
Abstract
Merkel cell carcinoma (MCC) is a highly invasive and metastatic skin cancer. While high expression of miR-375 is a characteristic of MCC, it seems not to contribute to the malignant phenotype of MCC cells. miR-375 enrichment in MCC-derived extracellular vesicles suggests its intercellular signaling function. Here, we demonstrate that horizontally transferred miR-375 causes fibroblast polarization toward cancer-associated fibroblasts (CAFs). The polarization is evidenced by phenotypic changes and induction of α-SMA, CXCL2, and IL-1β. Fibroblast polarization is inhibited by specific antagomirs and mimicked by experimental miR-375 expression. Mechanistically, miR-375 downregulates RBPJ and p53, two key players regulating fibroblast polarization. In clinical MCC samples, in situ hybridization located miR-375 in CAFs, which correlated with high α-SMA protein and low RBPJ and TP53 expression; single-cell RNAseq revealed a disparate fibroblast polarization negatively correlating with p53 pathway-related gene expression. Thus, the functional role of miR-375 in MCC is to generate a pro-tumorigenic microenvironment by inducing fibroblast polarization.
Similar content being viewed by others


Introduction
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine (NE) skin cancer with an estimated mortality rate of ~40% [1]. In most cases, MCC carcinogenesis is associated with the oncogenic Merkel cell polyomavirus (MCPyV), while in the other cases, it is UV-driven [1]. MCCs are heterogeneous tumors that are often infiltrated by various immune and stromal cells [2]. While the role of tumor-infiltrating lymphocytes has been studied in detail [3,4,5], the characteristics of tumor-associated fibroblasts in the MCC microenvironment are largely unexplored. Fibroblasts are very plastic cells that may contribute to tumor progression and metastasis depending on their polarization and activation states [6]. Hence, tumor-promoting fibroblasts are also termed cancer-associated fibroblasts (CAFs). CAFs are characterized by a spindle shape with elongated cytoplasmic processes [7], the expression of markers such as alpha-smooth muscle actin (α-SMA) [7, 8] and upregulation of pro-inflammatory genes such as C-X-C motif chemokine 2 (CXCL2) and interleukin 1 beta (IL-1β) [9, 10]. Fibroblast polarization is triggered via intrinsic and extrinsic mediators; however, this process is not fully understood [6]. Nevertheless, cancer cell-derived stimuli are assumed to be the major factors inducing CAF polarization. Their effect can be conveyed via direct cell-cell contact [11] or via soluble factors [12] or molecules that are mainly transported within extracellular vesicles (EVs), such as apoptotic blebs, microvesicles, or exosomes [13]. Exosomes are 30–130 nm in diameter, are actively released by the fusion of multivesicular endosomes with the plasma membrane and can stably persist in blood, lymph, and other bodily fluids [14]. Exosomes can contain a plethora of functional molecules, including proteins, DNAs, mRNAs, and non-coding RNAs. Thus, exosomes are important for endocrine and paracrine cell-cell communication [15]. microRNAs (miRNAs) are particularly common in exosomes because of their small size of 19-24 nucleotides. Notably, some miRNAs are selectively enriched in exosomes [16]; these miRNAs were classified as exosome-shuttle miRNAs [17]. The intercommunications between tumor and stromal cells via exosome-shuttle miRNAs impacts on tumor proliferation [18], angiogenesis [19, 20], metastasis [21, 22], immunity [23], as well as CAF activation [24, 25]. It should be noted, that the distinction of exosomes from other EVs is not trivial; thus, to be prudent we rather use the term EVs within this manuscript.
One of the most abundant miRNAs in MCC cells is miR-375 [26,27,28,29]. Its function in MCC has not yet been completely characterized, but it might promote NE differentiation of MCC cells [26]. Overexpression of truncated versions of the MCPyV-encoded large T antigen or the NE linage transcription factor ATOH1 induces miR-375 expression [30]. In addition to being expressed in MCC, miR-375 is expressed in medullary thyroid carcinoma [31], prostate cancer [32] and NE lung cancers [33]. The physiological function of miR-375 is best described by its role in pancreatic beta cells, in which it is an important regulator of insulin secretion [34]. miRNAs function by interfering with the stability of mRNA or inhibiting its translation; experimentally confirmed target genes of miR-375 are recombination signal binding protein for immunoglobulin kappa J region (RBPJ) and TP53 [26, 35]. In most cancers, miR-375 has been deemed a tumor suppressor [36], but it also functions as an oncogenic miRNA depending on the cellular context [32, 33]. We recently reported that knockdown of miR-375 in MCC cell lines has—at best—minimal effects on cell survival, proliferation and morphology [37]. Highly efficient knockdown did not alter any of the signaling pathways involving miR-375 target genes [37], thus posing the question about the functional role of this highly abundant miRNA in MCC. Based on our observation that miR-375 is present in MCC conditioned cell culture medium as well as sera of MCC patients [27], it may function as an exosome-shuttle miRNA. Here, we provide evidence that horizontally transferred miR-375 is important for the polarization of fibroblasts toward a CAF phenotype in MCC.
Results
Fibroblasts in MCC tumors exhibit a CAF-like phenotype
Based on our previous results [27, 37], we suggested that miR-375 acts in the intercellular signaling process via exosomal shuttling and can polarize stromal cells in the tumor microenvironment. Thus, we characterized fibroblasts in the MCC microenvironment by evaluating the expression of other fibroblast markers such as TE-7, Caveolin-1 (CAV1), α-SMA, and S100A4, using multiplex immunohistochemistry (mIHC) staining (n = 10, Supplementary Tables S1 and S2, example results shown in Fig. 1a and Supplementary Fig. S1). TE-7 was expressed at low levels in most stromal cells, with a few exceptions. α-SMA was expressed by fibroblast-like cells and pericytes in the vicinity of blood vessels and was often colocalized with CAV1. Unlike α-SMA, S100A4 was expressed mainly in single cells distributed throughout the tumor, with only some degree of co-localization with α-SMA. Notably, we did not observe obvious differences with respect to the fibroblast polarization depending on the presence or absence of MCPyV in the tumor. These morphological analyses demonstrate that MCC-associated fibroblasts are heterogeneous, although most exhibit a CAF-like phenotype.

a FFPE sections of MCC tissues were stained for the fibroblast markers TE-7 (red), Caveolin1 (yellow), α-SMA (green), and S100A4 (orange), as well as for the MCC tumor marker CK20 (light blue). Nuclei were stained with DAPI (dark blue). A representative MCC tissue is depicted. The scale bar represents 50 µm. b Fibroblasts visualized in a UMAP plot. Cells were annotated by mRNA expression profiles for known marker genes in three MCC tumors. c A CAF signature score was generated for fibroblasts in three tumors based on the mRNA expression of selected CAF marker genes (details are given in the “Material and Methods” section).
Next, we performed single-cell RNA sequencing (scRNAseq) of three MCC tumors using 10x Genomics Chromium. T-distributed stochastic neighbor embedding (tSNE) clustered these cells into MCC tumor cells, fibroblasts, T lymphocytes, and endothelial cells (data not shown). Notably, tSNE separated the MCC-associated fibroblasts into two clusters based on their expression profiles in all three tumors (Fig. 1b). Most of the fibroblasts expressed well-established CAF markers, such as fibroblast activation protein; Thy-1 cell surface antigen; smooth muscle actin alpha 2 (α-SMA encoded by ACTA2); Caveolin 1 (CAV1); and different collagenases. However, these markers were expressed at different levels among the fibroblasts (Supplementary Fig. S2a). Thus, we generated a CAF score based on the expression profile of 88 CAF-associated genes that also identified two clusters of fibroblasts in all three tumors (Fig. 1c, Supplementary Table S3) [38]. In addition, combined fibroblast clusters from three tumors identified above were also classified into CAFlow and CAFhigh groups according to their respective CAF score values (Supplementary Fig. S2b, c). To determine whether these two groups of fibroblasts in each tumor were independently developed or just at different stages of CAF polarization, we calculated the RNA velocity, which predicts the future state of individual cells on a time scale of hours based on the relative ratio of spliced and unspliced transcripts [39]. This analysis revealed that these two fibroblast clusters in all three tumors were directed toward different future states, as indicated by the RNA velocity vector visualized in by a principal component analysis (PCA) plot (Supplementary Fig. S2d). In summary, MCC-associated fibroblasts showed substantial heterogeneity and different polarization states, as revealed by the results of both immunohistochemistry-based staining and scRNAseq. On that basis, we next investigated whether these effects are triggered by horizontal transfer of miR-375.
miR-375 is enriched in MCC-derived EVs and transferred into fibroblasts
We confirmed the abundance of miR-375 and its function as an exosome-shuttle miRNA in MCC cell lines by using quantitative real-time polymerase chain reaction (RT-qPCR) to compare the abundances of previously reported highly expressed miRNAs (miR-19b, miR-106b, miR-375, miR-200c, and miR-182) in WaGa cells and in conditioned medium (CM) from WaGa cells (Fig. 2a, b) [27, 28]. While all tested miRNAs were abundant in the cell lysate, only miR-375 was present in relevant amounts in the CM. The strong predominance of miR-375 in the cell-free supernatant suggests the active and selective release of miR-375 by WaGa cells, presumably via exosomal enrichment [40]. To test this hypothesis, we isolated EVs from the CM of classical miR-375-expressing MCC cell lines (WaGa, PeTa, MKL-1, and UM-MCC-13); these EVs were typically sized between 50 and 100 nm (Fig. 2c), and positive for CD63 and Tsg101, two common EV markers, and negative for Calnexin, a marker for the endoplasmic reticulum as identified by immunoblot (Fig. 2d). miR-375 was strongly enriched in these EVs compared to the corresponding CM but it could not be detected in EVs derived from the miR-375-negative cell lines MCC13 and SCL-2 (Fig. 2e). The MCC-derived EVs were stained with Exo-Red to investigate whether they could be taken up by fibroblasts (Fig. 2f). MRC-5 fibroblasts and Fibro1.12 primary skin fibroblasts uniformly incorporated the labeled EVs within 3 h (Fig. 2g). These observations strongly support the hypothesis that horizontal transfer of miR-375 from MCC cells to fibroblasts contributes to intercellular communication.

The expression of miR-375, miR-182, miR-106b, miR-19b and miR-200c in WaGa cells (a) and CM from WaGa cells (b) was determined by RT-qPCR. The amplification curves and normalized relative expression level of each miRNA are shown. c EVs were isolated from WaGa CM. The sizes were determined by comparison to nanobeads of a defined size (50, 100, and 200 nm). d Expression of CD63, Tsg101 and Calnexin in WaGa cells and EVs derived from WaGa CMs were determined via immunoblot. e Relative miR-375 expression in EVs derived from four classical MCC cell lines (WaGa, PeTa, MKL-1, UM-MCC13) and a variant MCC cell line (MCC13) and SCL-2 cutaneous squamous cell carcinoma cells served as the control. Cq values were normalized to spiked-in cel-miR-39. f EVs isolated from WaGa cells were stained with Exo-Red; unstained EVs served as negative controls. g EGFP-expressing MRC-5 cells (left panels) or primary skin fibroblasts (Fibro.1.12, right panels) were cultured alone or in the presence of Exo-Red-labeled WaGa cell-derived EVs. Representative overlay images are shown; the scale bar represents 10 µm. Experiments were biologically replicated trice and were performed in triplicates. The error bars indicate the SDs; *** indicates p < 0.001.
MCC-derived factors polarize fibroblasts toward a CAF phenotype
To explore the functional impact of MCC-derived factors on fibroblasts, we performed a series of experiments co-culturing miR-375 expressing WaGa, PeTa cells, or miR-375-negative MCC13 cells with fibroblasts. Three different coculture conditions were tested: with direct cell-cell contact (DC), in a Transwell chamber system (TC) and in MCC CM. For all subsequent experiments, we used both the fibroblast cell line MRC-5 and primary fibroblasts. Although primary skin fibroblasts are biologically more relevant, they are characterized by a limited expansion capacity; thus, primary skin fibroblasts from different donors had to be used to ensure both reproducibility and biological relevance. Due to the different growth patterns of fibroblasts (adherent) and classical MCC cells (in suspension), they can be easily separated before subsequent analysis. However, the variant MCC cell line MCC13 does not exhibit the classical MCC cell growth pattern, and therefore, this cell line was not used in the DC experiments. The first discovery from these experiments was that either culture condition caused the new appearance of miR-375 in both MRC-5 and primary fibroblasts (Fig. 3a, c). The amount of detected miRNA varied substantially, with the largest amount under DC and the lowest under CM conditions (Fig. 3a, c). Since neither of the fibroblast lines expressed miR-375 when cultured alone, and none of the culture conditions induced pri-miR-375 (Fig. 3b, d), miR-375 expression was not induced in fibroblasts; rather, miR-375 was transferred from MCC cells to fibroblasts.

MRC-5 cells (a, b, e, f, i, k, m) or primary skin fibroblasts (Fibro1.4) (c, d, g, h, j, l, n) were cultured in MCC conditioned medium (CM), in a Transwell chamber system (TC) or in direct cell-cell contact (DC) with MCC13, WaGa or PeTa cells. a- d The relative expression levels of miR-375 (a, c) and pri-miR-375 (b, d) in fibroblasts were determined by RT-qPCR. e–h The relative ACTA2, CXCL-2, and IL1B mRNA expression levels were determined in fibroblasts under TC conditions (e, g) or DC conditions (f, h) by RT-qPCR. i, j α-SMA protein expression in fibroblasts cocultured with MCC cells was determined by immunoblotting; β-tubulin served as loading control. IF staining of α-SMA (green) in MRC-5 (k) and Fibro1.4 cells (l) after direct coculture with MCC cells. Cellular membranes were stained with WGA (red); nuclei, with DAPI (blue). The graphs show the values of cell aspect ratio (length/ width) of MRC-5 (m) and Fibro1.4 (n) under indicated conditions (n = 50), presented as box-and-whisker plots. The scale bars represent 50 µm. For RT-qPCR experiments, Cq values were normalized to U6 or HPRT expression and compared to the ΔCq value of untreated MRC-5 or Fibro1.4 cells, respectively. Experiments were biologically replicated trice and were performed in triplicates. The error bars indicate the SDs; * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001.
Next, we analyzed the functional effects of the EVs’ cargo transferred from MCC cells, including miR-375, on fibroblasts by measuring the mRNA expression of CAF markers, i.e., ACTA2, CXCL2 and IL1B. Consistent with the highest miR-375 uptake, CAF markers, especially ACTA2, also exhibited highest expression in fibroblasts under DC conditions (Fig. 3e to h). In addition, α-SMA expression was induced at the protein level, as evidenced by immunoblotting of cell lysates and by immunofluorescence (IF) staining (Fig. 3i–j). The latter assay also revealed substantial morphological changes consistent with a CAF-like phenotype in fibroblasts, such as the development of an elongated, stellate shape, under coculture conditions (Fig. 3k, l). It was previously described that fibroblast polarization is characterized by change in the cell’s aspect ratio [41]. Indeed, quantification of the cell aspect ratio under the respective culture condition confirmed such a change which was most prominent for both MRC-5 cells and primary fibroblasts if direct cell-cell contact was possible (Fig. 3m, n).
miR-375 alone causes fibroblast polarization
Since many factors transferred from MCC cells may cause fibroblast polarization, we next evaluated the extent to which miR-375 causes the observed effect. Thus, we experimentally induced miR-375 in fibroblasts (Fig. 4a, c). Ectopic expression of miR-375 resulted in increased mRNA expression of CXCL2, IL1B and, predominantly, ACTA2 (Fig. 4b, d). α-SMA protein expression was also induced upon ectopic expression of miR-375, as determined by immunoblotting (Fig. 4e, f) and IF staining (Fig. 4g, h). In addition, the morphology of these fibroblasts changed toward a CAF phenotype but to a lesser extent than in fibroblasts cocultured with miR-375-expressing MCC cells (Fig. 4g, h; Supplementary Fig. S3). Thus, these results provide conclusive evidence that miR-375 is sufficient for fibroblast polarization.

miR-375 was overexpressed in MRC-5 cells (a, b, e, g, i, j, m) or primary skin fibroblasts (Fibro1.12) (c, d, f, h, k, l, n). a, c: The relative miR-375 expression levels in untreated, miR negative control (NC)-transfected, and miR-375 mimic-transfected fibroblasts were determined by RT-qPCR. b, d The relative ACTA2, CXCL-2 and IL1B mRNA expression levels in untreated, miR-NC-transfected and miR-375 mimic-transfected fibroblasts were determined by RT-qPCR. e, f α-SMA protein expression in untreated, miR-NC-transfected and miR-375 mimic-transfected fibroblasts was determined by immunoblotting; β-tubulin served as loading control. Quantification was performed using ImageJ. g, h IF staining of α-SMA (green) in untreated, miR-NC-transfected and miR-375 mimic-transfected fibroblasts. Cellular membranes were stained with WGA (red) and nuclei with DAPI (blue). Scale bars represent 50 µm. i- l The relative TP53 and RBPJ mRNA expression levels in untreated, miR-NC-transfected and miR-375 mimic-transfected fibroblasts were determined by RT-qPCR. m, n p53 and RBPJ protein expression in untreated, miR-NC-transfected and miR-375 mimic-transfected fibroblasts was determined by immunoblotting; β-tubulin served as loading control. Quantification was performed using ImageJ. For RT-qPCR experiments, Cq values were normalized to U6 or HPRT expression and compared to the ΔCq value of untreated MRC-5 or Fibro1.4 cells, respectively. Experiments were biologically replicated trice and were performed in triplicates. The error bars indicate the SDs; * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001.
Most miRNAs function by interfering with the stability of mRNA or inhibiting its translation; RBPJ and TP53 are target genes of miR-375 [26, 35] and for both reduced expression is associated with fibroblast polarization [42,43,44]. Here, we indeed observed significant downregulation of RBPJ and TP53 mRNA and protein expression upon experimental induction of miR-375 expression (Fig. 4i–n). These observations indicate that miR-375-mediated inhibition of RBPJ and p53 is at least one of the mechanisms causing MCC-induced fibroblast polarization.
miR-375 antagomirs attenuate MCC-induced fibroblast polarization
To confirm the relevance of miR-375 for MCC-induced fibroblast polarization, we repeated our coculture experiments after introduction of miR-375 antagomirs either into MCC cells or fibroblasts. The miR-375 antagomirs in MCC cells dramatically reduced the endogenous miR-375 expression causing an equally reduced content of miR-375 in corresponding CMs and isolated EVs (Supplementary Fig. S4a–c). Thus, introduction of miR-375 antagomirs into MCC cells, reduced their impact on transferring miR-375 to fibroblasts by either DC or TC conditions, which was associated with reduced induction of α-SMA protein and ACTA2, CXCL2 and IL1B mRNA in the cocultured fibroblasts; the change in protein and mRNA expression was more pronounced under DC (Fig. 5a–f) as compared to TC conditions (Supplementary Fig. S5a–f). The observed effects of the miR-375 knockdown in MCC cells on their capacity of CAF polarization was directly explained by their reduced impact on RBPJ and TP53 mRNA and protein in fibroblasts after coculture which was again more evident under DC (Fig. 5g–j) than TC conditions (Supplementary Fig. S5g–j).

MRC-5 cells (a, b, e, g, i) or Fibro 1.12 primary skin fibroblasts (c, d, f, h, j) were directly cocultured with WaGa or PeTa cells transfected with miR-375 antagomirs for 72 h. a, c The relative miR-375 expression level in fibroblasts under the indicated conditions was determined by RT-qPCR. b, d α-SMA protein expression in fibroblasts under the indicated conditions was determined by immunoblotting; β-tubulin served as loading control. e, f The relative ACTA2, CXCL2 and IL1B expression level in fibroblasts under the indicated conditions was determined by RT-qPCR. g, h Relative expression of TP53 mRNA (left) and protein (right) level in fibroblasts under the indicated conditions was determined by RT-qPCR and immunoblotting. i, j Relative expression of RBPJ mRNA (left) and protein (right) level in fibroblasts under the indicated conditions was determined by RT-qPCR and immunoblotting. For all RT-qPCR experiments, Cq values were normalized to HPRT mRNA expression and compared to the ΔCq of the corresponding untreated MRC-5 or Fibro.1.12 cells transfected. Experiments were biologically replicated trice and were performed in triplicates. The error bars indicate the SDs; * indicates p < 0.05, ** indicates p < 0.01.
The introduction of the miR-375 antagomirs into fibroblasts substantially reduced the detectable amount of miR-375 transferred from MCC cells under TC or DC conditions (Supplementary Fig. S4d, e). However, the defang capacity of the antagomirs was less efficient; indeed, even in the presence of the antagomirs amounts of miR-375 detectable in fibroblasts under DC conditions exceeded the amounts observed under TC conditions in the absence of the antagomirs. Accordingly, the reversal of the coculture induced fibroblast polarization by antagomir expression in the fibroblasts was less striking (data not shown). Still, the induction of ACTA2 mRNA and α-SMA protein expression under TC conditions was significantly reduced in the presence of the antagomirs (Fig. 6a–d). Again, expression of RBPJ and TP53 mRNA was partially rescued by the miR-375 antagomirs (Fig. 6e–h).

MRC-5 cells (a, c, e, g) or Fibro 1.12 primary skin fibroblasts (b, d, f, h) were transfected with miR-375 antagomirs or control miRNA prior to coculture with WaGa or PeTa cells in a Transwell culture system. a, b: The relative ACTA2 expression level in fibroblasts under the indicated conditions was determined by RT-qPCR. c, d: α-SMA protein expression in fibroblasts under the indicated conditions was determined by immunoblotting; β-tubulin served as loading control. e- h: The relative TP53 and RBPJ mRNA expression levels in fibroblasts under the indicated conditions were determined by RT-qPCR. For all RT-qPCR experiments, Cq values were normalized to HPRT mRNA expression and compared to the ΔCq of the corresponding untreated MRC-5 or Fibro.1.12 cells transfected with antagomir control (anta-con). Experiments were biologically replicated trice and were performed in triplicates. The error bars indicate the SDs; * indicates p < 0.05, ** indicates p < 0.01.
Presence of miR-375 in tumor cells and CAFs from MCC tumor tissue in situ
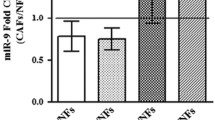
To translate our in vitro observations into the clinical setting, we performed in situ hybridization (ISH) with a miR-375 probe on a series of MCC tumor samples (n = 6, Supplementary Table S1). miR-375 was predominantly expressed in MCC cells but was also present in stromal cells with a fibroblast morphology (Fig. 7a, b; fibroblast-like stromal cells are indicated by arrows). The miR-375-positive stromal cells were characterized by the same morphological appearance and distribution pattern as the α-SMA-expressing cells. Thus, we established a relative α-SMA staining score in 20 MCC lesions and correlated this score with miR-375 expression as determined by RT-qPCR, demonstrating a significant positive correlation (Fig. 7c, d). Furthermore, the expression of RBPJ and TP53 was negatively correlated with miR-375 expression (Fig. 7e, f). Gene set enrichment analysis (GSEA) of the scRNAseq data for a primary MCC described above (Fig. 1b; Supplementary Fig. S2c) comparing the CAFhigh and CAFlow fibroblast clusters based on the established CAF score demonstrated that the p53-related pathway is more strongly inhibited in CAFhigh fibroblasts than in CAFlow fibroblasts (Fig. 7g). These in situ and ex vivo findings strongly support the idea that the horizontal transfer of miR-375 from MCC cells to fibroblasts triggers the polarization of the latter toward a CAF phonotype via the downregulation of RBPJ and p53.

a FFPE MCC tumor tissues were hybridized with a scrambled control probe (upper) or a miR-375-specific probe (lower). Whole slide scans of one representative MCC tissue are shown. b Two representative tumor areas are shown at high resolution to demonstrate the presence of miR-375 in fibroblasts (indicated by the arrows); the scale bars represent 10 µm. c MCC tissues stained for α-SMA and other fibroblast and MCC markers (same as Fig. 1a), and one representative MCC tissue is shown for α-SMA expression pattern. Spearman rank correlations of α-SMA staining scores (d), RBPJ (e) and TP53 (f) mRNA expression levels with the miR-375 expression levels. Statistical analyses were performed in R using the ggpubr package. g GSEA was performed using scRNAseq data from two fibroblast clusters, and p53-related signaling was revealed.
Discussion
In solid cancers, stromal cells have an impact at all stages of cancer progression. However, to date, fibroblasts in MCC have not been examined in detail. Here, we reveal the heterogeneity of fibroblasts in the stroma of MCC lesions, which reflects a spectrum of polarization toward a CAF phenotype. In most cancers, fibroblast polarization is triggered by tumor-derived factors such as TGF-β, PDGF, or IL-6 [6], which exhibit only low or no expression in classical MCC cell lines and tumors (data not shown). Thus, questions regarding alternative mechanisms of fibroblast polarization in MCCs are raised.
Regarding these questions, horizontal transfer of miRNAs contained within EVs has been reported to promote functional and metabolic reprogramming of fibroblasts [45, 46]. Here, we demonstrate that miR-375 transferred via EVs from MCC cells to fibroblasts targets RBPJ and p53, causing downregulation of their expression, which in turn is associated with fibroblast polarization. RBPJ, also called CSL, is a Notch mediator that suppresses CAF polarization [43, 44]. p53 downregulation allows fibroblasts to overcome cell senescence induced by RBPJ downregulation and thus enhances CAF polarization [42, 44]. Both RBPJ and TP53 have been reported to be target genes of miR-375 [26, 35], and this hypothesis was confirmed by our study: Experimental expression of miR-375 reduced the expression of RBPJ and p53, which was rescued by specific antagomirs. Indeed, miR-375 expression was inversely correlated with RBPJ and TP53 expression in MCC tumor tissues. Unfortunately, the mRNA expression of RBPJ and TP53 in fibroblasts from MCC tumors was below the detection limit of scRNAseq. However, p53-related signaling pathways were strongly inhibited in fibroblasts with a high CAF score.
Different MCC/fibroblast coculture conditions, i.e., CM, TC, and DC, resulted in increasing effects on CAF polarization in that order. This phenomenon can be explained by the amount of miR-375 transferred to fibroblasts via EVs. In addition, direct coculture may also allow transfer through tunneling nanotubes between tumor cells and fibroblasts [47]. In addition to miR-375, other factors horizontally transferred by EVs from MCC cells to fibroblasts may induce fibroblast polarization. Secretome analysis of MCC-derived exosomes revealed more than 160 proteins [48]. Hence, importantly, ectopic expression of miR-375 alone was sufficient to polarize fibroblasts, and miR-375 antagomirs decreased phenotypic changes in fibroblasts triggered by coculture with MCC cells. Thus, our findings indicate that miR-375 plays a major role in MCC-mediated CAF polarization.
We further demonstrated the presence of miR-375 in stromal cells with morphological characteristics of fibroblasts by in situ hybridization. Quantitative differences in miR-375 levels might be a cause of CAF heterogeneity in MCC, as evidenced by the mIHC-based staining and scRNAseq results. However, different cellular origins, e.g., tissue-resident fibroblasts or recruited mesodermal stem cells, prior activation stages or additional environmental factors may contribute to the fibroblast heterogeneity in MCC [6, 49]. The importance of CAF heterogeneity in cancer progression, prognosis, and therapy has been demonstrated in many caner types. Because of their heterogeneity, there is no single exclusive marker for CAFs, but—as demonstrated here—a set of markers consisting of morphologic characteristics together with specific gene and protein expression profiles is needed to characterize this cell type. Consequently, using a single marker for isolation and/or enrichment of CAFs hold the risk of selecting of a subpopulation of cells not reflecting the complete diversity of CAFs. Although we demonstrated the uptake of EVs by fibroblasts, other stromal cells, such as lymphocytes, macrophages and endothelial cells in the microenvironment, are also likely to be affected. As reported previously, mesenchymal stromal cell-derived exosomes altered the mRNA expression and function of B lymphocytes [50] and endothelial cells [51]. Notably, transferred miR-375 enhanced tumor-associated macrophage migration and infiltration into tumor spheroids in breast cancer [52].
In summary, we established the role of miR-375 in intercellular communication between MCC tumor cells and stromal fibroblasts. We demonstrated that miR-375 derived from MCC cells or endogenously expressed miR-375 can induce fibroblast polarization in vitro and that this event probably also occurs in vivo in MCC patients. These observations suggest that miR-375 is an attractive target for therapeutic interventions; indeed, the therapeutic feasibility of targeting miRNA is currently being investigated for miR-155 in cutaneous T cell lymphoma [53].
Material and methods
Tumor tissues
Formalin-fixed, paraffin-embedded (FFPE) MCC tumor samples from the Department of Dermatology, University Hospital Essen were used for immunohistochemistry (IHC), IF staining and miRNA in situ hybridization. One fresh primary MCC tumor was used for scRNAseq analysis. All tumor samples were excised for diagnostic or therapeutic reasons and were confirmed to be MCC according to established histological and immune-histochemical diagnostic guidelines. The study was conducted in accordance with ethical guideline provided in the ‘Declaration of Helsinki’. All studies on human material were approved by the ethics committee of the University Duisburg-Essen (11-4715; 17-7538-BO). Informed consent was obtained for use in research.
Multiplex immunohistochemistry (mIHC)
mIHC was performed using an Opal 7-color kit (PerkinElmer, Velbert, Germany) according to the manufacturer’s instructions. In brief, FFPE tissue sections were incubated at 60 °C for 1 h, deparaffinized in xylene and rehydrated with 100%, 96% and 70% ethanol for 2 min each. Sections were then cross-linked to slides by incubation in 10% formalin for 10 min before two 5-min washes in distilled water. Before incubation with each antibody, antigen retrieval was performed by microwaving the sections at 100 W in a suitable Opal antigen retrieval buffer with a pH of 9 (AR9) or a pH of 6 (AR6) for 15 min. After cooling at room temperature in the dark for 20 min, sections were blocked with antibody blocking solution for 10 min before incubation for 30 min at room temperature with the appropriate primary antibody (Supplementary Table S2). After three washes in TBS+ 0.05% Tween 20 (TBST), sections were incubated with Opal Polymer HRP Mouse and Rabbit reagent for 10 min at room temperature. After three washes in TBST, the appropriate Opal fluorophore was added at a dilution of 1:50 and incubated for 10 min at room temperature. After three washes in TBST, the process was repeated for reaction with the next antibody. The order in which the antibodies were added to the sections is summarized in Supplementary Table S2. After the final antibody incubation, nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min, and sections were embedded in ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, Oberhausen, Germany) and incubated at room temperature in the dark for 24 h before imaging with a Mantra quantitative pathology workstation (PerkinElmer).
Multiplex scRNAseq and data analysis
Tissue sections of in total three samples (MCC_1, MCC_2, MCC_3) were dissociated into single-cell suspensions and barcoded using the 10x Genomics Chromium v2.0 platform (10x Genomics, Leiden, Netherland) [54]. Library preparation was performed according to the 10x Genomics protocol, and the library was sequenced on an Illumina HiSeq 4000 platform (Illumina, Eindhoven, Netherland). Cell Ranger Single-Cell Software Suite version 2.1.1 (http://10xgenomics.com/) was used to align cDNA reads to the hg19 human reference genome. Cell types were annotated using marker genes (proliferating cells: MKI67 and TOP2A, fibroblasts: VIM, FN1, and S100A4; T cells: CD3E and CD8A; MCC cells: CHGA, ENO2, KRT20, and CD44; and endothelial cells: VIM, VWF, and CAV1). Afterwards, fibroblasts of all three samples were extracted and other cell types discarded. In total, we found 71 single fibroblast cells in MCC#1, 610 in MCC#2 and 120 in MCC#3. Normalized and raw expression counts for the fibroblast single cell analysis were uploaded to Figshare (https://doi.org/10.6084/m9.figshare.13095293), together with the respective cell meta data. This includes the CAF signature score which was computed using the Seurat AddModuleScore function and based on a list of 88 CAF-associated genes published by Tirosh et al. [38] (Supplementary Table S3). For normalization, dimension reduction, clustering, and visualization, we used Seurat R package v2 [55]. In order to correct for batch effects between the samples we applied the harmony package and adjusted the PCs calculated with Seurat [56]. Differentially expressed genes were called using Seurat’s FindMarkers function with the default settings. RNA velocity estimates were calculated using the corresponding velocyto R package with a gene-relative model based on PCA, and cell-cell distances calculated by Seurat [39]. Extrapolated cell states were projected onto PCA plot with the speed vectors that summarizes the transcriptional variability in the data in a lower-dimensional space using the “pca.velocity.plot“ function. Inferred trajectories are depicted as arrows, with the base of the arrow indicating the current cell state and the tip of the arrow indicating the extrapolated cell state.
Cell culture
All MCC cell lines, the cutaneous squamous cell carcinoma cell line SCL-2 [57] and the fibroblast cell line MRC-5, along with their culture conditions and growth patterns, have been described before [27]. Primary skin fibroblasts were isolated and cultured in our laboratory. All cell lines are regularly authenticated by short tandem repeat analysis (last performed in June 2019) and mycoplama test was performed monthly in our lab.
CM was generated by culturing cells (2 × 106) in fresh RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, PAN-Biotech, Aidenbach, Germany) and 1% penicillin/streptomycin (P/S, PAN-Biotech) for 72 h. Culture supernatants were centrifuged to remove floating cells and cell debris. CM was added to 70% confluent fibroblasts and incubated for 72 h.
For Transwell chamber system coculture (TC), 5×105 MRC-5 cells or 2 × 105 primary skin fibroblasts were seeded in the lower chambers of a 6-well plate with 0.4 µm pore size polyester membrane inserts (Corning, Hagen, Germany). After 24 h, 1 × 106 MCC cells were seeded in the inserts. Fibroblasts were harvested after 72 h of coculture.
For direct coculture (DC), MCC cells were added to 70% confluent, adherent, growing fibroblasts. After 72 h of coculture, the supernatants and floating cells were carefully removed, and the remaining adherent cells were washed at least 3 times with PBS to remove all floating cells. Adherent fibroblasts were harvested.
Generation of primary skin fibroblasts
Fibro1.4 and Fibro1.12 primary skin fibroblasts were generated from biopsies of healthy skin. Biopsies were cut into small pieces and incubated with 130 µM dispase II (Sigma-Aldrich, Darmstadt, Germany) in Hank’s balanced salt solution at 4 °C overnight to separate the dermis from the epidermis. The dermis was then transferred into fresh medium 1:1 DMEM + DMEM/F-12 (PAN-Biotech) supplemented with 1% P/S, 15% FBS, 10 µM amphotericin B (AMP-B, Sigma-Aldrich) and 75 µM ciprofloxacin (CIPRO, Sigma-Aldrich) and incubated overnight in a humidified incubator at 37 °C. The medium was replaced after 24 h and 48 h, gradually reducing the AMP-B and CIPRO concentrations to 5 µM and 35 µM, respectively. Then, the medium was changed weekly until fibroblasts began to form confluent patches. At this point, the excess tissues were discarded, and the remaining adherent fibroblasts were cultured in 1:1 DMEM + DMEM/F-12 supplemented with 1% P/S and 15% FBS. The pcDH_EGFP_puro plasmid and helper plasmids (pHCMV-G, pRSV rev and pMDLg/pRRE) were used to generate EGFP-labeled fibroblasts. Lentiviral supernatants were produced from HEK293T cells two days following transfection, and virus-containing supernatants were harvested and filtered through 0.45 μm pore size filters. Polybrene was added (5 μg/ml) for infection. After 16 h of incubation, the target cells were washed twice with culture medium and subjected to puromycin selection (1 μg/ml).
EVs isolation and characterization
EVs were isolated from 2 ml of CM using an ExoQuick-TC kit (System Biosciences, Palo Alto, CA, USA) following the manufacturer’s instructions. Isolated fractions of EVs were analysed in the violet side scatter channel with a CytoFLEX flow cytometer (Beckman Coulter, Brea, California, USA). Standardized nanoparticles of 50 nm, 100 nm and 200 nm were used to determine the sizes of the isolated EVs. The isolated EVs were stained with Exo-Red RNA fluorescent dye (System Biosciences) according to the manufacturer’s instructions.
miRNA overexpression
MRC-5 and Fibro1.12 cells were transfected with miR-375 mimics (Mission microRNA Mimic, HMI0537, Sigma-Aldrich) or negative control miRNA (Mission miRNA negative control 1, HMC0002, Sigma-Aldrich). Lipofectamine 3000 (Thermo Fisher Scientific) was used as the transfection reagent. The final miRNA mimic concentration used for transfection was 20 nM, and transfection was performed according to the protocol provided by the manufacturer.
miRNA in situ hybridization
Locked nucleic acid-based miRNA in situ hybridization was performed on FFPE MCC tissue sections by Bioneer in situ hybridization services (Hørsholm, Denmark). MCC sections were incubated with a specific probe against miR-375 (20 nM; DIG-TCACGCGAGCC GAACGAAA-DIG) or a scrambled control probe (20 nM; DIG-TGTA ACACGTCTATACGC CCA-DIG) at 58 °C in accordance with a previously described protocol [58].
RT-qPCR
Total RNA from cell lines and tumor tissues was isolated using a PeqGOLD total RNA Kit (VWR/Peqlab, Erlangen, Germany) and transcribed to cDNA using SuperScript IV reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer’s instructions. To isolate RNAs from cell free supernatant/EVs, the miRNeasy Mini Kit (Qiagen, Hilden, Germany) was used according to the manufacturer’s instructions; cel-miR-39 (ID000200) served as spike-in control. RT-qPCR was performed in a CFX Real-Time PCR system (Bio-Rad Laboratories, Düsseldorf, Germany). HPRT was used as the endogenous control, expression was detected using the SYBR Green method, and relative expression levels were calculated by the 2-ΔΔCq method using the corresponding untreated control or as otherwise indicated. The primer sequences are listed in Supplementary Table S4.
For miRNA analysis, Applied Biosystems TaqMan MicroRNA Assays (Thermo Fisher Scientific) were performed according to the manufacturer’s instructions. Pre-designed TaqMan microRNA assays for miR-182 (ID002334), miR-106b (ID000442), miR-19b (ID000396), miR-200c (ID002300), and miR-375 (ID000564) were used. The quantification cycle threshold (Cq) values of the analyzed miRNAs were normalized to the expression of the small nucleolar RNA RNU6B (ID001093) for cellular miRNAs or spike-in cel-miR-39 (ID000200) for miRNAs in cell free supernatant/EVs; the expression relative to the corresponding comparator was calculated using the 2-ΔΔCq method.
Immunoblot analysis
Cell lysis and protein extraction were performed using RIPA buffer (Sigma-Aldrich) supplemented with complete Mini protease inhibitor cocktail (Sigma-Aldrich). Proteins were separated by SDS-PAGE on 4–12% precast gradient gels (Thermo Fisher Scientific) and transferred to nitrocellulose membranes using an iBlot dry blotting system (Thermo Fisher Scientific). After blocking with 3% milk, membranes were incubated overnight at 4 °C with specific primary antibodies (Supplementary Table S5) After three washes, the appropriate peroxidase-conjugated secondary antibodies were added (Dako), and immunoreactions were visualized using ECL Western Blotting Substrate (Pierce, Rockford, USA) in a chemiluminescence imager (Amersham Imager 600, GE Healthcare, New York, USA). Quantification of immunoblot data was performed with ImageJ and GraphPad software (GraphPad Software Inc., San Diego, CA, USA).
IF staining
Fibroblasts were cultured on cover slips (Thermo Fisher Scientific) in 12-well plates. Before staining, cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with PBS and 0.2% Tween 20 for 15 min and blocked with 1% BSA in PBS for 1 h. Then, a primary antibody specific for α-SMA (clone 1A4, Dako), diluted 1:200 in PBS containing 1% BSA, was added and incubated overnight at 4 °C. After two washes, an Alexa Fluor 546-labeled goat-anti-mouse secondary antibody (1:200, Thermo Fisher Scientific) was added for one hour. After two washes, cells were incubated with 5 µg/ml wheat germ agglutinin (WGA, conjugated to Alexa Fluor 647, Thermo Fisher Scientific) for 30 min, and DAPI (1:2000, Thermo Fisher Scientific) was added to visualize nuclei. After three washes, cells were embedded in ProLong Diamond Antifade Mountant (Thermo Fisher Scientific) and then evaluated with an Axio Observer.Z1 microscope (Zeiss, Oberkochen, Germany).
Immunohistochemical staining
Immunohistochemical staining was performed on FFPE MCC tissue sections after deparaffinization. Antigen retrieval was performed using EDTA retrieval buffer (Sigma-Aldrich, pH 9) in a steamer at 95 °C for 20 min. After cooling for 20 min, samples were washed twice with PBS and incubated for 10 min with peroxidase blocking solution (3% H2O2 in methanol) at room temperature. Sections were then incubated for 30 min at room temperature with an antibody specific for α-SMA (clone 1A4, Dako) diluted 1:100 in PBS containing 3% BSA. After incubation with a horseradish peroxidase (HRP)-polymer anti-mouse antibody (POL2DS-006, Zytomed, Eching, Germany), detection was performed using Permanent HRP Green stain (Zytomed). After nuclei were counterstained with haematoxylin, sections were dehydrated and mounted in Vitro-Clud mounting medium (R. Langenbrinck, Emmendingen, Germany). The stained sections were then scored for α-SMA-positive fibroblasts using the Mantra quantitative pathology workstation with the inForm analysis software (PerkinElmer). In detail, the instrument was trained by manually categorizing the tissue sections into tumor and stromal segments, followed by automated cell segmentation via haematoxylin-stained nuclei. The system was then trained to automatically distinguish between cell phenotypes by manually identifying 100 tumor and 100 stromal cells. The final α-SMA staining score was calculated as the percentage of α-SMA-positive stromal cells. The cut-off for α-SMA-positive cells was set at an optical intensity intensity of greater than 0.26. For each section, four areas at the border of the tumor and four areas in the middle of the tumor were selected and analyzed as explained above. The means of the values for the tumor border and of those for the tumor middle were calculated for the final scoring.
GSEA
GSEA version 3.0 was used to conduct GSEA [59]. Pre-ranked analyses were performed by sorting differentially expressed genes according to their fold changes without prior filtering on significance or effect size. Enrichment was then tested with the hallmarks of cancer gene set [60].
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software Inc.) Experiments containing two groups of data were analyzed using the Mann–Whitney U test. Experiments containing more than two groups of data were analyzed using the Kruskal-Wallis test for unpaired nonparametric samples. Correlation analyses were performed in R Studio using the ggpubr R package. A p value < 0.05 was considered significant; the corresponding p values are indicated in the figures as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
References
Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbe C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Prim. 2017;3:17077.
Wardhani LO, Matsushita M, Iwasaki T, Kuwamoto S, Nonaka D, Nagata K, et al. Expression of the IDO1/TDO2-AhR pathway in tumor cells or the tumor microenvironment is associated with Merkel cell polyomavirus status and prognosis in Merkel cell carcinoma. Hum Pathol. 2019;84:52–61.
Paulson KG, Iyer JG, Simonson WT, Blom A, Thibodeau RM, Schmidt M, et al. CD8+ lymphocyte intratumoral infiltration as a stage-independent predictor of Merkel cell carcinoma survival a population-based study. Am J Clin Pathol. 2014;142:452–8.
Walsh NM, Fleming KE, Hanly JG, Hache KD, Doucette S, Ferrara G, et al. A morphological and immunophenotypic map of the immune response in Merkel cell carcinoma. Hum Pathol. 2016;52:190–6.
Wheat R, Roberts C, Waterboer T, Steele J, Marsden J, Steven NM, et al. Inflammatory cell distribution in primary Merkel cell carcinoma. Cancers. 2014;6:1047–64.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99:186–96.
Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–83.
Dror S, Sander L, Schwartz H, Sheinboim D, Barzilai A, Dishon Y, et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat Cell Biol. 2016;18:1006–17.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–47.
Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84.
Nagarsheth N, Wicha MS, Zou WP. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–72.
van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharm Rev. 2012;64:676–705.
Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–89.
Kourembanas S. Exosomes: vehicles of Intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol. 2015;77:13–27.
Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat Rev Clin Oncol. 2018;15:617–38.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–72.
Kogure T, Yan IK, Lin WL, Patel T. Inter-cellular nanovesicle mediated microrna transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1278a–1279a.
Cui H, Seubert B, Stahl E, Dietz H, Reuning U, Moreno-Leon L, et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene. 2015;34:3640–50.
Liu Y, Luo F, Wang BR, Li HQ, Xu Y, Liu XL, et al. STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells. Cancer Lett. 2016;370:125–35.
Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–15.
Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011;71:3792–801.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. 2019;70:241–58.
Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015;106:1362–9.
Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191.
Abraham KJ, Zhang X, Vidal R, Pare GC, Feilotter HE, Tron VA. Roles for miR-375 in neuroendocrine differentiation and tumor suppression via Notch pathway suppression in Merkel cell carcinoma. Am J Pathol. 2016;186:1025–35.
Fan K, Ritter C, Nghiem P, Blom A, Verhaegen ME, Dlugosz A, et al. Circulating cell-free miR-375 as surrogate marker of tumor burden in Merkel cell carcinoma. Clin Cancer Res. 2018;24:5873–82.
Renwick N, Cekan P, Masry PA, McGeary SE, Miller JB, Hafner M, et al. Multicolor microRNA FISH effectively differentiates tumor types. J Clin Invest. 2013;123:2694–702.
Xie H, Lee L, Caramuta S, Hoog A, Browaldh N, Bjornhagen V, et al. MicroRNA expression patterns related to Merkel cell polyomavirus infection in human Merkel cell carcinoma. J Invest Dermatol. 2014;134:507–17.
Fan KJ, Gravemeyer J, Ritter C, Rasheed K, Gambichler T, Moens U, et al. MCPyV large T antigen-induced Atonal Homolog 1 Is a lineage-dependency oncogene in Merkel Cell Carcinoma. J Invest Dermatol. 2020;140:56–65.
Hudson J, Duncavage E, Tamburrino A, Salerno P, Xi L, Raffeld M, et al. Overexpression of miR-10a and miR-375 and downregulation of YAP1 in medullary thyroid carcinoma. Exp Mol Pathol. 2013;95:62–67.
Szczyrba J, Nolte E, Wach S, Kremmer E, Stohr R, Hartmann A, et al. Downregulation of Sec23A protein by miRNA-375 in prostate carcinoma. Mol Cancer Res. 2011;9:791–800.
Nishikawa E, Osada H, Okazaki Y, Arima C, Tomida S, Tatematsu Y, et al. miR-375 is activated by ASH1 and inhibits YAP1 in a lineage-dependent manner in lung cancer. Cancer Res. 2011;71:6165–73.
Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, et al. miR-375 maintains normal pancreatic alpha- and beta-cell mass. P Natl Acad Sci USA. 2009;106:5813–8.
Liu Y, Xing R, Zhang X, Dong W, Zhang J, Yan Z, et al. miR-375 targets the p53 gene to regulate cellular response to ionizing radiation and etoposide in gastric cancer cells. DNA Repair. 2013;12:741–50.
Yan JW, Lin JS, He XX. The emerging role of miR-375 in cancer. Int J Cancer. 2014;135:1011–8.
Fan K, Zebisch A, Horny K, Schrama D, Becker JC. Highly expressed miR-375 is not an intracellular oncogene in Merkel cell polyomavirus-associated Merkel cell carcinoma. Cancers. 2020;12:529.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560:494–8.
Bhome R, Del Vecchio F, Lee GH, Bullock MD, Primrose JN, Sayan AE, et al. Exosomal microRNAs (exomiRs): Small molecules with a big role in cancer. Cancer Lett. 2018;420:228–35.
Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat Cell Biol. 2011;13:1457–U1178.
Arandkar S, Furth N, Elisha Y, Nataraj NB, van der Kuip H, Yarden Y, et al. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc Natl Acad Sci USA. 2018;115:6410–5.
Goruppi S, Procopio MG, Jo S, Clocchiatti A, Neel V, Dotto GP. The ULK3 kinase is critical for convergent control of cancer-associated fibroblast activation by CSL and GLI. Cell Rep. 2017;20:2468–79.
Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17:1193–204.
Mitra AK, Zillhardt M, Hua YJ, Tiwari P, Murmann AE, Peter ME, et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Disco. 2012;2:1100–8.
Yan W, Wu XW, Zhou WY, Fong MY, Cao MH, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol. 2018;20:597–609.
Thayanithy V, Dickson EL, Steer C, Subramanian S, Lou E. Tumor-stromal cross talk: direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl Res. 2014;164:359–65.
Konstantinell A, Bruun JA, Olsen R, Aspar A, Skalko-Basnet N, Sveinbjornsson B, et al. Secretomic analysis of extracellular vesicles originating from polyomavirus-negative and polyomavirus-positive Merkel cell carcinoma cell lines. Proteomics. 2016;16:2587–91.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Disco. 2019;18:99–115.
Khare D, Or R, Resnick I, Barkatz C, Almogi-Hazan O, Avni B. Mesenchymal stromal cell-derived exosomes affect mRNA expression and function of B-lymphocytes. Front Immunol. 2018;9:3053.
Liang X, Zhang L, Wang S, Han Q, Zhao RC. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci. 2016;129:2182–9.
Frank AC, Ebersberger S, Fink AF, Lampe S, Weigert A, Schmid T, et al. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat Commun. 2019;10:1135.
Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br J Haematol. 2018;183:428–44.
Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–96.
Popp S, Waltering S, Herbst C, Moll I, Boukamp P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352–60.
Lindahl LM, Fredholm S, Joseph C, Nielsen BS, Jonson L, Willerslev-Olsen A, et al. STAT5 induces miR-21 expression in cutaneous T cell lymphoma. Oncotarget. 2016;7:45730–44.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25.
Acknowledgements
This work was funded by DKTK ED03 from Deutsches Krebsforschungszentrum (DKFZ) including funding for KF. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
CR is now an employee of Merck Serono. Thilo Gambichler reports receiving speakers and/or advisory board honoraria from BMS, Sanofi-Genzyme, MSD, Novartis Pharma, Roche, Abbvie, Almirall, Janssen, Lilly, Pfizer, Pierre-Fabre, MerckSerono, outside the submitted work. NØ has received advisory honoraria from Micreos and Zealand Pharma. DS has received honoraria from Bristol-Myers Squibb, Roche, Novartis, Regeneron, Sanofi, MerckSerono, Amgen, 4SC, Merck Sharp and Dohme, Array, Pierre Fabre, Philogen, Incyte, and Pfizer, and research funding from Bristol-Myers Squibb and Novartis. SU declares research support from Bristol Myers Squibb and MerckSerono; speakers and advisory board honoraria from Bristol Myers Squibb, Merck Sharp and Dohme, Merck Serono, Novartis and Roche, as well as travel support from Bristol Myers Squibb, Merck Sharp and Dohme. JCB has received speaker honoraria from Amgen, MerckSerono, Pfizer, and Sanofi advisory board honoraria from 4SC, Amgen, CureVac, eTheRNA, MerckSerono, Novartis and InProTher, as well as research funding from Alcedis, Boehringer Ingelheim, Bristol-Myers Squibb, IQVIA, and MerckSerono; he also received travel support from 4SC and Incyte. All other authors state no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, K., Spassova, I., Gravemeyer, J. et al. Merkel cell carcinoma-derived exosome-shuttle miR-375 induces fibroblast polarization by inhibition of RBPJ and p53. Oncogene 40, 980–996 (2021). https://doi.org/10.1038/s41388-020-01576-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01576-6
- Springer Nature Limited
This article is cited by
-
Combining serum microRNAs and machine learning algorithms for diagnosing infectious fever after HSCT
Annals of Hematology (2024)
-
Engineered small extracellular vesicles as a novel platform to suppress human oncovirus-associated cancers
Infectious Agents and Cancer (2023)
-
The HDAC inhibitor domatinostat induces type I interferon α in Merkel cell carcinoma by HES1 repression
Journal of Cancer Research and Clinical Oncology (2023)
-
Intercellular crosstalk between cancer cells and cancer-associated fibroblasts via extracellular vesicles
Cancer Cell International (2022)
-
Small extracellular vesicles as key players in cancer development caused by human oncogenic viruses
Infectious Agents and Cancer (2022)