
Abstract
Serotonin type-3 receptor (5-HT3R) antagonists show potential as a treatment for cognitive deficits in schizophrenia. CVN058, a brain-penetrant, potent and selective 5-HT3R antagonist, shows efficacy in rodent models of cognition and was well-tolerated in Phase-1 studies. We evaluated the target engagement of CVN058 using mismatch negativity (MMN) in a randomized, double-blind, placebo-controlled, cross-over study. Subjects were stable outpatients with schizophrenia or schizoaffective disorder treated with antipsychotics. Subjects were not permitted to use other 5-HT3R modulators or serotonin reuptake inhibitors. Each subject received a high (150 mg) and low (15 mg or 75 mg) oral dose of CVN058 and placebo in a randomized order across 3 single-day treatment visits separated by at least 1 week. The primary pre-registered outcome was amplitude of duration MMN. Amplitude of other MMN deviants (frequency, intensity, frequency modulation, and location), P50, P300 and auditory steady-state response (ASSR) were exploratory endpoints. 19 of 22 randomized subjects (86.4%) completed the study. Baseline PANSS scores indicated moderate impairment. CVN058 150 mg led to significant improvement vs. placebo on the primary outcome of duration MMN (p = 0.02, Cohen’s d = 0.48). A significant treatment effect was also seen in a combined analysis across all MMN deviants (p < 0.001, d = 0.57). Effects on location MMN were independently significant (p < 0.007, d = 0.46). No other significant effects were seen for other deviants, doses or EEG measures. There were no clinically significant treatment related adverse effects. These results show MMN to be a sensitive target engagement biomarker for 5-HT3R, and support the potential utility of CVN058 in correcting the excitatory/inhibitory imbalance in schizophrenia.
Similar content being viewed by others


Introduction
Schizophrenia is a major public health problem that affects ~1% of the population worldwide, and is associated with both positive and negative symptoms and neurocognitive deficits [1,2,3,4,5]. Antipsychotics are the primary treatment for schizophrenia, but in addition to significant side effects [6, 7], marketed antipsychotics have limited efficacy for neurocognitive deficits [8], indicating the need for alternative approaches. A key challenge in the development of novel treatments for schizophrenia is the need for target engagement biomarkers, which facilitate dose selection and initial proof-of-mechanism assessment [9,10,11,12,13,14,15]. Here, we evaluate the utility of auditory mismatch negativity (MMN) as a target engagement biomarker for development of serotonin type-3 receptor (5-HT3R) antagonists in the treatment of persistent neurocognitive impairments in schizophrenia.
Neurocognitive impairments in schizophrenia are increasingly conceptualized as reflecting impairments of excitatory/inhibitory balance [16]. Excitation is mediated primarily by pyramidal (glutamatergic) neurons acting through N-methyl-d-aspartate-type glutamate receptor (NMDAR) and non-NMDAR glutamate receptors. Inhibitory activity is modulated by several classes of GABAergic interneurons. The most widely studied GABAergic interneuron classes in schizophrenia are parvalbumin (PV) and somatostatin (SOM) interneurons, that target primarily axons and dendrites of pyramidal neurons, respectively.
A third class of GABAergic interneurons is distinguished by expression of 5-HT3R. Unlike other 5-HTR [17], 5-HT3R are ionotropic and thus mediate fast neuronal excitation [18]. 5-HT3R GABAergic interneurons are further subdivided by their expression of vasoactive intestinal peptide (VIP) and cholecystokinin (CCK) [19,20,21]. 5-HT3R activate VIP/CCK interneurons leading to GABA release [22, 23] and inhibition of glutamatergic pyramidal neurons [24, 25] and SOM- and PV-expressing GABAergic interneurons [26]. VIP/CCK interneurons are modulated by excitatory thalamic glutamatergic and subcortical noradrenergic, serotonergic and cholinergic efferents [19]. Thus, inhibition of VIP/CCK interneurons by 5-HT3R antagonists may help reverse impairments caused by deficits in pyramidal glutamatergic and SOM/PV interneurons [27, 28].
5-HT3R are relatively concentrated in areas important for cognition, including the auditory cortex, hippocampus, and amygdala [29,30,31]. Preclinically, 5-HT3R antagonists attenuate neurocognitive effects induced by NMDAR antagonists such as PCP [32] or MK-801 [33] and may also modulate glutamatergic neurotransmission via 5-HT3R expressed on GABAergic VIP/CCK interneurons [22, 34,35,36]. Moreover, olanzapine and clozapine, two of the most efficacious antipsychotics [7], are potent antagonists at the 5-HT3R [37, 38].
Selective 5-HT3R antagonists, most commonly ondansetron, have also been evaluated as potential adjunctive agents to antipsychotics with encouraging results for both total and negative symptoms [39,40,41,42]. Several studies have also assessed effects of 5-HT3R antagonists on cognition [43,44,45], finding significant improvements in visual memory and cognitive symptoms [46]. In general, however, 5-HT3R antagonists used to date have been primarily tool compounds with poor non-brainstem CNS penetrance [47], off target effects at the nicotinic α7 receptor (nα7R) [48] and unclear in vivo target engagement.
Here, we investigate the target engagement of CVN058, a novel brain-penetrant, highly potent and selective 5-HT3R antagonist [49]. Unlike most other 5-HT3R antagonists, CVN058 is virtually without activity at the nα7R. CVN058 has shown efficacy in rodent models of cognition and was safe and well-tolerated in Phase-1 studies [49]. Because several antipsychotics, especially clozapine and olanzapine, are functional antagonists at the 5-HT3R [38, 50], we limited enrollment to patients on antipsychotics and other psychotropics with minimal 5-HT3R engagement (see Supplemental Table 1).
We utilized MMN as our predesignated, primary target engagement biomarker. MMN is elicited most commonly in an auditory oddball paradigm in which a sequence of repetitive standard stimuli is interrupted infrequently by physically or conceptually distinct “oddball” stimuli. In schizophrenia, deficits in MMN are highly related to impaired early auditory processing (EAP) and poor functional outcome [51, 52]. MMN activity maps primarily within the theta frequency range and thus serves as a putative index of interactions between pyramidal interneurons and SOM-type GABA interneurons [16, 53, 54]. MMN generation is reliably inhibited by NMDAR antagonists across human [12, 55,56,57,58,59], monkey [54, 60, 61] and rodent [62, 63] models. MMN has previously been used as a sensitive and reliable [64] measure of target engagement for NMDAR [65,66,67,68,69], and to a lesser extent, nα7R [70, 71] based compounds.
In addition, 5-HT3R antagonists are reported to improve P50 gating in schizophrenia [48, 72], which was included as an additional exploratory target engagement measure in the present study. Finally, we included other potential measures, including auditory P300 and auditory steady-state response (ASSR). As opposed to MMN, P300 reflects attention-dependent processing primarily within higher-order brain regions, and is sensitive to multiple neurochemical influences [73]. By contrast, ASSR is generated within primary auditory cortex, but reflects primarily high-frequency pyramidal to PV interneuron interactions [74].
MMN can be elicited by a range of deviant types. Deficits in response to duration deviant stimuli are most widely replicated in schizophrenia [75]. Duration MMN was therefore pre-registered as the primary target engagement measure. Nevertheless, responses to other deviant types such as location [76, 77], frequency [65,66,67], and intensity [78] are also well established. These were therefore designated as exploratory endpoints, with anticipated analysis across deviant types.
CVN058 was tested across at low- and high-doses to evaluate dose dependence. Doses were selected based on pharmacokinetic scaling. A low dose of 15 mg was initially selected, but raised to 75 mg after a pre-specified interim evaluation suggested lack of engagement. The high dose remained at 150 mg throughout the study.
Materials and methods
Subjects
This was a Phase 1b, randomized, placebo-controlled, double-blind, cross-over investigation conducted at Columbia University Medical Center/New York State Psychiatric Institute (CUMC/NYSPI). The study was approved by the New York State Psychiatric Institute Institutional Review Board, and conducted between November 2018 and February 2020. Written informed consent was obtained from all participants prior to participation. The trial protocol can be found in the Supplement.
Enrollment criteria included medically healthy male and female subjects diagnosed with schizophrenia or schizoaffective disorder, aged 18–50, medically stable, PANSS total score of <95 with no recent (within 4 weeks) exposure to other investigational medications or devices.
To minimize potential pharmacological confounds, subjects were required to be on a stable dose of risperidone, haloperidol, quetiapine, aripiprazole, paliperidone, lurasidone or ziprasidone (all of which have a low risk of impacting 5-HT3R) and were not permitted to be on other 5-HT3R modulators nor primarily serotonergic antidepressants (Supplemental Table 1).
Design
After providing informed consent, and medical/psychiatric screening to confirm eligibility, subjects underwent a tone matching task (TMT) to assess baseline EAP [79].
Each subject completed three treatment visits in a double-blind, randomized order, each visit separated by a washout period of 7–10 days. Each treatment visit included a single dose of study medication, serial PK samples, EEG assessments and end of day ratings. We initially tested two dose levels (15 and 150 mg); after 13 subjects, the 15 mg dose level showed a lack of effect in a preplanned blinded interim analysis and was replaced by a 75 mg dose. A randomization list was produced by the study biostatistician.
The study was conducted in accordance with Good Clinical Practice. All ERP analyses were conducted on blinded data and all extracted values were entered into the study database and locked prior to breaking of the study blind. No adjustments were made to extracted values subsequent to database lock.
Electrophysiology
The primary outcome measure was amplitude of MMN elicited by duration deviants, with other deviants (pitch, intensity, frequency modulation, location) and EEG measures (P50 inhibition, ASSR, and P300) exploratory using previously described methods [70, 80, 81]. EEG collection began ~1.5 h post dose, during the expected peak serum levels.
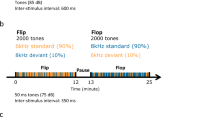
As previously [81], for MMN, auditory stimuli consisted of a sequence of tones presented in random order with a stimulus onset asynchrony (SOA) of 500 ms. Standard stimuli (45% sequential probability) were harmonic tones composed of three superimposed sinusoids (500, 1000 and 1500 Hz) 100 ms in duration with 5 ms rise/fall time presented at ~85 dB.
Six deviants were used i.e., pitch, duration, and intensity (10% probability each). The deviants were 10% higher in pitch, 50 ms longer in duration, 45% lower in intensity, respectively, and frequency modulated (FM; at 2 Hz with modulation index of 300) deviant (10% probability). All the above tones were presented binaurally with apparent location in the center midline. Two location deviants were included (7.5% probability each) that gave the percept of stimulus movement to the left vs. right hemifield (named respectively as RL and LR) based on an interaural delay time of 700 microseconds between ears in the appropriate direction. Seven runs of 5 mins each (600 stimuli/run) were presented as the subjects listened to the tones while watching a silent movie as a distractor. Full details on data analysis can be found in [81].
Behavioral assessments
Symptoms were assessed with the PANSS at baseline and after each treatment visit. The TMT was assessed at baseline. Safety was assessed with the Columbia-Suicide Severity Rating Scale [82] at each visit.
Pharmacokinetics
On each treatment day, plasma CVN058 level was assessed pre-dose (within 15 min prior to dosing), 1 h post-dose (pre-EEG), and 5 h post-dose (post-EEG).
Statistical analysis
Demographics and baseline characteristics were summarized for the overall sample using means and SDs for continuous variables, and proportions and frequencies for categorical variables.
The predesignated primary outcome was amplitude of MMN to duration deviants, with the predesignated primary comparison between high dose (150 mg) and placebo. Exploratory analysis was conducted across all MMN deviants, doses and secondary EEG outcomes. The MMN for duration deviant was measured as mean amplitude over 120–260 ms interval at electrode Fz. For frequency, intensity and FM, deviants were measured as mean amplitude over 100–200 ms interval at electrode Fz, and for location deviants the mean amplitudes were measured for 80–130 ms interval at left hemisphere electrode FC3 and right hemisphere electrode FC4. The predesignated primary analysis was designed to support internal decision making by the study sponsor, and is fully described in the supplemental statistical methods. The results of the predesignated primary analysis are presented in Supplemental Table 2.
For the present report, additional analyses were conducted. Linear, mixed effects models were used to accommodate correlated responses of the five MMN deviants. This model was fit for outcomes from each of the MMN deviants (i.e., duration, pitch, intensity, frequency modulation, location and across deviants). Exploratory EEG (P50, ASSR, and P300) was analyzed with repeated measure ANOVA.
Fixed factors for treatment (high dose, low dose, and placebo), sequence order (e.g., counterbalanced order of treatment of placebo, low-dose and high-dose CVN058), deviant-type and low dose type (15 or 75 mg) were used as appropriate. Intercept was included as a random factor in the mixed effects model. For these analyses, the 15 and 75 mg doses were combined as low dose unless otherwise specified.
Descriptive statistics were produced for adverse events and for plasma drug concentrations. Effect sizes for comparisons of CVN058 to placebo use Cohen’s d. Values in text are mean ± SD. All analyses presented in this manuscript, including Supplemental Table 2a, are two-tailed and interpreted based on an a priori cut-off α value for significance of p < 0.05 (two-tailed).
Power analysis
The study was powered based on previous MMN target engagement studies [66, 67], with the planned sample size of 20 completers estimated to provide ~80% power to detect a mean effect size of Cohen’s d = 0.5 with an error rate of alpha = 0.10.
Results
Sample
Twenty-two subjects (Supplemental Fig. 1, Table 1) were randomized and had at least one treatment visit. 19 subjects completed all three visits, including 19 for both the placebo and 150 mg doses, 13 for the 15 mg dose and 6 for the 75 mg dose. The study was stopped after 19 completers during the COVID-19 pandemic.
Baseline PANSS (65.4 ± 12.8) scores were consistent with mild to moderate baseline impairment. Baseline TMT scores were available for 20 subjects, with 40% exhibiting impairments [79].
MMN
In the prespecified primary analysis, high dose CVN058 (150 mg) treatment led to a significant improvement vs. placebo for the primary outcome of duration MMN (p = 0.02). Full results conducted with the predesignated plan are presented in Supplemental Table 2a.
In the mixed model analysis, high dose CVN058 (150 mg) treatment led to a significant improvement vs. placebo for the primary outcome of duration MMN (F1,26 = 4.47, p = 0.044, d = 0.48, Fig. 1). Because this was a within subject design, the sequence order for testing placebo, low-dose and high-dose CVN058 was counterbalanced across individuals. There was no significant effect of sequence order (F5,26 = 1.6, p = 0.19). The treatment by sequence order effect (F5,26 = 0.9, p = 0.47) was also not significant. Waveforms and voltage headmaps for duration MMN are presented in Fig. 2 and for the other deviants in Supplemental Fig. 2.

*p < 0.05, High dose vs. placebo in both predesignated model and confirmatory mixed model analysis.

Left: Duration MMN waveforms by treatment group, with line showing the analyzed latency window. Right: Scalp topographies by treatment group, over the analyzed latency window.
In an exploratory mixed model analysis, a highly significant treatment effect was also seen across all MMN deviants (F1,195 = 12.09, p < 0.001, d = 0.57, Fig. 3left). When separate analyses were performed for individual deviant types, there was also a significant treatment effect for MMN to location (F1,91 = 7.5, p = 0.007, d = 0.46, Fig. 3right). There were no significant effects for other deviants individually (Fig. 3right).

Left: Bar graph of model estimated mean ± standard error for combined MMN. Right: Bar graph of model estimated mean ± standard error for secondary MMN. **p < 0.01 and ***p < 0.001, high dose vs. placebo in confirmatory mixed model analysis.
When low doses were included in the model, the overall treatment effect remained significant for duration MMN (F2,16 = 9.3, p = 0.002), location MMN (F2,184 = 8.6, p < 0.001) and across all deviants (F2,408 = 11.5, p < 0.001). There was no significant effect of lower doses vs. placebo, but a trend toward significance was seen for MMN to intensity (F1,8 = 4.2, p = 0.07, d = 0.68).
No significant effects were observed on ERP responses to standard stimuli (Supplemental Table 2b and Supplemental Fig. 3).
Other exploratory outcome measures
No significant effects were seen in the other exploratory outcome measures (P50 gating, P300, ASSR). Full results conducted with the predesignated plan are presented in Supplemental Table 2a.
Pharmacokinetics
CVN058 levels were assessed per schedule (Table 2), and showed the expected dose dependent linear kinetics and compliance with study procedures, with no detectable carry-over between treatment visits.
Safety measures
No clinically significant side effects attributable to study drug were observed, and in general, the study drug was well-tolerated. All non-serious adverse events were mild with the exception of abnormal blood glucose and sodium in one subject, which were considered not related to treatment. Somnolence, dizziness, headache, diarrhea and throat irritation were the only other side effects reported in more than 5% in the active groups (Supplemental Table 3). There were no clinically significant changes in the PANSS or vital signs (Supplemental Tables 4 and 5), nor were there significant correlations of MMN with symptom change.
Three subjects did not complete. Two subjects withdrew consent after treatment visit 1. One subject was removed after treatment visit 1 after the study team’s discovery of a suicide attempt that occurred after consent but prior to randomization and any study treatment. The subject was hospitalized, and the suicide attempt was noted as a serious adverse event not considered related to study treatment.
Discussion
Despite the availability of numerous FDA approved antipsychotics, the majority of schizophrenia patients remain permanently disabled. The principal findings of the present report are that the novel 5-HT3R antagonist CVN058 shows both dose dependent target engagement using MMN as a physiological readout and improvement of MMN deficits in schizophrenia. The study thus supports both the utility of MMN as a target engagement biomarker and of 5-HT3R antagonists as potential novel treatments for cognitive impairment in schizophrenia.
Deficits in MMN generation were first demonstrated in schizophrenia almost 30 years ago [83]. MMN was subsequently shown to be sensitive to effects of NMDAR antagonists both during intracortical infusion in non-human primates and IV infusion in healthy human volunteers. Both sets of findings have been extensively replicated [59, 75]. In schizophrenia, MMN deficits are strongly associated with functionally relevant EAP deficits, characterized by elevated thresholds for detecting physical differences in auditory stimuli. In turn, EAP deficits are associated with cognitive deficits in more complex information processing [79], such as reading [84, 85] or auditory emotion recognition [86,87,88]. Similarly, a large cross-sectional study [89] supports a direct link between MMN, EAP and cognition. In previous schizophrenia studies [66, 67], improvement in MMN have been predictive of symptomatic and cognitive improvements, further supporting the clinical relevance of MMN. Thus, agents that reverse MMN deficits in schizophrenia may be of potential benefit in treatment both of cognitive impairments and persistent negative symptoms, and might improve long-term outcome.
The potential relevance of 5-HT3R for modulation of MMN is supported by the following lines of evidence: First, 5-HT3R are expressed prominently in areas known to generate MMN [90], including the auditory cortex [30] and the medial geniculate nucleus [91], and are integral for the regulation of basal, non-potentiated transmission [30]. Second, 5-HT3R antagonists can attenuate NMDAR antagonist-induced cognitive impairment preclinically [32, 33] and may also modulate glutamatergic neurotransmission via 5-HT3R expressed on GABAergic VIP/CCK interneurons [22, 34,35,36]. As recently reviewed [53], MMN is dependent on VIP/CCK interneuron modulation of both SOM interneurons and pyramidal neurons. As schizophrenia has localized deficits in both SOM interneurons and pyramidal neurons, 5-HT3R antagonists may help restore excitatory-inhibitory balance and improve MMN [92, 93].
In this study, the effects were broadest and most robust with the 150 mg dose, which was the highest tested dose. No significant effect was seen at the lower doses, suggesting dose dependent target engagement. The present results are thus encouraging of future phase II parallel group studies with CVN058 at the 150 mg dose incorporating both clinical measures and MMN. Further dose escalation studies may also be desirable as no significant safety concerns emerged even at the highest dose tested. In the FAST-FAIL approach [9,10,11,12,13,14,15], demonstration of target engagement, as in the present study, validates the compound, although it remains to be determined whether or not treatment through this mechanism (5-HT3R antagonism) will ultimately lead to clinical benefit.
Finally, the study provides some technical guidance in application of MMN as a biomarker for early stage clinical trials. Because MMN responses are based on individual stimulus features, rather than their conjunction, adding additional deviants to an MMN sequence does not lead to interference among deviants. Thus, relative to 1000 Hz, 100 ms standard stimuli, a 1000 Hz, 150 ms stimulus serves as frequency “standard” even though it serves as a duration deviant.
In general, even in “optimal” paradigms such as the one applied here, MMN analyses are usually performed on each deviant type individually, both by ourselves [66, 67] and others [94,95,96]. Our secondary analysis suggests that a multivariate approach across deviant types may increase sensitivity to treatment effects and thus increase statistical power. In addition, location MMN has been studied less extensively in schizophrenia than other MMN types, although it has been found to be consistently reduced both in schizophrenia [75, 76] and a clinical high risk group [81]. The finding of its significant sensitivity to CVN058 argues for location MMN’s greater inclusion in multivariate paradigms.
In addition, speech-related stimuli also elicit MMN that are sensitive to ketamine administration [95]. Our FM deviant stimulus, which showed a numeric but not statistically significant effect, may capture information related to both speech- and emotion-related stimuli [86, 87]. In prior studies using optimal [95] or speech-related [93] paradigms, nα7 agonists did not induce a significant overall change in MMN amplitude, but did revert low and high MMN amplitudes toward the mean.
Similarly, in a prior study using the same paradigm, we did not observe any significant effect of the nα7 agonist AVL-3288 on MMN to either individual deviants or across MMN types [70]. Similarly, a study of the nα7 agonist EVP-6124 produced nonsignificant MMN changes in a single deviant paradigm [71]. Thus, our findings with a 5-HT3R targeted treatment are novel relative to prior studies with nα7 agonists, and parallel prior results with NMDAR-glycine site modulators [65,66,67,68].
Some limitations of the present report should be acknowledged. Our design of three single-dose treatments on a narrow range of concomitant medications (Supplemental Table 1) restricted our assessments of both tolerability and efficacy to acute effects. This design may have also limited our ability to assess relationships between clinical changes and MMN. In prior studies using NMDAR-glycine site modulators for 4–6 weeks, MMN has significantly correlated with clinical improvements [65, 67].
In addition, we did not replicate previous positive P50 findings with 5-HT3R antagonists [48, 72]. We attempted to replicate the features of the specific device that was used in those prior studies [48, 72], but did not use the device itself. Similarly, in a recent study of the novel nα7R agonist AVL-3288 [70], we also did not observe target engagement with the P50 paradigm. These negative results thus may reflect limitations of our implementation of the P50 paradigm, rather than a more general failure of the approach.
Enhancement of NMDAR mediated neurotransmission remains a priority in schizophrenia drug development. The present approach suggests that this may be accomplished in part through restoration of excitatory-inhibitory balance among different interneuron classes, and that 5-HT3R-expressing interneurons may be an effective target. Overall, the present findings encourage further studies with both CVN058 as a potential cognition-enhancing agent and multivariate MMN as a target-engagement biomarker. While the results were clearest for the 150 mg dose, future work is required to delineate the dose dependent target engagement and clinical effects of multiple doses across a wider range of concomitant medications.
References
McCleery A, Green MF, Hellemann GS, Baade LE, Gold JM, Keefe RS, et al. Latent structure of cognition in schizophrenia: a confirmatory factor analysis of the MATRICS Consensus Cognitive Battery (MCCB). Psychol Med. 2015;45:2657–66.
Keefe RS, Harvey PD. Cognitive impairment in schizophrenia. In: Geyer MA, Gross G (eds). Novel Antischizophrenia Treatments, 2012/10/03 edn. Springer Berlin Heidelberg: Berlin, Heidelberg, 2012, pp 11–37.
Kern RS, Gold JM, Dickinson D, Green MF, Nuechterlein KH, Baade LE, et al. The MCCB impairment profile for schizophrenia outpatients: results from the MATRICS psychometric and standardization study. Schizophr Res. 2011;126:124–31.
Keefe RS, Fox KH, Harvey PD, Cucchiaro J, Siu C, Loebel A. Characteristics of the MATRICS Consensus Cognitive Battery in a 29-site antipsychotic schizophrenia clinical trial. Schizophr Res. 2011;125:161–8.
Kantrowitz JT. Managing Negative Symptoms of Schizophrenia: how far have we come? CNS Drugs. 2017;31:373–88.
Meftah AM, Deckler E, Citrome L, Kantrowitz JT. New discoveries for an old drug: a review of recent olanzapine research. Postgrad Med. 2020;132:80–90.
Huhn M, Nikolakopoulou A, Schneider-Thoma J, Krause M, Samara M, Peter N, et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. Lancet. 2019;394:939–51.
Ohi K, Muto Y, Sugiyama S, Shioiri T. Safety and Efficacy in Randomized Controlled Trials of Second-Generation Antipsychotics Versus Placebo for Cognitive Impairments in Schizophrenia: a meta-analysis. J Clin Psychopharmacol. 2020. https://doi.org/10.1097/JCP.0000000000001232.
Grabb MC, Cross AJ, Potter WZ, McCracken JT. Derisking Psychiatric Drug Development: the NIMH’s Fast Fail Program, A Novel Precompetitive Model. J Clin Psychopharmacol. 2016;36:419–21.
Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development. Nat Rev Drug Disco. 2018;18:82–4.
Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of Imaging-Based Biomarkers for Glutamate-Targeted Drug Development in Psychotic Disorders: a Randomized Clinical Trial. JAMA Psychiatry. 2018;75:11–9.
Javitt DC, Schoepp D, Kalivas PW, Volkow ND, Zarate C, Merchant K, et al. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3:102mr2.
Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Disco. 2008;7:68–83.
Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS, et al. Proof of mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: RCTs of pomaglumetad and TS-134 on ketamine-induced psychotic symptoms and pharmacoBOLD in healthy volunteers. Neuropsychopharmacology. 2020;45:1842–50.
Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J Jr, Lisanby SH, et al. A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating kappa-opioid antagonism as a treatment for anhedonia. Nat Med. 2020;26:760–68.
Javitt DC, Siegel SJ, Spencer KM, Mathalon DH, Hong LE, Martinez A, et al. A roadmap for development of neuro-oscillations as translational biomarkers for treatment development in neuropsychopharmacology. Neuropsychopharmacology. 2020;45:1411–22.
Kantrowitz JT. Targeting Serotonin 5-HT2A Receptors to Better Treat Schizophrenia: rationale and current approaches. CNS Drugs. 2020;34:947–59.
Yakel JL, Jackson MB. 5-HT3 receptors mediate rapid responses in cultured hippocampus and a clonal cell line. Neuron 1988;1:615–21.
Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B. The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci. 2010;30:16796–808.
Férézou I, Cauli B, Hill EL, Rossier J, Hamel E, Lambolez B. 5-HT3 receptors mediate serotonergic fast synaptic excitation of neocortical vasoactive intestinal peptide/cholecystokinin interneurons. J Neurosci. 2002;22:7389–97.
Tremblay R, Lee S, Rudy B. GABAergic Interneurons in the Neocortex: from Cellular Properties to Circuits. Neuron 2016;91:260–92.
Riga MS, Sanchez C, Celada P, Artigas F. Involvement of 5-HT3 receptors in the action of vortioxetine in rat brain: focus on glutamatergic and GABAergic neurotransmission. Neuropharmacol. 2016;108:73–81.
Kawaguchi Y, Kubota Y. Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neurosci. 1998;85:677–701.
Wall NR, De La Parra M, Sorokin JM, Taniguchi H, Huang ZJ, Callaway EM. Brain-Wide Maps of Synaptic Input to Cortical Interneurons. J Neurosci. 2016;36:4000–9.
Zhou X, Rickmann M, Hafner G, Staiger JF. Subcellular Targeting of VIP Boutons in Mouse Barrel Cortex is Layer-Dependent and not Restricted to Interneurons. Cereb Cortex. 2017;27:5353–68.
Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–4.
Volk DW, Sampson AR, Zhang Y, Edelson JR, Lewis DA. Cortical GABA markers identify a molecular subtype of psychotic and bipolar disorders. Psychol Med. 2016;46:2501–12.
Javitt DC, Sweet RA. Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nat Rev Neurosci. 2015;16:535–50.
Koyama Y, Kondo M, Shimada S. Building a 5-HT3A Receptor Expression Map in the Mouse Brain. Sci Rep. 2017;7:42884.
Lee KKY, Soutar CN, Dringenberg HC. Gating of long-term potentiation (LTP) in the thalamocortical auditory system of rats by serotonergic (5-HT) receptors. Brain Res. 2018;1683:1–11.
Acevedo-Triana CA, Leon LA, Cardenas FP. Comparing the Expression of Genes Related to Serotonin (5-HT) in C57BL/6J Mice and Humans Based on Data Available at the Allen Mouse Brain Atlas and Allen Human Brain Atlas. Neurol Res Int. 2017;2017:7138926.
Pehrson AL, Pedersen CS, Tolbol KS, Sanchez C. Vortioxetine Treatment Reverses Subchronic PCP Treatment-Induced Cognitive Impairments: a Potential Role for Serotonin Receptor-Mediated Regulation of GABA Neurotransmission. Front Pharmacol. 2018;9:162.
Boast C, Bartolomeo AC, Morris H, Moyer JA. 5HT antagonists attenuate MK801-impaired radial arm maze performance in rats. Neurobiol Learn Mem. 1999;71:259–71.
Dale E, Grunnet M, Pehrson AL, Frederiksen K, Larsen PH, Nielsen J, et al. The multimodal antidepressant vortioxetine may facilitate pyramidal cell firing by inhibition of 5-HT3 receptor expressing interneurons: an in vitro study in rat hippocampus slices. Brain Res. 2018;1689:1–11.
Faust TW, Assous M, Tepper JM, Koos T. Neostriatal GABAergic Interneurons Mediate Cholinergic Inhibition of Spiny Projection Neurons. J Neurosci. 2016;36:9505–11.
Riga MS, Sanchez C, Celada P, Artigas F. Sub-chronic vortioxetine (but not escitalopram) normalizes brain rhythm alterations and memory deficits induced by serotonin depletion in rats. Neuropharmacol. 2020;178:108238.
Bymaster FP, Falcone JF, Bauzon D, Kennedy JS, Schenck K, DeLapp NW, et al. Potent antagonism of 5-HT(3) and 5-HT(6) receptors by olanzapine. Eur J Pharm. 2001;430:341–9.
Eisensamer B, Uhr M, Meyr S, Gimpl G, Deiml T, Rammes G, et al. Antidepressants and antipsychotic drugs colocalize with 5-HT3 receptors in raft-like domains. J Neurosci. 2005;25:10198–206.
Juza R, Vlcek P, Mezeiova E, Musilek K, Soukup O, Korabecny J. Recent advances with 5-HT3 modulators for neuropsychiatric and gastrointestinal disorders. Med Res Rev. 2020;40:1593–678.
Kishi T, Mukai T, Matsuda Y, Iwata N. Selective serotonin 3 receptor antagonist treatment for schizophrenia: meta-analysis and systematic review. Neuromolecular Med. 2014;16:61–9.
Zheng W, Cai DB, Zhang QE, He J, Zhong LY, Sim K, et al. Adjunctive ondansetron for schizophrenia: a systematic review and meta-analysis of randomized controlled trials. J Psychiatr Res. 2019;113:27–33.
Kulkarni J, Thomas N, Hudaib AR, Gavrilidis E, Gurvich C. Ondansetron - a promising adjunctive treatment for persistent schizophrenia. J Psychopharmacol. 2018;32:1204–11.
Akhondzadeh S, Mohammadi N, Noroozian M, Karamghadiri N, Ghoreishi A, Jamshidi AH, et al. Added ondansetron for stable schizophrenia: a double blind, placebo controlled trial. Schizophr Res. 2009;107:206–12.
Levkovitz Y, Arnest G, Mendlovic S, Treves I, Fennig S. The effect of Ondansetron on memory in schizophrenic patients. Brain Res Bull. 2005;65:291–5.
Zhang ZJ, Kang WH, Li Q, Wang XY, Yao SM, Ma AQ. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: a double-blind, randomized, placebo-controlled study. Schizophr Res. 2006;88:102–10.
Kay S, Fiszbein A, Opler L. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–76.
Tyers MB, Freeman AJ. Mechanism of the anti-emetic activity of 5-HT3 receptor antagonists. Oncology. 1992;49:263–8.
Koike K, Hashimoto K, Takai N, Shimizu E, Komatsu N, Watanabe H, et al. Tropisetron improves deficits in auditory P50 suppression in schizophrenia. Schizophr Res. 2005;76:67–72.
Brice N, Dawson LA, Margolin DH, Schiffer HH, Monenschein H, Ray WJ, et al. Identification of CVN058, a novel brain penetrant, selective 5-HT3 receptor antagonist that shows efficacy in pre-clinical cognition models. Biol Psychiatry. 2021;89:S26.
Rammes G, Eisensamer B, Ferrari U, Shapa M, Gimpl G, Gilling K, et al. Antipsychotic drugs antagonize human serotonin type 3 receptor currents in a noncompetitive manner. Mol Psychiatry. 2004;9:846–58.
Light GA, Swerdlow NR, Thomas ML, Calkins ME, Green MF, Greenwood TA, et al. Validation of mismatch negativity and P3a for use in multi-site studies of schizophrenia: characterization of demographic, clinical, cognitive, and functional correlates in COGS-2. Schizophr Res. 2015;163:63–72.
Javitt DC, Freedman R. Sensory processing dysfunction in the personal experience and neuronal machinery of schizophrenia. Am J Psychiatry. 2015;172:17–31.
Javitt DC, Lee M, Kantrowitz JT, Martinez A. Mismatch negativity as a biomarker of theta band oscillatory dysfunction in schizophrenia. Schizophr Res. 2018;191:51–60.
Lakatos P, O’Connell MN, Barczak A, McGinnis T, Neymotin S, Schroeder CE, et al. The Thalamocortical Circuit of Auditory Mismatch Negativity. Biol Psychiatry. 2020;87:770–80.
Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–47.
Gunduz-Bruce H, Reinhart RM, Roach BJ, Gueorguieva R, Oliver S, D’Souza DC, et al. Glutamatergic modulation of auditory information processing in the human brain. Biol Psychiatry. 2012;71:969–77.
Heekeren K, Daumann J, Neukirch A, Stock C, Kawohl W, Norra C, et al. Mismatch negativity generation in the human 5HT2A agonist and NMDA antagonist model of psychosis. Psychopharmacol (Berl). 2008;199:77–88.
Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-d-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. 2015;116:57–67.
Rosburg T, Kreitschmann-Andermahr I. The effects of ketamine on the mismatch negativity (MMN) in humans - A meta-analysis. Clin Neurophysiol. 2016;127:1387–94.
Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci USA. 1996;93:11962–7.
Gil-da-Costa R, Stoner GR, Fung R, Albright TD. Nonhuman primate model of schizophrenia using a noninvasive EEG method. Proc Natl Acad Sci USA. 2013;110:15425–30.
Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, et al. Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull. 2010;83:147–61.
Ehrlichman RS, Maxwell CR, Majumdar S, Siegel SJ. Deviance-elicited changes in event-related potentials are attenuated by ketamine in mice. J Cogn Neurosci. 2008;20:1403–14.
Light GA, Swerdlow NR. Future clinical uses of neurophysiological biomarkers to predict and monitor treatment response for schizophrenia. Ann N Y Acad Sci. 2015;1344:105–19.
Greenwood LM, Leung S, Michie PT, Green A, Nathan PJ, Fitzgerald P, et al. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr Res. 2018;191:61–9.
Kantrowitz JT, Epstein ML, Beggel O, Rohrig S, Lehrfeld JM, Revheim N, et al. Neurophysiological mechanisms of cortical plasticity impairments in schizophrenia and modulation by the NMDA receptor agonist D-serine. Brain. 2016;139:3281–95. Pt 12
Kantrowitz JT, Epstein ML, Lee M, Lehrfeld N, Nolan KA, Shope C, et al. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: correlation with symptoms. Schizophr Res. 2018;191:70–9.
Lavoie S, Murray MM, Deppen P, Knyazeva MG, Berk M, Boulat O, et al. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology. 2008;33:2187–99.
Gilleen J, Nottage J, Yakub F, Kerins S, Valdearenas L, Uz T, et al. The effects of roflumilast, a phosphodiesterase type-4 inhibitor, on EEG biomarkers in schizophrenia: a randomised controlled trial. J Psychopharmacol. 2020:269881120946300.
Kantrowitz JT, Javitt DC, Freedman R, Sehatpour P, Kegeles LS, Carlson M, et al. Double blind, two dose, randomized, placebo-controlled, cross-over clinical trial of the positive allosteric modulator at the alpha7 nicotinic cholinergic receptor AVL-3288 in schizophrenia patients. Neuropsychopharmacology. 2020;45:1339–45.
Preskorn SH, Gawryl M, Dgetluck N, Palfreyman M, Bauer LO, Hilt DC. Normalizing Effects of EVP-6124, an Alpha-7 Nicotinic Partial Agonist, on Event-Related Potentials and Cognition: a Proof of Concept, Randomized Trial in Patients with Schizophrenia. J Psychiatr Pr. 2014;20:12–24.
Adler LE, Cawthra EM, Donovan KA, Harris JG, Nagamoto HT, Olincy A, et al. Improved p50 auditory gating with ondansetron in medicated schizophrenia patients. Am J Psychiatry. 2005;162:386–8.
Hamilton HK, Perez VB, Ford JM, Roach BJ, Jaeger J, Mathalon DH. Mismatch Negativity But Not P300 Is Associated With Functional Disability in Schizophrenia. Schizophr Bull. 2018;44:492–504.
O’Donnell BF, Vohs JL, Krishnan GP, Rass O, Hetrick WP, Morzorati SL. The auditory steady-state response (ASSR): a translational biomarker for schizophrenia. Suppl Clin Neurophysiol. 2013;62:101–12.
Avissar M, Xie S, Vail B, Lopez-Calderon J, Wang Y, Javitt DC. Meta-analysis of mismatch negativity to simple versus complex deviants in schizophrenia. Schizophr Res. 2018;191:25–34.
Fisher DJ, Labelle A, Knott VJ. Alterations of mismatch negativity (MMN) in schizophrenia patients with auditory hallucinations experiencing acute exacerbation of illness. Schizophr Res. 2012;139:237–45.
Perrin MA, Kantrowitz JT, Silipo G, Dias E, Jabado O, Javitt DC. Mismatch negativity (MMN) to spatial deviants and behavioral spatial discrimination ability in the etiology of auditory verbal hallucinations and thought disorder in schizophrenia. Schizophr Res. 2018;191:140–47.
Friedman T, Sehatpour P, Dias E, Perrin M, Javitt DC. Differential relationships of mismatch negativity and visual p1 deficits to premorbid characteristics and functional outcome in schizophrenia. Biol Psychiatry. 2012;71:521–9.
Donde C, Martinez A, Kantrowitz JT, Silipo G, Dias EC, Patel GH, et al. Bimodal distribution of tone-matching deficits indicates discrete pathophysiological entities within the syndrome of schizophrenia. Transl Psychiatry. 2019;9:16022.
Lee M, Sehatpour P, Dias EC, Silipo GS, Kantrowitz JT, Martinez AM, et al. A tale of two sites: differential impairment of frequency and duration mismatch negativity across a primarily inpatient versus a primarily outpatient site in schizophrenia. Schizophr Res. 2018;191:10–7.
Sehatpour P, Avissar M, Kantrowitz JT, Corcoran CM, De Baun HM, Patel GH, et al. Deficits in Pre-attentive Processing of Spatial Location and Negative Symptoms in Subjects at Clinical High Risk for Schizophrenia. Front Psychiatry. 2020;11:629144.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–77.
Shelley AM, Ward PB, Catts SV, Michie PT, Andrews S, McConaghy N. Mismatch negativity: an index of a preattentive processing deficit in schizophrenia. Biol Psychiatry. 1991;30:1059–62.
Revheim N, Corcoran CM, Dias E, Hellmann E, Martinez A, Butler PD, et al. Reading deficits in schizophrenia and individuals at high clinical risk: relationship to sensory function, course of illness, and psychosocial outcome. Am J Psychiatry. 2014;171:949–59.
Donde C, Martinez A, Sehatpour P, Patel GH, Kraut R, Kantrowitz JT, et al. Neural and functional correlates of impaired reading ability in schizophrenia. Sci Rep. 2019;9:16022.
Kantrowitz JT, Leitman DI, Lehrfeld JM, Laukka P, Juslin PN, Butler PD, et al. Reduction in tonal discriminations predicts receptive emotion processing deficits in schizophrenia and schizoaffective disorder. Schizophr Bull. 2013;39:86–93.
Kantrowitz JT, Hoptman MJ, Leitman DI, Moreno-Ortega M, Lehrfeld JM, Dias E, et al. Neural Substrates of Auditory Emotion Recognition Deficits in Schizophrenia. J Neurosci. 2015;35:14909–21.
Gold R, Butler P, Revheim N, Leitman DI, Hansen JA, Gur RC, et al. Auditory emotion recognition impairments in schizophrenia: relationship to acoustic features and cognition. Am J Psychiatry. 2012;169:424–32.
Thomas ML, Green MF, Hellemann G, Sugar CA, Tarasenko M, Calkins ME, et al. Modeling Deficits From Early Auditory Information Processing to Psychosocial Functioning in Schizophrenia. JAMA Psychiatry. 2017;74:37–46.
Lee M, Sehatpour P, Hoptman MJ, Lakatos P, Dias EC, Kantrowitz JT, et al. Neural mechanisms of mismatch negativity dysfunction in schizophrenia. Mol Psychiatry. 2017;22:1585–93.
Mitchell EA, Pratt JA. Neuroanatomical structures involved in the action of the 5-HT3 antagonist ondansetron: a 2-deoxyglucose autoradiographic study in the rat. Brain Res. 1991;538:289–94.
Karnani MM, Jackson J, Ayzenshtat I, Hamzehei Sichani A, Manoocheri K, Kim S, et al. Opening Holes in the Blanket of Inhibition: localized Lateral Disinhibition by VIP Interneurons. J Neurosci. 2016;36:3471–80.
Takesian AE, Bogart LJ, Lichtman JW, Hensch TK. Inhibitory circuit gating of auditory critical-period plasticity. Nat Neurosci. 2018;21:218–27.
Choueiry J, Blais CM, Shah D, Smith D, Fisher D, Illivitsky V, et al. CDP-choline and galantamine, a personalized alpha7 nicotinic acetylcholine receptor targeted treatment for the modulation of speech MMN indexed deviance detection in healthy volunteers: a pilot study. Psychopharmacol (Berl). 2020;237:3665–87.
de la Salle S, Shah D, Choueiry J, Bowers H, McIntosh J, Ilivitsky V, et al. NMDA Receptor Antagonist Effects on Speech-Related Mismatch Negativity and Its Underlying Oscillatory and Source Activity in Healthy Humans. Front Pharmacol. 2019;10:455.
Knott V, Impey D, Choueiry J, Smith D, de la Salle S, Saghir S, et al. An acute dose, randomized trial of the effects of CDP-Choline on Mismatch Negativity (MMN) in healthy volunteers stratified by deviance detection level. Neuropsychiatr Electrophysiol. 2015;1:1.
Acknowledgements
Nathaniel Heintz at Rockefeller University and the Howard Hughes Medical Institute provided critical insight into the selection of 5-HT3R as a therapeutic target for cognitive impairment associated with schizophrenia. Ricardo Soto and Emily Marschok at Halloran Consulting Group assisted with operational oversight of study conduct. Central operations for the study (e.g., data monitoring and integration) were managed by Clinilabs, Inc. Pharmacokinetic analyses were performed by PPD Laboratories. At Cerevance, Andrew Ayscough assisted with study drug importation; Rob Middlebrook managed budgets and contracting; and Brad Margus provided strategic guidance. Patricio O’Donnell (Takeda, Inc.) provided useful advice on study design. Stephanie Brazis, Megan Mayer, Natalie De-La-Garrigue, Melissa Conant, Anna Gwak, Beth Vayshenker, James Gangswich, Juliana Glasser and Amir Meftah assisted in the conduct of the study at NYSPI.
Funding
Funded by a grant by Cerevance to DCJ and JTK, and R01 MH49334 to DCJ. Drs DHM, MC, and NB are full-time employees of Cerevance. Dr JTK reports having received consulting payments within the last 24 months from Alphasights, Charles River Associates, Medscape, Putnam, techspert.io, Third Bridge, MEDACorp, Parexel, GroupH, Simon Kucher, ECRI Institute, ExpertConnect, Parexel, Schlesinger Group, CelloHealth, Acsel Health, Strafluence, Guidepoint, L.E.K. and Smart Analytic and System Analytic. He serves on the MedinCell Psychiatry and Karuna Mechanism of Action Advisory Boards. He has conducted clinical research supported by the NIMH, Sunovion, Roche, Alkermes, Cerevance, Corcept, Takeda, Taisho, Lundbeck, Boehringer Ingelheim, NeuroRX and Teva within the last 24 months. Dr JTK was a co-investigator on a study that receives lumeteperone and reimbursement for safety testing for an investigator-initiated research from Intra-Cellular Therapies Inc. He owns a small number of shares of common stock from GSK. Dr DCJ reports having received consulting payments within the last 2 years from Autifony, Biogen, SK Life Sciences, Boehringer Ingelheim, and Biogen. He holds intellectual property rights for use of NMDA modulators in treatment of neuropsychiatric disorders; for parcel-guided TMS treatment of depression; and for EEG-based diagnosis of neuropsychiatric disorders. He holds equity in Glytech, AASI, and NeuroRx.
Author information
Authors and Affiliations
Contributions
Dr JTK, DCJ, and PS had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors reviewed the final submission and gave final approval of the submitted version. Substantial contributions to conception and design: JTK, DCJ, PS, Marl-C, DHM, NB, MC. Acquisition, analysis, or interpretation of data: JTK, DCJ, HMdeB, AB, PS, and Marl-C. Drafting of the paper: PS, JTK, DCJ, DHM. Critical revision of the paper for important intellectual content: JTK, DCJ, PS, HMdeB.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sehatpour, P., Javitt, D.C., De Baun, H.M. et al. Mismatch negativity as an index of target engagement for excitation/inhibition-based treatment development: a double-blind, placebo-controlled, randomized, single-dose cross-over study of the serotonin type-3 receptor antagonist CVN058. Neuropsychopharmacol. 47, 711–718 (2022). https://doi.org/10.1038/s41386-021-01170-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01170-8
- Springer Nature Switzerland AG