the in vitro methylation assay. GST-tagged GAR, NONO, and R251K were incubated with purified GST-PRMT1 in the presence or absence of 0.6 μM SAM. Proteins were separated on SDS–PAGE and subjected to western blotting analysis and Coomassie blue staining. GAR, glycine- and arginine-rich N-terminal region of fibrillarin. e NONO R251K mutation reduces cell proliferation. KM12 (1 × 104) and HCT8 (2 × 104) cells were seeded on day 0 and counted on days 2 and 4. NONO R251K mutation inhibited KM12 cell migration in the wound-healing assay (f) and invasion in the transwell assay (g). For experiments shown in e–g, KM12 cells were transfected with pCDH-CMV-Flag-NONO (WT) or pCDH-CMV-Flag-NONO-R251K (R251K mutant) for 24 h before the indicated assay. Scale bar, 400 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
R251 methylation had no influence on NONO expression (Supplementary Fig. S3b), but we investigated it’s impact on the oncogenic function of NONO. The results of the cell proliferation, wound-healing, and transwell assays revealed that compared to WT NONO, CRC cells expressing hypomethylated NONO (R251K) showed reduced growth, migration, and invasion (Fig. 4e–g). Thus, PRMT1-mediated R251 methylation promotes the oncogenic function of NONO in CRC.
PRMT1 has an oncogenic function that is correlated with poor outcome in CRC
Given that PRMT1 was overexpressed in CRC tissue and PRMT1-mediated arginine methylation of NONO enhanced tumor growth and metastasis, we hypothesized that tumor progression induced by NONO arginine methylation was initiated by PRMT1. PRMT1 knockdown inhibited cell proliferation, whereas its overexpression had the opposite effect (Fig. 5a). The wound-healing and transwell assays showed that PRMT1 silencing suppressed KM12 and HCT8 cell migration and invasion (Fig. 5b, c). Conversely, PRMT1 overexpression enhanced these malignant behaviors (Fig. 5d, e). PRMT1 transcript level was higher in CRC than in normal tissue in TCGA (Supplementary Fig. S4a). Immunohistochemical (IHC) analysis of CRC tissue confirmed that PRMT1 was highly expressed in the CRC zone (Supplementary Fig. S4b). Moreover, elevated PRMT1 expression was correlated with advanced TNM stage (P = 0.040; Supplementary Table S3) and shorter overall survival (P = 0.006; Fig. 5f) in CRC patients.

a PRMT1 promotes CRC cell proliferation. KM12 (1 × 104) and HCT8 (1.8 × 104 or 2 × 104) cells were seeded on day 0 and counted every 2 days. PRMT1 knockdown inhibited and PRMT1 overexpression promoted, KM12 cell migration in the wound-healing assay (b, d) and invasion in the transwell assay (c, e). KM12 cells were transfected with pCDH-CMV-Flag-PRMT1 (PRMT1-OE) or empty vector (EV) for 24 h before the indicated assay. EV empty vector (pCDH-CMV) transfected cells, NONO-OE NONO-overexpressing cells (transfected with pCDH-CMV-Flag-NONO). Scale bar, 400 μm. f Elevated PRMT1 expression is correlated with shorter overall survival in CRC patients. The 97 patients were divided into PRMT1 low (n = 61) and high (n = 36) expression subgroups. *P < 0.05, **P < 0.01, ***P < 0.001.
PRMT1 inhibition reduces NONO arginine methylation and suppresses CRC progression
To further confirm the role of PRMT1 in promoting CRC progression, we used the PRMT1-selective small-molecule inhibitors AMI-1, MS023, and C7280948 to prevent substrate recognition or binding by PRMT1 [28,29,30]. The Duolink PLA showed that NONO aDMA level was significantly reduced in AMI-1–treated KM12 and HCT8 CRC cells compared to control cells (Fig. 6a and Supplementary Fig. S4c) and in the xenograft model, mice treated with AMI-1 had smaller tumors (Fig. 6b). MS023 and C7280948 also reduced NONO aDMA level in KM12 and HCT8 cells, as demonstrated with western blotting assay (Fig. 6c), and suppressed cell proliferation, migration, and invasion (Fig. 6d–f). However, these effects were not observed in NONO-depleted KM12 and HCT8 cells treated with the inhibitors (Fig. 6d–f), indicating that NONO mediates the oncogenic effects of PRMT1 in CRC. Taken together, these findings suggest that inhibiting NONO arginine methylation by PRMT1 can prevent the malignant transformation of CRC irrespective of KRAS mutation status.

a Treatment with the PRMT1 inhibitor AMI-1 reduced NONO aDMA level. KM12 and HCT8 cells treated with AMI-1 (1.2 and 0.6 mM) or DMSO for 48 h were subjected to Duolink PLA with anti-ADMA antibody. b AMI-1 treatment reduced tumor xenograft weight (n = 6 mice per group). c Treatment with the PRMT1 inhibitors MS023 and C7280948 decrease aDMA NONO level. KM12 and HCT8 cells were treated with 10 μM MS023 and 40 μM C7280948, respectively, for 48 h, and subjected to NONO IP and western blotting analysis with anti-ADMA antibody. d Treatment with MS023 or C7280948 inhibits CRC cell proliferation. KM12 WT/NONO-KO (2 × 104) and HCT8 WT/NONO-KO (4 × 104) cells were seeded on day 0 and counted on days 2 and 4 after treatment with the indicated inhibitor. KM12 and HCT8 WT/NONO-KO cells treated with MS023 and C7280948 were subjected to the wound-healing assay (e) and transwell assay (f), respectively. For (d–f), MS023, C7280948, or DMSO was added at the time the cells were seeded. Scale bar, 400 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Anti-EGFR/vascular endothelial growth factor receptor-based targeted therapy as well as immunotherapy involving programmed death (PD)−1/programmed death ligand (PD-L)1 blockade have shown survival benefits in advanced CRC patients with metastasis [31]. Clarifying the molecular mechanisms of metastasis can reveal novel therapeutic targets. In this study we found that NONO was overexpressed and arginine-methylated in CRC tissue, regardless of KRAS mutation status. We demonstrated that the R251 residue of NONO was asymmetrically dimethylated by PRMT1 to promote the proliferation, migration, and invasion of CRC cells. Aberrant expression of PRMT1 and NONO was correlated with shorter overall survival in CRC patients. Suppressing PRMT1 by knocking down its expression or using pharmacological inhibitors abrogated the increases in CRC cell proliferation, migration, and invasion mediated by R251-methylated NONO, an effect that was independent of KRAS mutation status (Fig. 7).

In CRC cells, PRMT1-mediated methylation of NONO at R251 promotes tumor cell proliferation and metastasis. Malignant progression induced by NONO arginine methylation may be abrogated by treatment with small-molecule inhibitors of PRMT1 (AMI-1, MS023, and C7280948).
Aberrant expression of NONO is correlated with malignant phenotypes in various cancers [32, 33]. NONO increases lncRNA Prostate cancer gene expression marker (PCGEM)1 expression via upregulation of androgen receptor [34], resulting in castration-resistant prostate cancer [35]. The NONO/PSF complex is a critical regulator of radiation-induced DNA double-stranded breaks or ultraviolet-induced DNA damage and confers resistance to radiotherapy in melanoma cells [36]. Knockdown of NONO/RALY complex components reversed YB-1 overexpression-induced oxaliplatin resistance in CRC cells [37], while NONO silencing modulated the cellular response to ultraviolet-induced DNA damage in melanoma cells [38]. However, NONO is a nuclear protein that cannot be directly targeted by currently available antibody drugs or small-molecule inhibitor compounds. Therefore, other related molecules are alternative therapeutic targets for CRC treatment. We showed here that PRMT1-mediated arginine methylation is essential for the oncogenic function of NONO, and that expressing the R251K arginine methylation mutant of NONO in CRC cells attenuated their malignant behaviors. Similar effects were observed in vitro and in vivo by blocking NONO arginine methylation with the PRMT1-specific inhibitors AMI-1, MS023, and C7280948. Thus, therapeutic targeting of PRMT1-mediated arginine methylation is a promising strategy for CRC treatment.
R251K mutation largely abolished arginine methylation of NONO by PRMT1, indicating that the R251 residue is critical for the oncogenic function of NONO in CRC. Pharmacological inhibition of PRMT1 blocked NONO arginine methylation and abrogated the oncogenic phenotypes associated with aDMA NONO. Interestingly, R251 is not located in the glycine- and arginine-rich motif, a consensus sequence for PRMTs [23]. Several proteins including signal transducer and activator of transcription (STAT)1 [39], peroxisome proliferator-activated receptor gamma coactivator (PGC)−1α [40], and runt-related transcription factor (RUNX)1 [41] are methylated by PRMTs at nonconsensus sites; meanwhile, other proteins containing a GAR motif such as TR3 interact with but are not a substrate of PRMT1 [42], suggesting that the motif is not be essential for PRMT1-mediated arginine methylation.
We previously reported that PRMTs may share substrates and have distinct roles in different tumor types. In CRC, methylation of R198/200 in the extracellular domain of EGFR by PRMT1 enhanced receptor dimerization and cell proliferation, resulting in tumor cell resistance to the anti-EGFR monoclonal antibody cetuximab [26]. However, in breast cancer cells, abolishing PRMT5-mediated EGFR Arg1175 methylation increased EGF-induced extracellular signal-regulated kinase (ERK) activation by preventing the recruitment of Src homology 2 domain-containing protein tyrosine phosphatase (SHP)1 to EGFR, leading to enhanced cell proliferation, migration, and invasion of EGFR-overexpressing tumor cells [43]. In the present work, we found that PRMT1 but not other PRMTs was overexpressed in CRC tissue relative to adjacent normal tissue, and catalyzed arginine methylation of NONO. PRMT3 and PRMT4 also catalyzed arginine methylation of NONO, but at residues other than R251 (Supplementary Fig. S5). A previous study reported that NONO was methylated by PRMT4 at R357, R365, and R378 [19]. Therefore, NONO R251 may be an arginine residue specifically methylated by PRMT1. Notably, PRMT1 knockdown enhanced the antitumor efficacy of cetuximab in both KRAS mutant and WT CRC cells, suggesting that targeting PRMT1 may sensitize CRC cells to cetuximab regardless of KRAS mutation status [26]. We also found that PRMT1 enhanced CRC cell proliferation and metastasis via an EGFR-independent mechanism of asymmetric dimethylation of NONO at R251 that was independent of the presence of KRAS mutation, as the effect was observed in both KM12 cells expressing WT KRAS and KRAS-mutant HCT8 cells.
Arginine methylation regulates signal transduction cascades [44]. In order to elucidate the mechanism underlying the arginine methylation-mediated oncogenic function of NONO, we carried out RNA sequencing analysis of CRC cells expressing WT and R251K-mutant NONO. The results indicated that the cGMP-dependent protein kinase or protein kinase G and cAMP pathways are potential targets of arginine-methylated NONO (Supplementary Fig. S6a–c). NONO is required for cAMP-dependent activation of CREB target genes [45], and NONO-mediated cAMP signaling is involved in tumor growth [15, 46] or metastasis [14]. In this context, our current findings imply that aDMA R251 is responsible for the tumor-enhancing effects of NONO in CRC, including increased cell migration and invasion, which are independent of classic epithelial-to-mesenchymal transition signaling pathways (Supplementary Fig. S6d, e) but may be mediated by cAMP signaling.
In summary, we demonstrated that PRMT1-mediated asymmetric dimethylation of NONO at R251 promotes CRC progression by enhancing tumor cell proliferation, migration, and invasion. These results imply that small-molecule inhibitors of PRMT1 have clinical potential for the treatment of advanced CRC.
Materials and methods
More details are provided in the Supporting Information.
Tissue specimens
Four sets of human CRC samples were used in this study. NONO, PRMT1, PRMT3, PRMT4, PRMT5, PRMT6, and PRMT8 mRNA and protein expression in tumor and adjacent normal tissues was evaluated in cohort I (28 CRC patients) and cohort II (29 CRC patients), respectively, by quantitative (q)PCR and western blotting, respectively. Tumor specimens from cohort III (93 locally advanced CRC patients) and cohort IV (97 locally advanced CRC patients) were used to analyze the correlation between clinical outcome and NONO and PRMT1 expression detected by immunohistochemistry. Locally advanced CRC was defined as T3–4/N + M0 (stage II/III) according to the American Joint Committee on Cancer TNM Classification System (8th Edition) [47], and was diagnosed based on retrospective review of contrast-enhanced pelvic magnetic resonance imaging. CRC patients with distant metastasis were excluded. All cases received standard treatment of neoadjuvant chemoradiotherapy followed by radical surgery and adjuvant chemotherapy. Tissue samples were obtained from the Tissue Bank of the Sixth Affiliated Hospital of Sun Yat-sen University with approval from the Human Medical Ethics Committee of Sun Yat-sen University and with written informed consent from all patients for the use of their data. Clinicopathologic parameters and follow-up information were obtained from the Follow-up Database of the Sixth Affiliated Hospital of Sun Yat-sen University.
Plasmid construction and lentiviral infection
Human Flag-NONO, Myc-NONO, HA-PRMT1, Flag-PRMT1 and its truncated mutants (1–343 aa, 32–343 aa, and 32–353 aa) were cloned into pCDH-CMV for transient transfection. Using the pCDH-CMV-Flag-NONO as a template, Flag-NONO R-to-K mutants (R251K, R287K, R364K, R383K, R251/287K, R364/383K, 3RK, 4RK) were developed using mutagenesis kit (cat. no. SMK-101; Toyobo, Osaka, Japan). Human PRMT1, NONO, NONO-R251K, and GAR were cloned into pGEX-6P-1 for protein purification.
To knockout endogenous NONO, LentiCRISPR-Dual-sgNONO plasmid was constructed by inserting a fragment containing sgRNA#1-scaRNA-pH1 (H1 promoter)-sgRNA#2 into lentiCRISPR vector (cat. no. 98290; Addgene, Watertown, MA, USA). To knockdown endogenous PRMT1, pLKO.1-shPRMT1 were constructed by inserting the shRNA of PRMT1 into pLKO.1 (cat. no. 8453; Addgene) according to the Addgene website protocol (http://www.addgene.org/protocols/plko/).
For virus production and infection, plasmids were cotransfected with psPAX2 (cat. no. 12260; Addgene) and pMD2.G (cat. no. 12259; Addgene) into HEK 293T cells for 48 h. The culture medium was collected, supplemented with polybrene (10 μg/ml) and incubated with KM12 and HCT8 cells for 48 h. Cells were selected with puromycin (2 μg/ml) to generate stable cell lines.
The sequences of all primers were listed in Supplementary Table S4.
RNA extraction and qPCR
Total RNA was isolated from tissues or cells using TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions, and reverse-transcribed to cDNA using qPCR RT Master Mix (cat. no. FSQ-301; Takara, Shiga, Japan). qPCR analyses were performed using SYBR Green (cat. no. QPK-201; Takara) on a LightCycler 480 instrument (Roche, Basel, Switzerland). Gene expression was normalized to U6 for tissues and GAPDH for cells. The primer sequences used in the present study were listed in Supplementary Table S4.
Western blotting assay
Western blotting assay was performed as described previously [38]. The following antibodies were used: Flag (cat. no. ab49763; abcam, Cambridge, MA, USA), HA (cat. no. ab1265; abcam), Myc (cat. no. ab9106; abcam), GAPDH (cat. no. 60004–1-Ig; Proteintech, Chicago, IL, USA), NONO (cat. no. 611279; BD Bioscience, Franklin lakes, NJ, USA), PRMT1 (cat. no. 2449; Cell Signaling Technology, CST, Beverly, MA, USA), PRMT5 (cat. no. D160716; Sangon Biotech, Shanghai, China), pan-ADMA (cat. no. 13522; CST), pan-MMA (cat. no. 8015; CST). Images were captured with ChemiDocTM Imaging System (Bio-Rad, Hercules, CA, USA), and the band intensity were analyzed using Image J software (National Institutes of Health, Bethesda, MD, USA).
IHC staining
IHC for NONO (1:1500; cat. no. 611279; BD Bioscience) and PRMT1 (1:1000; cat. no. 11279–1-AP; Proteintech) were performed on CRC tissue. The IHC score ranging from 0 to 3 according to the ToGA trial [48] was used to determine the intensity and the percentage of stained cancer cells. According to IHC score, sections were ranked into two groups: low expression (H score was 0 and 1) and high expression (H score was 2 and 3).
Cell counting, migration, and invasion assays
Cell counting assay was used to analyze the proliferation of cells. KM12 (1~2 × 104) or HCT8 (1.8~4 × 104) cells were seeded in 24-well plates with triplicate. Cell numbers were counted every two days.
Wound-healing assay was used to examine the migration capacity of tumor cells in vitro. KM12 (8 × 104) or HCT8 (1 × 105) cells were cultured in 2 well culture-inserts (cat. no. 80209; iBidi, Martin Reid, Germany) overnight for wound generating. The wounds were captured every 12 h with microscope (Olympus, Tokyo, Japan) and were measured by Adobe Photoshop CC 2017 (San Jose, CA, USA).
Transwell assay was performed to analyze the migration and invasive capacity of tumor cells in Cell Culture Inserts of 24 well with 8.0 μm pore size (Falcon, BD Bioscience). For migration assay, KM12 (4 × 104) or HCT8 (8 × 104) cells in 100 μl serum-free medium were seeded in the top chamber. Medium containing 10% FBS was used as a chemoattractant in the lower chamber. For inhibitor treatment, 0.6–1.2 mM AMI-1 (cat. no. 7884), 10 μM MS023 (S8112), 40 μM C7280948 (cat. no. S6737; all inhibitors form Selleck chemicals, Houston, Texas, USA) or equal volume of Dimethyl sulfoxide (DMSO, cat. no. D8418; Sigma-Aldrich, St. Louis, MO, USA) was added into the top chamber. After incubation for 24 h, cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet (cat. no. 0424A17; Leagene, Beijing, China). Cells were captured in five random fields with microscope (Olympus). For invasion assay, the membranes of inserts were coated with Matrigel (1:20, cat. no. 356234; Corning, NY, USA) to form matrix barriers before seeding cells.
Duolink PLA
aDMA NONO and the interaction between NONO and PRMT1 were detected using Duolink PLA kits (cat. no. DUO92014 and DUO92008; Sigma-Aldrich) according to the manufacturer’s instructions. The primary antibodies used for the assay were the same as those used for immunofluorescence analysis. KM12 cells (4 × 104) or HCT8 cells (8 × 104) were seeded on slides and cultured for 48 h. After fixation and blocking, the slides were incubated with primary antibodies for 2 h at room temperature, followed by PLUS (cat. no. DUO92001) and MINUS (cat. no. DUO92005) PLA probes (both from Sigma-Aldrich) for 1 h at 37 °C. The slides were incubated in ligation solution for 30 min at 37 °C and in amplification solution for 100 min at 37 °C. After nuclear staining with 4′,6-diamidino-2-phenylindole, fluorescent foci representing methylated NONO or NONO/PRMT1 complex were visualized and imaged with a confocal microscope (model TCS-SP8; Leica, Wetzlar, Germany).
CoIP assay
Cells were resuspended in radioimmunoprecipitation assay (RIPA) lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 1% Nonidet P-40, and 1 mM EDTA) with protease and phosphatase inhibitor cocktails (cat. no. 04693132001 and 04906837001, respectively; Roche, Basel, Switzerland). After incubation for 30 min on ice, the cells were centrifuged at 13,000 g for 15 min at 4 °C to remove cellular debris. Proteins in the whole-cell lysate were quantified with a bicinchoninic acid assay kit (cat. no. 23225; Thermo Fisher Scientific).
For immunoprecipitation (IP) of Flag- or Myc-tagged proteins, cleared lysate was mixed with anti-Flag magnetic beads (cat. no. M8823; Sigma-Aldrich) or anti-Myc magnetic beads (cat. no. B26301; Bimake, Houston, TX, USA) for 2–4 h at 4 °C. For IP of endogenous NONO or PRMT1 protein, cleared lysate was incubated overnight at 4 °C with 0.5–2 μg of anti-NONO antibody (cat. no. 611279; BD Bioscience), anti-PRMT1 antibody (cat. no. 2449; CST), or isotype-matched IgG. Dynabeads Protein G (Invitrogen, Carlsbad, CA, USA) were then added, and the tube was rotated for 2–4 h at 4 °C. The beads were washed three times with RIPA buffer and incubated with 0.1 M glycine HCl (pH 3.1) to release immunoprecipitated proteins. The supernatant was neutralized with 1 M NaOH and analyzed by western blotting.
In vitro methylation assay
Substrate protein (purified GST-GAR, GST-NONO, or GST-NONO-R251K; 10 μg) was incubated with 1 μg of GST-PRMT1 and 0.6 mM nonradioactive SAM (cat. no. B9003S; New England Biolabs, Ipswich, MA, USA) in 30 μl of methyltransferase reaction buffer (20 mM Tris-HCl [pH 8.0], 200 mM NaCl, and 0.4 mM EDTA) for 1 h at 37 °C. The reaction was terminated by adding 5× SDS loading buffer and heating for 10 min at 100 °C. Samples were divided into two equal parts: one was used for detection of the methylation signal by western blotting, and the other was subjected to Coomassie blue staining as the loading control.
Mouse xenograft model
Female BALB/c nude mice (5 weeks old) were purchased from GemPharmatech (Jiangsu, China) and maintained in a specific pathogen-free room on a 12:12-h light/dark cycle, and were fed autoclaved chow and water. KM12/HCT8 WT or NONO KO cells (1 × 106) were resuspended in 100 μl of phosphate-buffered saline (PBS) and then subcutaneously injected into the right or left posterior flank of each mouse. The animals were sacrificed 3 weeks later and tumors were excised and weighted.
For AMI-1 treatment, 2 weeks after implantation, AMI-1 (0.5 mg in 100 μl PBS) or 100 μl PBS was intratumorally injected once a day for 7 days [49]. On day 8, animals were sacrificed and the tumors were weighted. Experiments involving animals were used randomly and approved by the Institutional Animal Care and Use Committee of the Sixth Affiliated Hospital of Sun Yat-sen University.
Statistical analysis
All data are shown as mean ± standard deviation of at least three independent experiments. Differences between two subgroups were analyzed with the unpaired t test using Prism v7.0 software (GraphPad Inc, San Diego, CA, USA). Differences in NONO, PRMT1, PRMT3, PRMT4, and PRMT5 mRNA and protein levels between paired tumor and adjacent normal tissues were compared with the paired t test using Prism v7.0. Overall survival was analyzed with the Kaplan–Meier method and the log-rank test using SPSS v25.0 software (SPSS Inc, Chicago, IL, USA). P < 0.05 was considered significant.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Sauer R, Liersch T, Merkel S, Fietkau R, Hohenberger W, Hess C, et al. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. J Clin Oncol. 2012;30:1926–33.
Dienstmann R, Salazar R, Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J Clin Oncol. 2015;33:1787–96.
Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9.
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl J Med. 2009;360:1408–17.
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Petti E, Buemi V, Zappone A, Schillaci O, Broccia PV, Dinami R, et al. SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nat Commun. 2019;10:1001.
Mircsof D, Langouët M, Rio M, Moutton S, Siquier-Pernet K, Bole-Feysot C, et al. Mutations in NONO lead to syndromic intellectual disability and inhibitory synaptic defects. Nat Neurosci. 2015;18:1731–6.
Jaafar L, Li Z, Li S, Dynan WS. SFPQ•NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 2017;45:1848–59.
Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, et al. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–63.
Feng P, Li L, Deng T, Liu Y, Ling N, Qiu S, et al. NONO and tumorigenesis: more than splicing. J Cell Mol Med. 2020;24:4368–76.
Zhu Z, Zhao X, Zhao L, Yang H, Liu L, Li J, et al. p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP-1A. Oncogene. 2016;35:1399–410.
Yu T, Zhao Y, Hu Z, Li J, Chu D, Zhang J, et al. MetaLnc9 facilitates lung cancer metastasis via a PGK1-activated AKT/mTOR pathway. Cancer Res. 2017;77:5782–94.
Chen Z, Li JL, Lin S, Cao C, Gimbrone NT, Yang R, et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Investig. 2016;126:2267–79.
Proteau A, Blier S, Albert AL, Lavoie SB, Traish AM, Vincent M. The multifunctional nuclear protein p54nrb is multiphosphorylated in mitosis and interacts with the mitotic regulator Pin1. J Mol Biol. 2005;346:1163–72.
Deshar R, Yoo W, Cho EB, Kim S, Yoon JB. RNF8 mediates NONO degradation following UV-induced DNA damage to properly terminate ATR-CHK1 checkpoint signaling. Nucleic Acids Res. 2019;47:762–78.
Alfano L, Caporaso A, Altieri A, Costa C, Forte IM, Iannuzzi CA, et al. NONO ubiquitination is mediated by FBW7 and GSK3 β via a degron lost upon chromosomal rearrangement in cancer. J Cell Physiol. 2018;233:4338–44.
Hu SB, Xiang JF, Li X, Xu Y, Xue W, Huang M, et al. Protein arginine methyltransferase CARM1 attenuates the paraspeckle-mediated nuclear retention of mRNAs containing IRAlus. Genes Dev. 2015;29:630–45.
Iino K, Mitobe Y, Ikeda K, Takayama KI, Suzuki T, Kawabata H, et al. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020;111:148–59.
Herrmann F, Lee J, Bedford MT, Fackelmayer FO. Dynamics of human protein arginine methyltransferase 1(PRMT1) in vivo. J Biol Chem. 2005;280:38005–10.
Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65:8–24.
Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50.
Greenblatt SM, Liu F, Nimer SD. Arginine methyltransferases in normal and malignant hematopoiesis. Exp Hematol. 2016;44:435–41.
Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. 2014;25:21–36.
Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Investig. 2015;125:4529–43.
Snijders AP, Hautbergue GM, Bloom A, Williamson JC, Minshull TC, Phillips HL, et al. Arginine methylation and citrullination of splicing factor proline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. RNA. 2015;21:347–59.
Li X, Wang C, Jiang H, Luo C. A patent review of arginine methyltransferase inhibitors (2010-8). Expert Opin Ther Pat. 2019;29:97–114.
Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, et al. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem Biol. 2016;11:772–81.
Luo M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol. 2012;7:443–63.
Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell. 2018;34:1012–.e1013.
Hu Z, Dong L, Li S, Li Z, Qian Y, Li Y, et al. Splicing Regulator p54 nrb/Non-POU Domain-Containing Octamer-Binding Protein Enhances Carcinogenesis Through Oncogenic Isoform Switch of MYC Box-Dependent Interacting Protein 1 in Hepatocellular Carcinoma. Hepatology. 2020;72:548–68.
Schiffner S, Zimara N, Schmid R, Bosserhoff AK. p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis. 2011;32:1176–82.
Ho TT, Huang J, Zhou N, Zhang Z, Koirala P, Zhou X, et al. Regulation of PCGEM1 by p54/nrb in prostate cancer. Sci Rep. 2016;6:34529.
Yamamoto R, Osawa T, Sasaki Y, Yamamoto S, Anai M, Izumi K, et al. Overexpression of p54(nrb)/NONO induces differential EPHA6 splicing and contributes to castration-resistant prostate cancer growth. Oncotarget. 2018;9:10510–24.
Li S, Kuhne WW, Kulharya A, Hudson FZ, Ha K, Cao Z, et al. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009;37:6746–53.
Tsofack SP, Garand C, Sereduk C, Chow D, Aziz M, Guay D, et al. NONO and RALY proteins are required for YB-1 oxaliplatin induced resistance in colon adenocarcinoma cell lines. Mol Cancer. 2011;10:145.
Alfano L, Costa C, Caporaso A, Altieri A, Indovina P, Macaluso M, et al. NONO regulates the intra-S-phase checkpoint in response to UV radiation. Oncogene. 2016;35:567–76.
Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–41.
Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005;19:1466–73.
Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008;22:640–53.
Lei NZ, Zhang XY, Chen HZ, Wang Y, Zhan YY, Zheng ZH, et al. A feedback regulatory loop between methyltransferase PRMT1 and orphan receptor TR3. Nucleic Acids Res. 2009;37:832–48.
Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13:174–81.
Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–72.
Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, et al. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc Natl Acad Sci USA. 2007;104:20314–9.
Qin Y, Chen W, Jiang G, Zhou L, Yang X, Li H, et al. Interfering MSN-NONO complex-activated CREB signaling serves as a therapeutic strategy for triple-negative breast cancer. Sci Adv. 2020;6:eaaw9960.
Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen YJ, Ciombor KK, et al. Rectal cancer, version 2.2018, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2018;16:874–901.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97.
Ansorena E, García-Trevijano ER, Martínez-Chantar ML, Huang ZZ, Chen L, Mato JM, et al. S-adenosylmethionine and methylthioadenosine are antiapoptotic in cultured rat hepatocytes but proapoptotic in human hepatoma cells. Hepatology. 2002;35:274–80.
Acknowledgements
This study was supported by the Guangdong Science and Technology Project (No. 2017B090901065, No. 2019B030316003), Natural Science Foundation of China (No. 81872188, No. 81902867, No. 81903152), Natural Science Foundation of Guangdong Province (No. 2019A1515010901, No. 2020A1515010314), and China Postdoctoral Science Foundation (No. 2019M663301).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, XK., Wang, YL., Wang, F. et al. PRMT1 enhances oncogenic arginine methylation of NONO in colorectal cancer. Oncogene 40, 1375–1389 (2021). https://doi.org/10.1038/s41388-020-01617-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01617-0
- Springer Nature Limited
This article is cited by
-
PRMT1 in human neoplasm: cancer biology and potential therapeutic target
Cell Communication and Signaling (2024)
-
The pleiotropic nature of NONO, a master regulator of essential biological pathways in cancers
Cancer Gene Therapy (2024)
-
PRMT1-mediated PGK1 arginine methylation promotes colorectal cancer glycolysis and tumorigenesis
Cell Death & Disease (2024)
-
LPCAT2 inhibits colorectal cancer progression via the PRMT1/SLC7A11 axis
Oncogene (2024)
-
PRMT6-CDC20 facilitates glioblastoma progression via the degradation of CDKN1B
Oncogene (2023)