
Abstract
The phase 3 COMMODORE trial evaluated gilteritinib versus salvage chemotherapy (SC) in a predominantly Asian relapsed/refractory (R/R) FLT3-mutated (FLT3mut+) acute myeloid leukemia (AML) patient population. The primary endpoint was overall survival (OS); secondary endpoints included event-free survival (EFS) and complete remission (CR) rate. As of June 30, 2020 (interim analysis: 32.2 months after study initiation), 234 patients were randomized (gilteritinib, n = 116; SC, n = 118). Median OS was significantly longer with gilteritinib versus SC (9.6 vs. 5.0 months; HR 0.566 [95% CI: 0.392, 0.818]; p = 0.00211) with a median follow-up of 10.3 months. Median EFS was also significantly longer with gilteritinib (2.8 vs. 0.6 months; HR 0.551 [95% CI: 0.395, 0.769]; p = 0.00004). CR rates with gilteritinib and SC were 16.4% and 10.2%, respectively; composite CR rates were 50.0% and 20.3%, respectively. Exposure-adjusted grade ≥3 adverse event (AE) rates were lower with gilteritinib (58.38 events/patient-year [E/PY]) versus SC (168.30 E/PY). Common AEs with gilteritinib were anemia (77.9%) and thrombocytopenia (45.1%). Gilteritinib plasma concentration peaked ~4 h postdose; ~3-fold accumulation occurred with multiple dosing. The COMMODORE trial demonstrated that gilteritinib significantly improved OS and EFS in predominantly Asian patients, validating the outcomes of gilteritinib from the ADMIRAL trial in R/R FLT3mut+ AML.

Similar content being viewed by others

Introduction
Due to poor prognosis, additional treatment options for patients with FLT3-mutated (FLT3mut+) acute myeloid leukemia (AML) who are refractory to therapy or have relapsed (R/R) are needed globally. For patients with R/R FLT3mut+ AML, remission rates with salvage chemotherapy (SC) appear lower, duration of remission is shorter, and overall survival (OS) is decreased relative to patients without FLT3 mutations [1,2,3]. Activated FLT3 with internal tandem duplication (ITD) in and around the juxtamembrane domain is present in up to 35% of AML cases; tyrosine kinase domain (TKD) point mutations are present in 7–10% of AML cases [3]. Compared with patients without FLT3-ITD mutations, those with FLT3-ITD mutations have a higher relapse rate and reduced disease-free survival and OS [4,5,6,7,8]. FLT3-TKD mutations are associated with acquired resistance to quizartinib and sorafenib [9, 10].
Gilteritinib has activity against FLT3-ITD and FLT3-TKD mutations, is approved in multiple countries, including Japan and South Korea, and has recently received conditional approval in China for the treatment of R/R FLT3mut+ AML [11,12,13]. In the phase 3 ADMIRAL trial, superior survival benefit and favorable safety profile were shown for patients receiving gilteritinib versus those receiving SC (hazard ratio [HR] 0.64 [95% CI: 0.49, 0.83]; p < 0.001) in the R/R FLT3mut+ AML setting [14].
To date, published evidence related to gilteritinib in Asian patients with R/R FLT3mut+ AML is limited to uncontrolled, nonrandomized studies [15, 16] and a subgroup analysis of Japanese patients from the ADMIRAL trial [17]. Data from the Japanese subgroup of ADMIRAL were based on a total of 48 patients of whom 33 received gilteritinib and 15 received SC [17]. Larger randomized, controlled trials of outcomes in a predominantly Asian population were therefore required. Furthermore, the phase 3 COMMODORE study was initiated in China, Russia, Singapore, Thailand, and Malaysia. Per requirements of the Center for Drug Evaluation National Medical Products Administration in China, the clinical benefit of gilteritinib in Chinese patients with R/R FLT3mut+ AML from COMMODORE was used to support full approval for gilteritinib in China. The objective of the COMMODORE study was to analyze the efficacy and safety of gilteritinib versus SC, as well as the pharmacokinetics (PK) of gilteritinib in predominantly Asian patients with R/R FLT3mut+ AML.
Methods
Study design
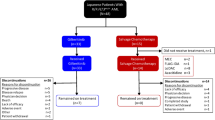
COMMMODORE (NCT03182244) is an ongoing phase 3, open-label, multicenter, randomized study in patients with R/R FLT3mut+ AML (Fig. S1). Patients with R/R FLT3mut+ AML from ~50 centers in China, Russia, Singapore, Thailand, and Malaysia were randomized 1:1 to gilteritinib or SC. Adults (aged ≥18 or per local regulations) with primary AML or AML secondary to myelodysplastic syndrome who were refractory to or had relapsed after first-line AML therapy (≥1 cycle of standard dose anthracycline containing induction therapy or other induction therapy considered the optimum choice per investigator assessment). Patients had an Eastern Cooperative Oncology Group (ECOG) performance status ≤2 and were positive for FLT3-ITD or FLT3-TKD D835 or I836 mutations in bone marrow or whole blood per central laboratory testing using previously described polymerase chain reaction methodology where a mutant-to-nonmutant allelic ratio ≥0.05 denoted FLT3 mutation positivity [14]. Patients diagnosed with acute promyelocytic leukemia, BCR::ABL1-positive leukemia, AML secondary to prior chemotherapy, or active central nervous system disease were excluded. Other exclusion criteria were current or history of New York Heart Association class 3 or 4 congestive heart failure, mean of triplicate Fridericia corrected QT interval >450 msec, long QT syndrome, coagulation abnormality (e.g., disseminated intravascular coagulation), hypokalemia or hypomagnesemia, active hepatitis B or C, human immunodeficiency virus, or other uncontrolled infection, active hepatic disorder, clinically significant graft-versus-host disease (GVHD) or systemic corticosteroid therapy for GVHD.
Before randomization, a chemotherapy regimen was investigator preselected for each patient from either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, high-dose cytarabine, and granulocyte colony-stimulating factor (FLAG); chemotherapy regimens are listed in Table S1. FLAG was chosen as an SC option over FLAG plus idarubicin (FLAG-Ida), which was administered in the ADMIRAL trial, based on the results of a survey of physicians across several Asian countries. Survey results indicated a preference for FLAG as the standard of care because it was considered less toxic than FLAG-Ida. Because azacitidine is not approved for AML in China, it was not included as a chemotherapy option in the COMMODORE trial. Patients randomized to MEC or FLAG received one cycle of therapy and were assessed for response on or after Day 15. The decision to continue treatment with MEC or FLAG or observe for recovery was based on bone marrow cellularity. Patients randomized to gilteritinib or LoDAC continued treatment until a discontinuation criterion was met. Patients in the SC arm could not cross over to the gilteritinib arm prior to the interim analysis. Gilteritinib was administered at a starting dose of 120 mg/day.
Randomization was stratified by response to first-line therapy and preselected chemotherapy. Patients had an end of treatment visit within 7 days after treatment discontinuation, followed by a 30-day follow-up, and subsequently entered a long-term follow-up period for collection of patient-reported outcomes, subsequent AML treatment, remission status, and survival. A total of 21 patients enrolled from designated sites in China who were randomized into the gilteritinib arm participated in the PK cohort. Patients in the PK cohort were administered the study drug in the same manner and underwent the same efficacy/safety assessments as other patients except for blood sampling for additional gilteritinib PK measurements.
Dose interruptions/reductions for gilteritinib were permitted according to prespecified criteria. Stepwise dose reduction to 80 or 40 mg/day was permitted after the patient experienced a clinical benefit or in cases of myelosuppression. Dose escalation to gilteritinib 200 mg per day was permitted based on bone marrow and hematology results in patients not achieving a composite complete remission (CRc; defined as combined complete remission [CR], CR with incomplete hematologic recovery [CRi], and CR with incomplete platelet recovery [CRp]) after Cycle 1. Patients in the gilteritinib arm could undergo hematopoietic stem cell transplantation (HSCT) without leaving the trial; gilteritinib was stopped and a pre-HSCT visit performed prior to starting the HSCT conditioning regimen. Following HSCT, patients could resume gilteritinib 30 to 90 days after transplantation if they had successful engraftment without relapse or no uncontrolled complications of transplantation. Treatment was administered over continuous 28-day cycles.
Endpoints and assessments
The primary endpoint was OS. Key secondary efficacy endpoints were event-free survival (EFS) and CR rate. Response to treatment was assessed according to modified International Working Group criteria [18] (Table S2) as was done in the ADMIRAL trial [14]. As such, CR was defined as the presence of bone marrow regenerating normal hematopoietic cells with achievement of a morphologic leukemia-free state, absolute neutrophil count (ANC) > 1 × 109/L, platelet count ≥100 × 109/L, normal marrow differential with <5% blasts, and red blood cell and platelet transfusion independence. The definition of CRp was fulfillment of all CR criteria except for platelet recovery (platelet count ≥100 × 109/L). The definition of CRi was fulfillment of all CR criteria except for hematologic recovery (ANC ≥ 1 × 109/L) with residual neutropenia, complete platelet recovery (platelet count ≥100 × 109/L), and red blood cell and platelet transfusion independence. EFS was defined as the time from the date of randomization until the date of documented relapse (including relapse after CR, CRp, and CRi), treatment failure (discontinuation of treatment without previous response), reported off-treatment relapse or initiation of a new AML therapy (excluding subsequent on-study HSCT), or all-cause death, whichever occurred first. For treatment failure, the event date was defined as the randomization date. Additional secondary efficacy endpoints were leukemia-free survival, duration of remission, and CRc rate; safety and PK were also evaluated. Other secondary and exploratory endpoints including pharmacogenomic, pharmacodynamic, and patient-reported outcomes, and healthcare resource utilization were evaluated, but are not reported herein.
For patients receiving gilteritinib or LoDAC (low-intensity regimen), bone marrow samples or biopsy were required during screening and on Day 1 of Cycles 2 and 3. Patients not achieving CRc had bone marrow assessments repeated at Day 1 of every two subsequent cycles. Patients achieving CRc had bone marrow sampling repeated 1 month after the date of remission and every three subsequent cycles or if there was suspicion of relapse. For the MEC and FLAG groups (high-intensity regimens), bone marrow samples were required during screening and at Day 1 of Cycle 2. An additional bone marrow sample was required at Cycle 1, Day 15 or later (per institutional guidelines) to evaluate the need for a second cycle. Blood sampling for PK analyses were obtained at prespecified time points (Table S3).
Statistical analyses
This study implemented a group-sequential design using the O’Brien-Fleming boundaries for OS and Pocock boundaries for EFS and CR rate as implemented by Lan-DeMets alpha spending method with one interim and one final analysis planned. An interim analysis, conducted by an independent data monitoring committee (IDMC), was planned when ~50% of deaths had occurred. EFS and CR rate were analyzed according to the sequential testing procedure on controlling overall type I error rate for multiple endpoints; EFS was tested when the null hypothesis of OS was rejected at the interim or final analyses, and CR rate was tested when the null hypotheses of both OS and EFS were rejected hierarchically at the interim or final analyses. Significance levels for EFS and CR rate were based on Pocock alpha spending function. All statistical tests of treatment effects were conducted at a two-sided 0.05 level of significance, unless otherwise specified. Assuming a 1:1 randomization ratio with a 10% dropout rate, a planned sample of ~318 patients would provide the trial with 90% power to detect a difference in median OS between the gilteritinib group (7.7 months) and SC (5.0 months) (HR for death of 0.65) on the basis of 230 deaths.
OS and EFS were analyzed using a stratified log-rank test and a stratified Cox proportional hazards model (with strata to control for response to first-line AML therapy and preselected SC) for all patients randomized to treatment (intention-to-treat population). Prespecified sensitivity analyses were performed for OS and EFS for all randomized patients with FLT3 mutation based on a central test (full analysis population). Subgroup analyses for the primary efficacy endpoints were planned. Response rates were analyzed with the Cochran-Mantel-Haenszel test (to control for response to first-line AML therapy and preselected SC) and duration of remission and leukemia-free survival with stratified Cox proportional hazards model in all randomized patients. Survival curves and median time-to-event variables were estimated using the Kaplan–Meier method and reported with corresponding 95% confidence intervals (CIs). Median follow-up duration was calculated according to a reverse Kaplan–Meier estimate.
Safety/tolerability were analyzed in all patients who received ≥1 dose of study treatment; data were summarized descriptively. Safety evaluations were based on adverse events (AEs; graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03), clinical laboratory values, vital signs, electrocardiogram results, and ECOG performance status. PK analyses were evaluated in patients with sufficient plasma concentration data available to facilitate derivation of ≥1 PK parameter and for whom the dosing time on the day of sampling was known. Non-compartmental analysis was performed after single and multiple dosing of gilteritinib from Chinese patients in the PK cohort. PK parameters were summarized using descriptive statistics.
Trial oversight
The trial protocol was approved by institutional review boards or independent ethics committees at participating sites. This trial was conducted in accordance with the study protocol, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines, and the applicable regulations and guidelines governing clinical study conduct and ethical principles with their origin in the Declaration of Helsinki. Written and signed informed consent were obtained from all patients or their guardian or legal representative prior to screening. The study sponsor ensured that the use and disclosure of protected health information obtained during the trial complied with federal and/or regional legislation related to the privacy and protection of personal information.
Results
Patient demographics and disposition
As of June 30, 2020 (interim analysis data cutoff date; 32.2 months from the date of study initiation), 234 patients were randomized (gilteritinib, n = 116; SC, n = 118). Of the 118 patients in the SC group, 57 received FLAG, 29 received MEC, and 18 received LoDAC. Most patients were of Asian origin (85.9%; n = 201/234), with 64.5% (n = 151/234) from China. Median age was 50.5 years in the total population, and 51.5 and 49.5 years in the gilteritinib and SC groups, respectively; most patients had not previously received FLT3 inhibitors (87.9% and 93.2%, respectively) (Table 1). Baseline FLT3 mutations in the gilteritinib versus SC groups were: FLT3-ITD (91.4% vs. 83.1%), FLT3-TKD (6.0% vs. 11.9%), and both FLT3-ITD and FLT3-TKD (2.6% vs. 5.1%). Based on the results of the interim analysis for efficacy demonstrating a favorable outcome for gilteritinib compared with SC, the IDMC recommended the study be stopped. The most common reasons for treatment discontinuation were disease relapse (25.9%, n = 30/116) in the gilteritinib group and lack of efficacy (33.1%, n = 39/118) in the SC group (Fig. 1). In patients receiving SC, the median duration of LoDAC was 28.0 days (range, 9–113 days), of MEC was 28.0 days (range, 2–97 days), and of FLAG was 28.0 days (8–73 days). The median duration of treatment with gilteritinib was 113.0 days (range, 7–746). Eighty-six patients received high-intensity chemotherapy (FLAG or MEC); of these patients, 66 (FLAG, n = 45; MEC, n = 21) received one treatment cycle and 20 (FLAG, n = 12: MEC, n = 8) received two treatment cycles.

Reasons for exclusion prior to randomization were negative FLT3 mutation status, unclear relapsed/refractory status, other inclusion criteria not met, patient withdrawal, other exclusion criteria met, investigator decision, medical records for hospitalization not available, death, and economic reasons. *Indicates patients on high-dose chemotherapy who completed one cycle of treatment with a composite complete remission and were taken off of treatment or completed two cycles of treatment. FLAG fludarabine, high-dose cytarabine, and granulocyte colony-stimulating factor, FLT3 FMS-like tyrosine kinase 3, MEC mitoxantrone, etoposide, and intermediate-dose cytarabine.
After first-line therapy, 55.6% of patients were primary refractory without HSCT (gilteritinib, 56.9%; SC, 54.2%) and 44.4% had other relapsed disease (gilteritinib, 43.1%; SC, 45.8%) (Table 1). Across both treatment groups, most patients had not received prior FLT3 inhibitor therapy (gilteritinib, 87.9%; SC, 93.2%) or prior HSCT (gilteritinib, 97.4%; SC, 95.8%). Among the 22 patients who received prior FLT3 inhibitor therapy (gilteritinib, n = 14; SC, n = 8), 19 had previously received sorafenib (gilteritinib, n = 11; SC, n = 8) and 3 (gilteritinib, n = 3; SC, n = 0) had previously received midostaurin.
Efficacy
As of the interim analysis data cutoff date of June 30, 2020, 124 deaths had occurred (62 deaths in each group). Median follow-up duration for OS was 10.3 months for the total study population, 11.2 months for the gilteritinib arm, and 8.1 months for the SC arm. Median OS was significantly longer with gilteritinib (9.6 months) versus SC (5.0 months; HR 0.566 [95% CI: 0.392, 0.818]; p = 0.00211; predetermined two-sided significance level of 0.00454 based on the number of observed OS events; Fig. 2A); 1-year survival rates were 35.1% and 25.1%, respectively. Median OS censored at the time of HSCT was also significantly longer in the gilteritinib arm compared with the SC arm (median OS 9.6 vs. 4.6 months; HR 0.506 [95% CI: 0.343, 0.745]; p = 0.00043). A consistent pattern of longer survival with gilteritinib compared with SC was also observed across multiple subgroups, including both high-intensity (FLAG or MEC) and low-intensity (LoDAC) chemotherapy subgroups (Fig. 2B). Notably, the survival benefit was greater in males (HR 0.354 [95% CI: 0.201, 0.625]) than in females (HR 0.822 [95% CI: 0.515, 1.312]).

A Kaplan–Meier curve of overall survival (primary endpoint). The intention-to-treat population includes all randomized patients. The predetermined two-sided significance level was 0.00454 based on the number of observed overall survival events. B Subgroup analyses of overall survival. Preselected high-intensity therapy included FLAG or MEC; preselected low-intensity chemotherapy consisted of LoDAC. CI confidence interval, CRc composite complete remission, ECOG Eastern Cooperative Oncology Group, FLAG fludarabine, high-dose cytarabine, and granulocyte colony-stimulating factor, FLT3 FMS-like tyrosine kinase 3, HR hazard ratio, HSCT hematopoietic stem cell transplantation, IRT interactive response technology, ITD internal tandem duplication, LoDAC low-dose cytarabine, MEC mitoxantrone, etoposide, and intermediate-dose cytarabine, NE not estimable, SC salvage chemotherapy, TKD tyrosine kinase domain.
Patients on gilteritinib had statistically significantly longer EFS than those receiving SC (median EFS 2.8 vs. 0.6 months; HR 0.551 [95% CI: 0.395, 0.769]; p = 0.00004; predetermined two-sided significance level of 0.04011; Fig. 3). CR rates with gilteritinib and SC were 16.4% and 10.2%, respectively (5.9% [95% CI: −2.7%, 14.6%]; p = 0.17690; predetermined two-sided significance level of 0.04087 based on the number of randomized patients). Rates of CR in patients with FLT3-ITD mutations only were 17.9% (n = 19/106) in the gilteritinib group and 9.2% (n = 9/98) in the SC group; corresponding CR rates were 0 (n = 0/7) and 14.3% (n = 2/14), respectively, in patients with FLT3-TKD mutations only and 0 (n = 0/3) and 16.7% (n = 1/6), respectively, in patients with both FLT3-ITD and FLT3-TKD mutations. The median duration of CR was 15.8 months in the gilteritinib group and could not be evaluated in the SC group given that only one patient relapsed during the treatment period, which was limited to one or two treatment cycles in the high-intensity subgroup of the SC arm. CRc rates were 50.0% for gilteritinib and 20.3% for SC (treatment difference 29.5% [95% CI: 17.8%, 41.3%]; p < 0.00001). Additional antileukemic response data are shown in Table 2. During the study, transplantation rates were 19.8% (n = 23) for gilteritinib and 5.9% (n = 7) for SC (treatment difference 13.9% [95% CI: 5.5%, 22.3%]; p = 0.00151). Best overall response rates before transplantation are shown in Table S4, with CR and CRc rates of 12.9% and 47.4%, respectively, with gilteritinib, and 10.2% and 20.3% with SC. Of the 23 patients in the gilteritinib arm who underwent transplantation during the study, 19 underwent on-study transplantation and 13 patients were eligible to resume gilteritinib maintenance treatment post-transplantation.

Kaplan–Meier curve of event-free survival (a key secondary endpoint). The predetermined two-sided significance level was 0.04011. CI confidence interval, HR hazard ratio.
Safety
The median duration of treatment exposure was 113 days in the gilteritinib group, 28 days in the LoDAC group, 28 days in the MEC group, and 28 days in the FLAG group; treatment exposure was 46.4 patient-years in the gilteritinib group and 10.0 patient-years in the SC group. Among patients in the gilteritinib group, 96.5% (n = 109/113) experienced dose interruptions, 34.5% (n = 39/113) experienced dose decreases, and 27.4% (n = 31/113) experienced dose increases. Gilteritinib dose increases due to lack of achievement of CRc occurred in 21.2% of patients (n = 24/113).
The AE incidence, including those considered by the investigator to be drug-related, were comparable between the gilteritinib (100.0% and 91.2%) and SC (100.0% and 91.3%) groups. Almost all patients (gilteritinib, 97.3% and SC, 100.0%) had AEs that occurred within the first 30 days of study treatment. Grade ≥3 AEs in the gilteritinib (97.3%) versus SC (94.2%) groups were also comparable; rates for serious AEs were higher for gilteritinib (74.3%) versus SC (61.5%). When adjusted for treatment exposure, AE rates were lower with gilteritinib (grade ≥3, 58.38 events/patient-year [E/PY]; serious, 6.19 E/PY) than with SC (grade ≥3, 168.30 E/PY; serious, 12.40 E/PY; Table S5). The most common AEs occurring in the gilteritinib group were anemia (77.9%), thrombocytopenia (45.1%), pyrexia (44.2%), and increased blood lactate dehydrogenase (44.2%). For SC, the most common AEs were anemia (64.4%), decreased white blood cell count (41.3%), hypokalemia (38.5%), and decreased platelet count (38.5%) (Table 3). Anemia was the only grade ≥3 AE occurring in over 50% of patients in the gilteritinib (71.7%) and SC (59.6%) groups, respectively. Common serious AEs included febrile neutropenia (gilteritinib 23.9% vs. SC 23.1%) and pneumonia (gilteritinib 14.2% vs. SC 6.7%). Differentiation syndrome (considered possibly related to study treatment) was reported in one (0.9%) patient in the gilteritinib group and no (0.0%) patients in the SC group. Additional detail on signs and symptoms of differentiation syndrome can be found in Table S6. A total of 10 patients in the gilteritinib group and none in the SC group had prolonged QT intervals; one of these patients experienced grade ≥3 QT prolongation. Overall, four patients in the gilteritinib group experienced dose reductions due to QT prolongation; in one patient, QT prolongation led to discontinuation of gilteritinib.
AEs leading to death occurred in 22 (19.5%) and 15 (14.4%) of patients receiving gilteritinib or SC, respectively. Drug-related AEs led to death in nine (8.0%) patients in the gilteritinib group and seven (6.7%) patients in the SC group; events occurring in more than one patient were neutropenic sepsis (n = 3) and febrile neutropenia (n = 2) in the gilteritinib group and septic shock (n = 3) in the SC group.
Pharmacokinetics
A second cutoff date of November 13, 2020, was used for PK analyses. The plasma concentration of gilteritinib reached the peak in ~4 h (Fig. S2) after dosing and declined with an estimated half-life ranging from 16.6 to 130 h (Table S7). The median trough concentration (Ctrough) of gilteritinib on Day 15 was 247 ng/mL. Accumulation was demonstrated to occur with multiple dosing, with an approximately 3-fold mean accumulation ratio (Table S8).
Discussion
Treatment options for patients with AML have expanded in the past decade with the emergence of several targeted therapies against FLT3, IDH1 and IDH2, and BCL-2 [19]. Gilteritinib was the first FLT3-targeted therapy to be approved for the treatment of R/R FLT3mut+ AML. The ADMIRAL trial established the benefit of gilteritinib in patients with R/R FLT3mut+ AML [14], which led to the approval of gilteritinib for this indication in Japan, the US, and the EU [20]. In February 2021, gilteritinib was granted conditional approval by the China National Medical Products Administration based on the results of the ADMIRAL trial [11]. To expand the evidence base in an Asian patient population, the COMMODORE study was conducted in a predominantly Asian population, which included 64.5% of patients from China. Preselected chemotherapy options in the SC group of the COMMODORE trial differed slightly from ADMIRAL, with FLAG being used instead of FLAG-Ida and the absence of azacitidine. The decision to use FLAG was based on a survey of Asian physicians that demonstrated a preference for FLAG, which was perceived to be less toxic than FLAG-Ida. Azacitidine was not included as a low-intensity chemotherapy option because it is not indicated for AML in China. By design, in both the COMMODORE and ADMIRAL trials, duration of exposure to gilteritinib was longer than exposure to SC [14]. Although the proportions of patients with FLT3-TKD mutations alone were balanced in the gilteritinib and SC arms (8.5% and 8.1%, respectively) of the ADMIRAL trial [14], a higher proportion of patients in the SC (11.9%) versus the gilteritinib group (6.0%) of COMMODORE had FLT3-TKD mutations. Although patients in COMMODORE were younger (median age, 50.5 years) than those in ADMIRAL (median age, 62.0 years) [14], the efficacy and safety profiles of gilteritinib compared with SC were consistent with and affirm the results from the ADMIRAL trial. Median OS for gilteritinib versus SC was 9.3 versus 5.6 months in the ADMIRAL trial [14] and 9.6 versus 5.0 months in the COMMODORE trial; risk of death was reduced by 36% and 43% in the respective trials. Of note, the HR for death in the subgroup of patients with primary refractory disease without HSCT was 0.437 (95% CI: 0.262, 0.727), in favor of gilteritinib over SC; this is in contrast to findings in ADMIRAL wherein the HR for death was 0.99 (95% CI: 0.63, 1.55) in the same subgroup [14]. COMMODORE data demonstrated efficacy of gilteritinib in patients with refractory disease. A greater survival benefit with gilteritinib was observed in male versus female patients; however, the reason for this difference is unclear. Median EFS for gilteritinib versus SC was 2.8 versus 0.7 months in ADMIRAL [14] and 2.8 versus 0.6 months in COMMODORE.
Although studies of gilteritinib in Asian populations are limited, the Japanese subgroup of the ADMIRAL trial demonstrated a median OS of 14.3 months with gilteritinib and 9.6 months with SC; however, the difference was not significant (HR 0.605 [95% CI: 0.236, 1.549]) [17]. Rates of CRc in the gilteritinib and SC arms of the Japanese subgroup were 57.6% and 20.0%, respectively, which were comparable to CRc rates in COMMODORE (50.0% and 20.3%, respectively) [17]. Interim results from a Japanese postmarketing surveillance of gilteritinib in real-world settings reported a CRc rate of 62.7% and median time to reach CRc of 1.6 months, which was comparable to the COMMODORE trial (median time to reach CRc, 1.9 months) [16].
As observed in ADMIRAL [14], exposure-adjusted AE rates for patients in the COMMODORE trial favored gilteritinib versus SC. AEs in patients in the COMMODORE trial were generally consistent with those of previous Japanese subgroups and with the overall FLT3mut+ AML population in prior clinical and real-world studies [14,15,16,17]. In the ADMIRAL Japanese subgroup analysis, the most common treatment-emergent AEs in patients receiving gilteritinib were febrile neutropenia (55%), increased aspartate aminotransferase (AST; 52%), increased alanine aminotransferase (ALT; 46%), anemia (39%), constipation (39%), increased blood creatinine phosphokinase (39%), decreased platelet count (39%), and nausea (36%); a higher incidence of drug-related grade ≥3 febrile neutropenia (36% vs. 21%), increased ALT (18% vs. 0%), and increased AST (18% vs. 0%) were seen with gilteritinib versus SC [17]. In this study with a broader population of patients in Asia, the incidence of increased ALT and AST appeared lower than previously reported, although rates of all grade events were higher than in the SC group. Rates of grade ≥3 increased ALT and AST appeared comparable between groups.
Exposure parameters after gilteritinib 120 mg administration in Chinese patients with R/R FLT3mut+ AML in the PK cohort were similar with those observed in other studies [15, 21, 22]. These results support that the same dose regimen is applicable to Chinese patients with R/R FLT3mut+ AML. The median Ctrough of gilteritinib in the COMMODORE PK cohort (247 ng/mL) was comparable to that observed with 120 mg gilteritinib monotherapy in patients with R/R FLT3mut+ AML in the ADMIRAL trial (279 ng/mL) and patients with R/R AML in the CHRYSALIS trial (227 ng/mL) [14, 22] but was lower than the value observed with 120 mg gilteritinib plus azacitidine or gilteritinib alone in newly diagnosed patients with FLT3mut+ AML in the phase 3 LACEWING trial (584.5 ng/mL) [23]. Although reasons for the disparity in Ctrough values between patients with R/R AML and those with newly diagnosed AML have not been elucidated, differences in patient age and duration of drug exposure may be contributing factors.
In conclusion, results of the COMMODORE trial further validate and affirm the clinical efficacy and safety data from the ADMIRAL trial in a predominantly Asian population, reinforcing the significant benefit of gilteritinib in R/R FLT3mut+ AML. In addition to the use of gilteritinib in the R/R AML setting, findings from a recent phase 1b study (NCT02236013) suggest that gilteritinib is well tolerated and clinically beneficial in the frontline setting when administered as part of induction and consolidation therapy for newly diagnosed AML, demonstrating a CRc rate of 89% and a median OS of 46.1 months in patients with FLT3 mutation [24]. Another ongoing trial is evaluating efficacy and safety of frontline gilteritinib versus midostaurin as part of induction and consolidation therapy in patients with FLT3mut+ AML (NCT03836209) [25]. An ongoing phase 1/2 trial (NCT04240002) is investigating safety, tolerability, and clinical response with gilteritinib plus induction chemotherapy in pediatric and young adult patients with R/R FLT3-ITD+ AML [26]. Evaluation of combination therapy with gilteritinib, venetoclax, and azacitidine in frontline and R/R AML settings is also underway in a phase 1/2 trial [27].
Data availability
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
References
Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301.
Chevallier P, Labopin M, Turlure P, Prebet T, Pigneux A, Hunault M, et al. A new Leukemia Prognostic Scoring System for refractory/relapsed adult acute myelogeneous leukaemia patients: a GOELAMS study. Leukemia. 2011;25:939–44.
Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312.
Moreno I, Martin G, Bolufer P, Barragan E, Rueda E, Roman J, et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica. 2003;88:19–24.
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–84.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia. 2005;19:1345–9.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9.
Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43.
Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3.
Astellas Pharma, Inc. Astellas’ XOSPATA® (gilteritinib) receives conditional approval by China’s National Medical Products Administration for relapsed or refractory acute myeloid leukemia with a FLT3 mutation. 2021. https://www.astellas.com/en/news/23246.
Astellas Pharma, Inc. Astellas announces approval in Japan for Xospata 40 mg tablets for the treatment of FLT3mut+ relapsed or refractory AML. 2018. https://www.astellas.com/system/files/news/2018-09/180921_3_En.pdf.
Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129:257–60.
Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381:1728–40.
Usuki K, Sakura T, Kobayashi Y, Miyamoto T, Iida H, Morita S, et al. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory acute myeloid leukemia: an open-label phase 1 study. Cancer Sci. 2018;109:3235–44.
Sugamori H, Lee T, Mitomi T, Yamagishi C. Interim results from a postmarketing surveillance study of patients with FLT3-mutated relapsed/refractory AML treated with the FLT3 inhibitor gilteritinib in Japan. Jpn J Clin Oncol. 2022;52:766–73.
Hosono N, Yokoyama H, Aotsuka N, Ando K, Iida H, Ishikawa T, et al. Gilteritinib versus chemotherapy in Japanese patients with FLT3-mutated relapsed/refractory acute myeloid leukemia. Int J Clin Oncol. 2021;26:2131–41.
Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21:4642–9.
Ahmadmehrabi K, Haque AR, Aleem A, Griffiths EA, Roloff GW. Targeted therapies for the evolving molecular landscape of acute myeloid leukemia. Cancers. 2021;13:4646.
Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79:331–9.
James AJ, Smith CC, Litzow M, Perl AE, Altman JK, Shepard D, et al. Pharmacokinetic profile of gilteritinib: a novel FLT-3 tyrosine kinase inhibitor. Clin Pharmacokinet. 2020;59:1273–90.
Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18:1061–75.
Wang ES, Montesinos P, Minden MD, Lee JH, Heuser M, Naoe T, et al. Phase 3 trial of gilteritinib plus azacitidine vs azacitidine for newly diagnosed FLT3mut+ AML ineligible for intensive chemotherapy. Blood. 2022;140:1845–57.
Pratz KW, Cherry M, Altman JK, Cooper BW, Podoltsev NA, Cruz JC, et al. Gilteritinib in combination with induction and consolidation chemotherapy and as maintenance therapy: a phase IB study in patients with newly diagnosed AML. J Clin Oncol. 2023;41:4236–46.
Clinicaltrials.gov. Gilteritinib vs midostaurin in FLT3 mutated acute myeloid leukemia. 2024. https://www.clinicaltrials.gov/study/NCT03836209?term=NCT03836209&rank=1.
ClinicalTrials.gov. A study of gilteritinib (ASP2215) combined with chemotherapy in children, adolescents and young adults with FMS-like tyrosine kinase 3 (FLT3)/internal tandem duplication (ITD) positive relapsed or refractory acute myeloid leukemia (AML). 2024. https://clinicaltrials.gov/study/NCT04240002?tab=results.
ClinicalTrials.gov. Azacitidine, venetoclax, and gilteritinib in treating patients with recurrent/refractory FLT3-mutated acute myeloid leukemia, chronic myelomonocytic leukemia, or high-risk myelodysplastic syndrome/myeloproliferative neoplasm. 2024. https://www.clinicaltrials.gov/study/NCT04140487.
Acknowledgements
Medical writing/editorial support was provided by Stephanie Phan, PharmD, and Cheryl Casterline, MA, from Peloton Advantage, LLC, an OPEN Health company, and funded by the study sponsor.
Funding
This study was funded by Astellas Pharma, Inc.
Author information
Authors and Affiliations
Contributions
JW was involved in the conception of the study design; all authors contributed to the acquisition of study data; JW, MY, TS, TK, MT, MK, XM, and NH analyzed the study data. JW, MY, TS, TK, MT, MK, XM, and NH participated in the interpretation of study data. All authors participated in the drafting of the manuscript, provided final approval of the submitted version, and agree to be held accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
MY, TS, TK, MT, MK, XM, and NH are all employees of Astellas. TS reports Astellas stocks. BJ, JL, LL, XD, HJ, LG, JT, LWLL, AK, and EM report consulting and honoraria fees from Astellas. JW, JH, and SB have nothing to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Jiang, B., Li, J. et al. Phase 3 study of gilteritinib versus salvage chemotherapy in predominantly Asian patients with relapsed/refractory FLT3-mutated acute myeloid leukemia. Leukemia (2024). https://doi.org/10.1038/s41375-024-02382-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41375-024-02382-9
- Springer Nature Limited