
Abstract
In chronic lymphocytic leukemia (CLL), survival of neoplastic cells depends on microenvironmental signals at lymphoid sites where the crosstalk between the integrin VLA-4 (CD49d/CD29), expressed in ~40% of CLL, and the B-cell receptor (BCR) occurs. Here, BCR engagement inside–out activates VLA-4, thus enhancing VLA-4-mediated adhesion of CLL cells, which in turn obtain pro-survival signals from the surrounding microenvironment. We report that the BCR is also able to effectively inside–out activate the VLA-4 integrin in circulating CD49d-expressing CLL cells through an autonomous antigen-independent BCR signaling. As a consequence, circulating CLL cells exhibiting activated VLA-4 express markers of BCR pathway activation (phospho-BTK and phospho-PLC-γ2) along with higher levels of phospho-ERK and phospho-AKT indicating parallel activation of downstream pathways. Moreover, circulating CLL cells expressing activated VLA-4 bind soluble blood-borne VCAM-1 leading to increased VLA-4-dependent actin polymerization/re-organization and ERK phosphorylation. Finally, evidence is provided that ibrutinib treatment, by affecting autonomous BCR signaling, impairs the constitutive VLA-4 activation eventually decreasing soluble VCAM-1 binding and reducing downstream ERK phosphorylation by circulating CLL cells. This study describes a novel anchor-independent mechanism occurring in circulating CLL cells involving the BCR and the VLA-4 integrin, which help to unravel the peculiar biological and clinical features of CD49d+ CLL.

Similar content being viewed by others


Introduction
CD49d, the alpha chain of the CD49d/CD29 (alpha4/beta1) integrin heterodimer (hereafter very-late-antigen-4, VLA-4), expressed in about 40% of chronic lymphocytic leukemia (CLL) cases, is one of the most relevant prognostic biomarkers in CLL [1,2,3]. Functionally, in the context of CLL-involved tissues, VLA-4 mediates both cell–cell and cell–matrix interactions by binding to its ligands vascular cell adhesion molecule-1 (VCAM-1), fibronectin [4], and Elastin MIcrofibriL INterfacer-1 (EMILIN-1) [5], thus delivering pro-survival signals and protecting CLL cells from drug-induced apoptosis [6,7,8,9].
VLA-4, present on the cell surface in an inactive conformation with low affinity for its ligand, can switch to a high-affinity/activated state in response to several stimuli, including those deriving from chemokine receptor or immuno-receptor engagement, according to a mechanism known as inside–out activation [10, 11].
The critical role played by the B-cell receptor (BCR) in CLL development and progression is demonstrated by the therapeutic efficacy of drugs blocking BCR signaling [12,13,14], as well as by the clinical impact in CLL of several molecular BCR features, including the mutational status of immunoglobulin heavy chain variable (IGHV) genes and the BCR stereotypy [15,16,17].
A link between BCR and VLA-4 activation in CLL has been recently identified in studies showing that in CD49d-expressing CLL the VLA-4 integrin can be inside–out activated upon BCR triggering, thus reinforcing the adhesive capacities of CLL cells [10]. This mechanism remains effective also when the BCR signaling is weakened by BTK inhibitors, resulting in a retention of BCR-mediated VLA-4 activation and VLA-4-mediated cell adhesion that translates, in CD49d+ CLL, in a reduced lymphocytosis and nodal response, and shorter progression-free survival (PFS) in the setting of BTK inhibitors therapy [18,19,20].
In CLL, BCR can also signal in an antigen-independent autonomous manner, as a consequence of BCR homotypic interactions occurring on CLL cell surface [21]. Interestingly, it was shown that distinct BCR stereotypes are characterized by specific homotypic BCR contacts resulting in qualitatively different cell-autonomous signals and peculiar clinical outcomes [22].
Here we describe a novel anchor-independent BCR-mediated mechanism of VLA-4 activation occurring in circulating CD49d+ CLL cells which may contribute to explain the negative prognostic impact of CD49d in CLL [1,2,3].
Materials and methods
CLL patients
The study included 1034 CD49d+ CLL from a consecutive series of 1985 CLL diagnosed and treated according to the current iwCLL guidelines [23], referred to the Clinical and Experimental Onco-Hematology Unit of the Centro di Riferimento Oncologico in Aviano for immunocytogenetic analyses between 2015 and 2020. Clinical and biological characterization of the cohort, as reported previously [2, 24,25,26,27], is summarized in Supplementary Table 1. CLL are considered CD49d+ as reported [1, 2]. For specific experiments samples from CD49d− CLL were used (n = 54).
The study also included CD49d+ CLL treated with ibrutinib as follows: (1) 12 refractory/relapsed CLL patients enrolled in a multicenter Italian-named patient program (NPP); (2) 14 out of 36 CLL patients enrolled in the prospective observational multicenter trial IOSI-EMA-001 (NCT02827617), chosen for the availability of CD49d expression data and frozen samples.
Studies were carried out in accordance with the Declaration of Helsinki, approved by the Institutional Review Boards of the Centro di Riferimento Oncologico in Aviano (Approvals Nos. IRB-05-2010, IRB-05-2015, and CRO-2017-31) and of the Azienda Ospedaliero-Universitaria of Novara (Approval No. CE034/2023) and informed consent was obtained from all subjects. All methods were performed in accordance with the relevant guidelines and regulations.
Functional flow cytometry assays
Constitutive VLA-4 activation in fresh whole blood samples was evaluated using anti-beta1/CD29 PE clone HUTS-21 monoclonal antibody (mAb), anti-CD19 BV421 or PerCpCy5.5 and anti-CD5 PE-Cy7. VLA-4 activation in thawed CLL cells was evaluated using the tri-peptide LDV, mimicking VLA-4 ligands [28], in the presence of anti-HUTS-21 PE, as reported [19, 29]. The mean fluorescence intensity (MFI) of labeled HUTS-21 mAb was measured, and VLA-4 receptor occupancy (RO) was determined ranging from 0.0 (no RO) to 1.0 (100% RO), as reported [19, 29]. Real-time kinetic of VLA-4 affinity on TKO cells (see below) was cytometrically obtained by using unlabeled and fluorochrome-conjugated LDV (LDV-FITC, 4 nM; TOCRIS, Bristol, UK), as previously reported [28]. To obtain the dissociation rate constants (Koff), the data were fitted to a one-phase exponential decay equation: Koff < 0.02 s−1, high affinity; Koff > 0.06 s−1, low affinity [28].
Levels of bound soluble (s)VCAM-1 were evaluated using anti-CD106/VCAM-1 PE Vio770 (Miltenyi Biotec, Bergisch Gladbach, Germany), anti-CD5 FITC, anti-CD19 APC, anti-HUTS-21 PE. VCAM-1 binding assay was performed using recombinant human (rh)VCAM-1-Fc protein (20 μg/ml, R&D, Minneapolis, MN, USA), anti-CD3 BUV395 and secondary antibody Alexa Fluor 647-labeled goat anti-human IgG (15 ng/µl, Jackson Immunoresearch, West Grove, PA, USA). Actin polymerization was evaluated using Alexa Fluor 488-conjugated phalloidin (Invitrogen, Waltham, MA, USA). Phosphoflow analyses on both whole blood and thawed samples were done using anti-ERK1/2 (pT202/pY204) BV421 or PE, anti-BTK (pY223)/Itk (pY180) BV421 or PE, anti-PLC-γ2 (pY759) Alexa Fluor 647 and anti-AKT (pS473) Alexa Fluor 647.
Cells were acquired on BD FACS CantoII or BD FACS Fortessa X-20 flow cytometers and analyzed with FACS DIVA software upon instrument calibration.
All mAbs and instruments were from BD Biosciences (San Jose, CA, USA) if not otherwise specified.
Triple-knockout (TKO) cells
TKO cells were retroviral-transduced to express CLL-derived BCR as previously reported [30, 31]. Cultured TKO cells [30, 31] were tested for VLA-4 affinity and calcium influx upon different stimuli (4-hydroxytamoxifen, 4-OHT, 4-OHT plus F(ab)2 anti-IgM or manganese, Mn2+) and/or pre-treatment with specific BCR inhibitors [30, 31].
Statistics
All statistical analyses were performed using MedCalc software and GraphPad Prism5. Normally distributed data were compared using two-sided t-tests (paired or unpaired, with Welch’s correction), otherwise two-sided Mann–Whitney test (independent) or Wilcoxon high-rank test (paired analysis) were used.
Additional experimental details are reported in the Supplementary Methods.
Results
VLA-4 can be constitutively activated in CLL
The activation state of the VLA-4 integrin was analyzed using the conformation-sensitive anti-beta1/CD29 mAb HUTS-21, which binds to a regulatory region of the beta1 integrin chain (hybrid domain) that is exposed when the conformation of the VLA-4 integrin changes from a resting/low-affinity state to an activated/high-affinity state (Fig. 1A) [29]. A prerequisite for HUTS-21 binding is the simultaneous binding of a ligand to the integrin molecule [29]. Therefore, staining was done in whole blood to exploit the presence of VLA-4 ligands in plasma. Control experiments detailed in Supplementary Methods and Supplementary Fig. 1 confirmed the specificity of HUTS-21 binding.

A Schematic representation of VLA-4 conformations. VLA-4 activation results in a change of conformation of the alpha4/beta1 chains of the VLA-4 integrin from a bent conformation, corresponding to a low affinity for the ligand, to an upright conformation with exposition of the hybrid domain in the beta1 chain which is recognized by the anti-CD29 HUTS-21 mAb in the presence of the ligand, and that corresponds to a high affinity for the ligand. B Fresh whole blood samples from a consecutive series of 1985 CLL were stained with anti-CD49d mAb; 1034 were identified as CD49d+ CLL, i.e. expression CD49d ≥ 30% (left graph), they were stained with the conformation-sensitive anti-CD29 HUTS-21 mAb, and analyzed by flow cytometry. The graph on the right shows the levels of HUTS-21 binding, reported as the percentage of positive cells over the unstained control in CD19+CD5+ cells. C Level of sVCAM-1 (ng/ml) in plasma samples from CD49d- (gray dots; n = 41) and CD49d+ (blue dots; n = 80) CLL cases. Data are presented as mean ± SD. The p value refers to Mann–Whitney test. D Correlation analysis between plasma sVCAM-1 levels (ng/ml) and binding of HUTS-21, evaluated as % HUTS-21 in CD19+CD5+ cells from CD49d+ CLL patients (n = 80). The correlation coefficient (r) and p value were obtained by the Spearman’s method. E Whole blood samples from CD49d+ CLL with moderate/high HUTS-21 binding levels (n = 15) were stained with anti-CD19, -CD5, -VCAM-1/CD106 and -HUTS-21 mAbs. VCAM-1 binding levels are reported in the context of CD19+CD5+ cells as a percentage of positive cells above the threshold set on plasma-depleted samples. Data are presented as mean ± SD. F HUTS-21 mean fluorescence intensity (MFI) in 23 CD49d+ CLL cases, evaluated in the context of VCAM-1pos (purple dots) and VCAM-1neg (gray dots) CLL cells. The p value refers to Wilcoxon test.
Out of 1985 consecutive CLL, 1034 (52%) were identified as CD49d+ CLL (Supplementary Table 1, Fig. 1B). In this cohort, HUTS-21 binding was mostly characterized by a single peak homogeneously shifted compared to unstained control, indicating HUTS-21 binding in the whole CLL clone, although with variable intensity; a bimodal pattern of HUTS-21 binding, characterized by the concomitant presence of a negative and positive sub-population, was also observed in some instances (Supplementary Fig. 2). The levels of HUTS-21 binding, evaluated as percentage of positive CLL cells, had a mean of 14.9% (±18.5 SD, range 0–98.4%), indicating an overall dim expression of activated VLA-4 (Fig. 1B).
Levels of soluble VCAM-1 inversely correlate with the degree of constitutive VLA-4 activation
Soluble VCAM-1 (sVCAM-1) was quantified in plasma samples from 41 CD49d− and 80 CD49d+ CLL, the latter group with HUTS-21 data available. A significantly higher amount of sVCAM-1 was found in plasma samples from CD49d+ CLL compared to CD49d− CLL (p < 0.0001; Fig. 1C), in keeping with previous data of ours [32]. Notably, among the CD49d+ CLL, we observed a significant inverse correlation between the amount of sVCAM-1 and HUTS-21 binding levels (r = −0.3, p = 0.005; Fig. 1D), that led us to hypothesize that a partial depletion of sVCAM-1 from plasma could be due to its binding to activated VLA-4. This hypothesis was tested by simultaneously staining cells with anti-VCAM-1/CD106 and HUTS-21 mAbs (n = 15). VCAM-1 was detected in a fraction of cells in all samples analyzed (Fig. 1E). Overall, significantly higher constitutive VLA-4 activation was observed in the context of VCAM-1-positive cells compared to VCAM-1-negative cells (p < 0.0001; Fig. 1F and Supplementary Fig. 3A). Control experiments confirmed the specificity of the assay (Supplementary Fig. 3B).
Constitutive VLA-4 activation is associated to increased phosphorylation levels of signaling proteins
The analysis of constitutive VLA-4 activation through the HUTS-21 binding highlighted intra-sample variability, with a Gaussian distribution of HUTS-21 binding levels indicating a variable range of VLA-4 activation within the same CLL cell population (Fig. 2A). We analyzed the phosphorylation state of key signaling proteins involved in CLL cell activation and survival in the context of CD49d+ CLL cells (n = 28) which stained negative or positive with the HUTS-21 mAb, according to HUTS-21 binding levels above or below the threshold set on the unstained control (hereafter identified as HUTS-21pos and HUTS-21neg, respectively).

A Representative histogram plot showing the analysis of the flow cytometry strategy used to split CLL cells within the same sample according to HUTS-21 binding levels; CLL cells (concomitantly expressing CD19 and CD5, not shown) were split in HUTS-21neg (green) and HUTS-21pos (pink) according to HUTS-21 binding levels below or above the threshold set on the unstained control (open histogram). B Expression values (MFI) of phospho (p)-ERK, p-AKT, p-BTK, and p-PLC-γ2 in the context of HUTS-21neg (green dots) and HUTS-21pos (pink dots) CLL cells, as identified in (A) from 28 CLL cases. C Expression values (MFI) of IgM in the context of HUTS-21neg (green dots) and HUTS-21pos (pink dots) CLL cells (n = 21), as identified in (A); the reported p values refer to Wilcoxon test. Data are presented as mean ± SD. D Gating strategy used for the analysis of HUTS-21 in circulating CLL cells with CD49d bimodal expression. The CLL cell population was first split in CD49d− (gray) and CD49d+ (blue) cells, and the CD49d+ cells were further split in HUTS-21neg (green) and HUTS-21pos (pink) according to HUTS-21 binding levels below or above the threshold set on the unstained control (not shown). E Expression values (MFI) of phospho (p)-ERK, p-AKT, p-BTK, and p-PLCγ2 in the context of CD49d- (gray dots), CD49d+HUTS-21neg (green dots) and CD49d+HUTS-21pos (pink dots) CLL cells, as identified in (D) from 12 CD49d bimodal CLL cases (9 samples for p-PLCγ2). The reported p values refer to Wilcoxon test. Data are presented as mean ± SD.
In all cases analyzed, HUTS-21pos cells vs HUTS-21neg cells displayed significantly higher phosphorylation of ERK, AKT, BTK and PLC-γ2 (Fig. 2B and Supplementary Fig. 4A). The higher levels of protein phosphorylation observed in HUTS-21pos cells compared to HUTS-21neg cells could not be attributed to integrin activation due to the simultaneous staining with the HUTS-21 mAb, as the addition of the anti-HUTS-21 mAb did not increase phosphorylation levels by itself (Supplementary Fig. 4B). Concomitant analysis of IgM expression highlighted higher levels of IgM in HUTS-21pos cells compared to HUTS-21neg cells (Fig. 2C).
Moreover, the same analysis, when carried out in CLL samples characterized by CD49d bimodal expression (n = 12) [20], highlighted comparable levels of protein phosphorylation in CD49d− and CD49d+/HUTS-21neg cells, while significantly higher levels were found in CD49d/HUTS-21pos cells (Fig. 2D, E).
Binding of sVCAM-1 increases actin polymerization and phospho-ERK levels
The effects of sVCAM-1 binding were analyzed in cell suspension from 28 thawed CD49d+ CLL samples, using rhVCAM-1-Fc. The binding of rhVCAM-1 to CLL cells was variable among samples, directly correlated with the expression of CD49d, separately assessed in the same samples (p < 0.0001, Fig. 3A), and was completely inhibited by pre-treatment of cells with the VLA-4 antagonist Firategrast, confirming the specificity of the binding (Fig. 3B).

A Thawed cells from 28 CD49d+ CLL samples were incubated with recombinant human (rh)VCAM-1-Fc protein and secondary Alexa Fluor 647-labeled goat anti-human-Fc antibody. VCAM-1 binding levels, analyzed on CD3− cells and reported as Alexa Fluor 647 MFI values (MFI rhVCAM-1) were correlated to CD49d expression (MFI values), assessed separately in the same thawed samples. The correlation coefficient (r) and p value were obtained by Spearman’s method. B VCAM-1 binding levels (MFI rhVCAM-1) of the same samples reported in (A) treated (black dots) or not (purple dots) with the VLA-4 integrin antagonist Firategrast. C F-actin polymerization was evaluated in ten CLL samples after incubation (1–5 min) with rhVCAM-1. Results are displayed as the average of the ratio of phalloidin MFI between cells incubated with rhVCAM-1 over cells without rhVCAM-1 (control). Mean intensity of F-actin (D) and F-actin circularity (E) in CLL cells not binding VCAM-1 (green) and in cells binding VCAM-1 (orange) as assessed by InFlow microscopy in six samples. Histogram overlays and sample images are shown for a representative CLL sample and representative cells not binding or binding VCAM-1. BF bright field; the merged images refer to the merging of CD49d/F-actin/VCAM-1. F Expression levels (MFI) of phospho (p)-ERK (n = 25) in CLL cells incubated with rhVCAM-1 pre-treated (black dots) or not (purple dots) with Firategrast. G Ratio of p-ERK MFI (n = 8) in CLL cells incubated for 24 h with rhVCAM-1 pre-treated (black dots) or not (purple dots) with Firategrast. H Expression levels (MFI) of p-BTK (n = 19) in CLL cells incubated with rhVCAM-1 pre-treated (black dots) or not (purple dots) with Firategrast. I Representative histogram plot showing the analysis of the flow cytometry strategy used to split CLL cells within the same sample according to the levels of rhVCAM-1-Fc binding; CLL cells (corresponding to CD3− cells) were split in VCAM-1neg (gray) and VCAM-1pos (purple) according to rhVCAM-1-Fc Alexa Fluor 647 expression below or above the threshold set on cells pre-treated with Firategrast (open histogram). Expression values (MFI) of p-ERK (n = 25, J) and p-BTK (n = 19, K) in the context of VCAM-1neg (gray dots) and VCAM-1pos (purple dots) CLL cells, as identified in (I). The reported p values refer to Wilcoxon test (B, F–H, J, K) and to one-way repeated measures ANOVA with multiple comparisons (C). Data are presented as mean ± SD.
Functionally, the addition of rhVCAM-1 significantly affected actin dynamics, by increasing rapid (1–5 min) actin polymerization in CLL cells (n = 10; Fig. 3C), as observed by conventional flow cytometry. Analysis by InFlow microscopy, which combines flow cytometry and fluorescence microscopy, confirmed higher levels of polymerized actin in CLL cells bound to VCAM-1 compared to cells not binding VCAM-1 (n = 6, p = 0.0084; Fig. 3D and Supplementary Figs. 5, 6). Cells bound to VCAM-1 also displayed a different spatial distribution of F-actin compared to cells not binding VCAM-1, with a group of cells characterized by F-actin polarization to a pole of the cell. Quantification of F-actin polarization was assessed by calculating the circularity values of the F-actin signal, where higher circularity values are associated with a homogeneous distribution of F-actin signal in round cells, whereas lower circularity values are associated to a polarized signal. Cells bound to VCAM-1 were characterized by significantly lower F-actin circularity than cells not binding VCAM-1 (p = 0.024; Fig. 3E). Of note, binding of VCAM-1 also induced a polarization of the CD49d molecules determining an overlapping of the CD49d/VCAM-1 signals (see the InFlow microscopy sample merge images in Fig. 3D and Supplementary Fig. 6).
Moreover, addition of rhVCAM-1 to thawed cells significantly increased p-ERK1/2 levels compared to cells pre-treated with Firategrast both in short-term (20′) experiments (n = 25, p = 0.0074; Fig. 3F) and after 24 h exposure to rhVCAM-1 (n = 8, p = 0.0156; Fig. 3G). Conversely, phosphorylation of BTK was not directly affected by addition of rhVCAM-1 (n = 19; Fig. 3H), in keeping with the notion that BTK is not part of the integrin downstream signaling [33].
Intra-sample analysis, by comparing cells capable to bind rhVCAM-1 (VCAM-1pos) vs cells not capable to bind rhVCAM-1 (VCAM-1neg) (Fig. 3I), showed significantly higher levels of p-ERK1/2 and p-BTK in VCAM-1pos vs VCAM-1neg cells (p = 0.0002; Fig. 3J, K), suggesting an increased ability to bind VCAM-1 by cells with an activated BCR signaling in turn capable to trigger VLA-4 integrin activation via inside–out mechanism [10].
Association between constitutive VLA-4 activation and BCR features
Given the link between BCR triggering and VLA-4 activation [19], we verified if specific BCR-related features were associated with different levels of constitutive VLA-4 activation, as investigated by HUTS-21 staining. IGHV analysis was available in 911 out of 1034 CD49d+ CLL (88.1% of cases), 874 cases (96%) showing a single productive rearrangement [34]. Among these cases, 364 (42%) were IGHV mutated (M), and 510 (58%) unmutated (UM). In line with previous data [35, 36], the most frequently used IGHV genes belonged to the IGHV3 family (n = 374, 42.8%), followed by the IGHV1 family (n = 254, 29.1%), the IGHV4 family (n = 181, 20.7%), and other less frequent families (n = 65, 7.4%) (Supplementary Table 2).
By comparing the levels of HUTS-21 mAb binding in CLL cases divided according to the IGHV mutational status, constitutive VLA-4 activation was significantly higher in M compared to UM CLL and, consistently, in cases from the IGHV3 and IGHV4 families compared to the IGHV1 family (Fig. 4A, B and Supplementary Fig. 7A). Analysis of the main BCR stereotypes, evaluated as previously reported [37], identified 16/19 major subsets cumulatively accounting for 118 cases (13.5%), subset#1 being the largest (38 cases), followed by subset#2 (18 cases: 16/18 carrying the IGL3-21 light chain, two cases with light chain not evaluated; 14/16 carrying the R110 mutation), and the other less represented subsets [38] (Supplementary Table 2). HUTS-21 binding highlighted higher, although variable, VLA-4 activation levels in CLL from subset#2 compared to CLL from subset#1 (p = 0.03; Fig. 4C). Consistent with the higher IgM levels found in HUTS-21pos cells compared to HUTS-21neg cells (see above), we observed again a positive correlation (r = 0.6644, p = 0.0036) between IgM and HUTS-21 binding levels (Fig. 4D), whereas IgD levels did not correlate with HUTS-21 (Supplementary Fig. 6B).

Levels of HUTS-21 binding evaluated as a percent of positive cells in CLL cases split according to the IGHV mutational status (M mutated vs UM unmutated; A), IGHV families (IGHV1, IGHV3 and IGHV4; B) and BCR stereotypy (subset#1 vs subset#2; C). P values refer to the unpaired t-test. D In the context of CLL from subset#2, the levels of HUTS-21 binding evaluated as percent of positive cells was correlated to IgM expression (%). The correlation coefficient (r) and p value were obtained by the Pearson’s method.
Constitutive VLA-4 activation is driven by the autonomous BCR signaling
The link between VLA-4 activation and BCR autonomous signaling was explored using the TKO cell model [21, 22, 31], highly expressing the VLA-4 integrin on the cell membrane (Supplementary Fig. 8A). TKO cells were transduced with eight different CLL-derived BCRs, four from CLL with high constitutive VLA-4 activation (hereafter TKO-high) and four from CLL with low constitutive VLA-4 activation (hereafter TKO-low). All CLL-derived BCR retained the original IGHV-D-J and IGLV-J structures and expressed the IgM constant region (Supplementary Table 3). These cellular models were employed to analyze the kinetics of formation and dissociation of the VLA-4 complex with its ligand [28] upon exposure to 4-OHT, to simulate BCR autonomous signaling, or 4-OHT plus anti-IgM, to simulate BCR activation by external antigens (Fig. 5A) [21, 22, 31].

A Flow cytometric real-time kinetic analysis of VLA-4 affinity state in triple-knockout (TKO) cells expressing the 4-hydroxytamoxifen-inducible (4-OHT-inducible) ERT2-SLP-65 fusion protein and eight different CLL-derived BCRs, four from CLL with high constitutive VLA-4 activation (TKO-high) and four from CLL with low constitutive VLA-4 activation (TKO-low). TKO cells were incubated with LDV-FITC and stimulated with PBS, used as negative control (gray line), 4-OHT, to simulate BCR autonomous signaling (red line), 4-OHT plus anti-IgM (+αIgM), to simulate BCR activation by external antigen (blue line), or manganese (Mn2+), used as a technical positive control (green line), resulting in a different equilibrium binding of LDV-FITC to VLA-4, depending on the respective integrin affinity state. The graph reports FITC MFI values vs time (seconds) analysis from a representative TKO cell line. Dissociation kinetics of the fluorescent LDV peptide from the cells is induced by 500-fold excess of unlabeled peptide. To obtain the dissociation rate constants (Koff), the data were fitted to a one-phase exponential decay equation. The calculated Koff values indicate the VLA-4 affinity state after stimulating the cells with the different substances; Koff < 0.02 s−1 high affinity, Koff > 0.06 s−1 low affinity. B Summary of the Koff values obtained at the different conditions in TKO-high (red dots) and TKO-low (green dots) cells. Individual symbols represent the mean of at least three experiments for each TKO cell line, run in triplicate. The inset shows flow cytometric real-time kinetic curves of representative TKO-high (red curve) and TKO-low (green curve) cell lines stimulated with 4-OHT. C Calcium response to 4-OHT and 4-OHT + αIgM in TKO-high (red) and TKO-low (green) cells. The pseudocolored dot plots show calcium influx over time (in seconds) in a representative TKO-high (upper plot, TKO UD4) and TKO-low (lower plot, TKO RIO128) cell line stimulated with 4-OHT (left) and with 4-OHT + αIgM (right). In all panels, data are presented as mean ± SD; p values refer to the unpaired t-test (A–C); ns not significant.
In the absence of any stimulation (PBS), both TKO-high and TKO-low bound the ligand with comparable low affinity (Fig. 6B), corresponding to unactivated VLA-4 [28]. Likewise, upon Mn2+ stimulation, corresponding to the maximum of VLA-4 activation (positive control), affinities in terms of Koff values were again similar in both TKO groups (Fig. 5B). However, upon stimulation with 4-OHT, Koff turned out significantly lower for TKO-high compared to TKO-low (p = 0.01; Fig. 5B), indicating a higher affinity of the VLA-4 integrin expressed in the context of TKO-high cells upon BCR autonomous signaling. Anti-IgM stimulation similarly increased VLA-4 affinity in both TKO groups (Fig. 5B).

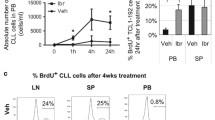
A VLA-4 receptor occupancy (RO) was evaluated in cells from two independent cohorts of CLL patients treated with ibrutinib in the context of a named patient program (NPP; green symbols, n = 12) and in the context of a clinical trial (orange symbols, n = 15), at pre-treatment (t0) and after 14, 30, 60, 90 days of ibrutinib therapy. B VLA-4 affinity in six CLL cases expressing CD49d, pre-treated (light blue dots) or not (purple dots) with ibrutinib in vitro (1 µM). Values are expressed as Koff fold change on the untreated condition. Data are presented as mean ± SD. Soluble recombinant human (rh)VCAM-1 binding levels (MFI rhVCAM-1-Fc) (C), expression values (MFI) of phospho (p)-BTK (D), and p-ERK (E) in CLL cells from nine cases pre-treated (light blue dots) or not (purple dots) with ibrutinib in vitro (1 µM). P values refer to the paired t-test (A, C–E) and ratio paired t-test (B).
A parallel analysis of calcium (Ca2+) release consistently showed a significantly higher Ca2+ influx in TKO-high vs TKO-low cell models following stimulation with 4-OHT (p = 0.0152; Fig. 5C), while the highest Ca2+ influx levels, similar in both conditions, were observed upon IgM stimulation (Fig. 5C).
Ig light chain substitution in two TKO cell lines from the TKO-high group (Supplementary Table 3) negatively affected BCR autonomous signaling, as evidenced by the reduction in Ca2+ release, and led to a transition from a high to low-affinity state of VLA-4 (p < 0.0001; Supplementary Fig. 8B).
Constitutive VLA-4 activation in CLL is impaired by ibrutinib exposure in vivo and in vitro
Serial samples from CLL patients treated in vivo with ibrutinib were collected at baseline and at different timepoints after the initiation of ibrutinib therapy. The impact of ibrutinib therapy on constitutive VLA-4 activation was determined by RO calculation, as described [19, 29]. The levels of constitutive VLA-4 activation were significantly lower after 14 days of ibrutinib treatment compared to pre-treatment (n = 14; Fig. 6A). Similarly, in a subgroup of CLL with samples available at pre-treatment and at days 30, 60, and 90 on ibrutinib (n = 12, 6, and 5), constitutive VLA-4 activation levels observed at pre-treatment progressively decreased starting from day 30, and reached the lowest levels at days 60 and 90 (Fig. 6A). Consistently, in vitro ibrutinib treatment on thawed CLL cells (n = 6) significantly impaired VLA-4 affinity (Fig. 6B).
With the aim of verifying if the impaired constitutive VLA-4 activation observed during ibrutinib treatment could affect sVCAM-1 binding, rhVCAM-1 binding assay was performed on CLL samples (n = 9) pre-treated or not with ibrutinib in vitro. In line with a decreased VLA-4 affinity observed in vivo and in vitro, treatment with ibrutinib significantly impaired rhVCAM-1 binding (Fig. 6C) and consequently also downstream phosphorylation of BTK and ERK (Fig. 6D, E).
BCR pathway inhibitors impair BCR autonomous signaling and VLA-4 affinity
Given the impairment of constitutive VLA-4 activation upon ibrutinib therapy, we evaluated the effect of ibrutinib and PI3K inhibitors (idelalisib and copanlisib) exposure in vitro on BCR autonomous signaling by taking advantage of the TKO cell model. All the BCR pathway inhibitors inhibited Ca2+ influx upon 4-OHT addition, while not affecting Ca2+ release following anti-IgM (Fig. 7A). Consistently, all the BCR pathway inhibitors significantly impaired VLA-4 affinity in all TKO cells tested, with the only exception of idelalisib, which however further potentiated the effect of ibrutinib when used in combination (Fig. 7B, C). Finally, ibrutinib did not alter the increase in VLA-4 affinity observed after BCR activation with anti-IgM (Fig. 7C), in keeping with previous observations [19].

A Calcium response in four TKO-high cell lines. Cells either untreated or pre-treated with ibrutinib, idelalisib, a combination of ibrutinib and idelalisib and copanlisib were stimulated with 4-OHT followed by anti-IgM (αIgM; black arrows). Each row represents a different cell line and each column corresponds to a different treatment. B Flow cytometric real-time kinetic curves of a representative TKO cell line (TKO RMPTV327) stimulated with 4-OHT at the following conditions (from left to right): untreated (black), pre-treated with ibrutinib (light blue), idelalisib (orange), combination ibrutinib/idelalisib (green), and copanlisib (pink). C VLA-4 affinity in TKO cell lines treated as reported in (B). For the ibrutinb condition, cells were also stimulated with 4-OHT + αIgM or Mn2+. Values are expressed as Koff fold change using for each cell line the untreated, 4-OHT stimulated condition as reference condition. Each dot represents the mean value of at least two experiments with two replicates for each TKO cell line. Data are presented as mean ± SD. P values refer to the ratio paired t-test; ns not significant.
Discussion
The main objectives of the present study were to assess whether: (i) the VLA-4 integrin is present in a constitutive activated state in circulating CLL cells; (ii) such a constitutive VLA-4 activated state has a functional meaning for circulating CLL cells; (iii) the constitutive VLA-4 activated state, as determined in circulating CLL cells, may result from an inside–out mechanism originating from the BCR via an antigen-independent autonomous signaling.
The first point was addressed by analyzing, in a large cohort of CLL expressing VLA-4, the presence of the active form of the integrin using the conformation-sensitive anti-beta1 integrin HUTS-21 mAb [29]. A wide degree of HUTS-21 binding was found, indicating the presence of variable levels of constitutively activated VLA-4 integrin detectable in circulating CD49d+ CLL cells.
Our approach, employed here on a large scale by analyzing more than 1000 cases, bears similarities to a previous study conducted in the context of multiple myeloma, in which, using the same antibody, the authors demonstrated that the activation state of integrin VLA-4 could be tightly regulated in the presence of a soluble form of VCAM-1 contained in the liquid fraction of bone marrow (BM) aspirates [39].
sVCAM-1 is present in plasma samples at significantly higher levels in CD49d+ CLL cases than in CD49d− CLL cases, as already observed by us [32] in line with previous studies indicating overexpression of VCAM-1 in the BM microenvironment of CD49d+ CLL [8]. Notably, we here describe a negative correlation between levels of activated VLA-4 integrin in circulating CLL cells and plasma levels of sVCAM-1, and were able to demonstrate the sequestration of sVCAM-1 by cells expressing a constitutively activated form of VLA-4.
The interaction between VLA-4 and soluble ligands in circulating CLL cells was previously hypothesized by us in a study in which we observed that circulating CLL cells expressing CD49d have a phospho-proteomic profile consistent with a continuous integrin engagement [32]. In other cellular contexts, the ability of VLA-4 to bind sVCAM-1 and deliver signals that control cellular functions has been demonstrated [40]. In our setting, CLL cells expressing a constitutively activated form of VLA-4 expressed higher levels of ERK and AKT suggesting the activation of pro-survival pathways; consistently, binding of sVCAM-1 to CLL cells in vitro confirmed ERK phosphorylation, as well as a more effective polymerization of actin. Indeed, treatment of CLL cells with the VLA-4 antagonist Firategrast, by preventing the binding of sVCAM-1 [41], lowered ERK phosphorylation, indicating this effect as the result of integrin pathway activation.
On the other hand, circulating CLL cells showing higher levels of p-BTK, displayed increased binding of sVCAM-1, suggesting that an activated BCR pathway is in turn able to activate the VLA-4 integrin via inside–out, thus promoting sVCAM-1 binding. In CLL, the interaction between the BCR pathway and VLA-4 has so far been studied only upon exogenous BCR stimulation, as it occurs in CLL tissue sites [18, 19]. In that context, it was shown that VLA-4-mediated CLL cell adhesion is enhanced by BCR-derived signals, and only partially impaired by ibrutinib, providing the functional basis for the lower recirculation lymphocytosis observed in CD49d+ CLL upon ibrutinib treatment [19].
In the present study, we investigated whether an autonomous BCR signaling may be involved in the constitutive VLA-4 activation of circulating CLL cells via inside–out mechanisms. Since cell-autonomous signaling is a consequence of homotypic inter/intra-cellular interactions between BCR molecules via specific self-epitopes, different binding kinetics and affinities have been demonstrated in CLL, with BCRs deriving from indolent cases showing stronger affinities than those from aggressive cases [22]. In keeping with these observations, a significant association was found here between a higher activation state of VLA-4 and specific BCR features, such as a M IGHV gene status, the use of IGHV3 and IGHV4 gene families, as well as of the subset #2 stereotype. This finding implies that certain BCR characteristics may directly or indirectly influence the activation status of VLA-4, including the different ability of BCRs to signal in a manner independent of external BCR stimulation, as presumably occurs in circulating CLL cells.
To test this hypothesis, we exploited the same TKO cellular model originally used to describe BCR autonomous signaling [21], given the high levels of VLA-4 expression found in these cells, as shown here. Experiments with TKO cells provide clear evidence of higher BCR autonomous signaling paralleled with increased activation/affinity of VLA-4 integrin molecules in TKO cells expressing a BCR derived from CLL cells with high constitutive VLA-4 activation than in cells expressing a BCR derived from CLL cells with low constitutive VLA-4 activation, clearly supporting our hypothesis of a link between BCR autonomous signaling and inside–out activation of VLA-4. In this context, we also showed that the proper coupling of Ig heavy/light chains is necessary for effective autonomous signaling capacity by BCR, since Ig light chain substitution resulted in a clear inhibition of autonomous signaling and consequent reduction of VLA-4 activation in keeping with studies pinpointing the importance of a defined Ig sequence in facilitating autonomous BCR signaling [42, 43].
It was recently described that autonomous BCR signaling requires the IgM isotype, and does not occur via the IgD isotype [44]. Although our case series has not been extensively characterized for IgD and IgM expression, we showed that, within the same CLL sample, IgM expression was higher in cells with higher levels of HUTS-21 staining, i.e. with activated VLA-4, than in cells with lower HUTS-21 staining, i.e. with non-activated VLA-4. Consistently, a direct correlation between IgM and HUTS-21 expression levels was also found in the context of subset#2 CLL.
In summary, here we provided compelling evidence that BCR and VLA-4, two canonical microenvironmental receptors, can operate and interact with each other also in the bloodstream in an anchor-independent manner. In particular, BCR signals delivered via autonomous stimulation can effectively activate VLA-4 molecules, which in turn may be engaged by soluble plasma components.
The interaction between sVCAM-1 with VLA-4 may contribute to keep CD49d+ CLL cells alive in the blood circulation, as witnessed by increased phosphorylation of ERK and AKT [45, 46], and to make them more prone to migrate outside the bloodstream, as shown by increased/more efficient actin polymerization, ultimately influencing recirculation and homing of circulating cells whose integrin receptor is engaged [33]. The increased survival and migratory capacity observed in CD49d+ CLL cells following plasma stimulation [32] as well as the clinical notion that CD49d+ CLL patients are more likely to have lymphadenopathy and/or are more prone to acquire lymphadenopathy over time [47] is consistent with these data.
In this study, we also demonstrated that, following exposure to ibrutinib in vivo, the levels of constitutive VLA-4 activation, expressed in circulating CD49d+ CLL cells, progressively decreased. This observation, in addition to corroborating the link between BCR signaling and constitutive VLA-4 activation, underlines the role of BTK inhibition in suppressing, at least partially, such a BCR-dependent VLA-4 activation occurring in circulating CD49d+ CLL cells [48, 49]. A by-pass mechanism involving phosphorylation of phosphatidylinositide 3-kinase (PI3K) that reactivates VLA-4 through the more effective antigen-dependent BCR stimulations has been described allegedly occurring in protective tissue compartments [18, 19]. A similar adaptation mechanism is apparently lacking in blood circulation where CLL cells only rely upon an autonomous (less effective?) BCR signaling to reactivate VLA-4.
Besides the association of constitutive VLA-4 activation with definite BCR characteristics (IGHV with M configuration, expression of the IGHV3/IGHV4 gene families or of the subset #2 stereotype) [24, 38, 50], constitutive VLA-4 activation was also accompanied with higher Ca2+ influx, according to the experiments with TKO cells, and increased ERK phosphorylation. Notably, all these features mostly associate to the so-called anergic state of CLL [51]. In this context, it could be speculated that anergic CLL cells expressing VLA-4 are particularly favored to remain alive in blood and, given the expression of a frequently activated integrin, are more prone to migrate to tissue sites where both BCR and VLA-4 can be more robustly engaged by their natural microenvironmental ligands. Several intrinsic factors responsible for the reversibility of the anergic state in CLL have been described [51,52,53]; here we propose the expression of VLA-4 and its constitutive activation occurring through tonic inside–out activation via BCR as an additional mechanism to prolong the survival of anergic CLL cells in the blood, as well as to promote their migration to tissues where they become prone to positive signals. The well-known negative prognostic impact of high CD49d expression in the context of M-CLL [1, 2, 54, 55] could be explained on the basis of these reasonings.
The anchor-independent process described here, allegedly aimed at keeping CLL cells alive in bloodstream, might resemble a similar mechanism occurring in solid tumors to counteract the phenomenon of apoptotic cell death by tissue detachment known as anoikis [56]. In this context, detached solid tumor cells exploit their own surface adhesion molecules to interact with soluble serum factors and promote their survival during bloodstream recirculation ultimately reaching their final sites of metastasis [56].
The functionally active interactions of BCR/VLA-4 on circulating CD49d+ CLL cells open up the possibility that similar cross-talks may operate in the bloodstream by exploiting other adhesion molecules and/or chemokine receptors, which in turn may interact with their respective soluble ligands present in plasma [57,58,59].
Data availability
The data generated in this study are available from the corresponding author on reasonable request.
References
Bulian P, Shanafelt TD, Fegan C, Zucchetto A, Cro L, Nückel H, et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol. 2014;32:897–904.
Gattei V, Bulian P, Del Principe MI, Zucchetto A, Maurillo L, Buccisano F, et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood. 2008;111:865–73.
Shanafelt TD, Geyer SM, Bone ND, Tschumper RC, Witzig TE, Nowakowski GS, et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: a prognostic parameter with therapeutic potential. Br J Haematol. 2008;140:537–46.
Ruoslahti E. Integrins. J Clin Investig. 1991;87:1–5.
Tissino E, Pivetta E, Capuano A, Capasso G, Bomben R, Caldana C, et al. Elastin MIcrofibriL INterfacer1 (EMILIN-1) is an alternative prosurvival VLA-4 ligand in chronic lymphocytic leukemia. Hematol Oncol. 2022;40:181–90.
Dal Bo M, Tissino E, Benedetti D, Caldana C, Bomben R, Del Poeta G, et al. Microenvironmental interactions in chronic lymphocytic leukemia: the master role of CD49d. Semin Hematol. 2014;51:168–76.
De la Fuente MT, Casanova B, Garcia-Gila M, Silva A, Garcia-Pardo A. Fibronectin interaction with α4β1 integrin prevents apoptosis in B cell chronic lymphocytic leukemia: correlation with Bcl-2 and Bax. Leukemia. 1999;13:266.
Zucchetto A, Benedetti D, Tripodo C, Bomben R, Dal Bo M, Marconi D, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009;69:4001–9.
Zucchetto A, Vaisitti T, Benedetti D, Tissino E, Bertagnolo V, Rossi D, et al. The CD49d/CD29 complex is physically and functionally associated with CD38 in B-cell chronic lymphocytic leukemia cells. Leukemia. 2012;26:1301–12.
Arana E, Harwood NE, Batista FD. Regulation of integrin activation through the B-cell receptor. J Cell Sci. 2008;121:2279–86.
Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42.
Wiestner A. BCR pathway inhibition as therapy for chronic lymphocytic leukemia and lymphoplasmacytic lymphoma. Hematol. Am. Soc. Hematol. Educ. Program. 2014;2014:125–34.
Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–84.
Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54.
Vlachonikola E, Sofou E, Chatzidimitriou A, Stamatopoulos K, Agathangelidis A. The significance of B-cell receptor stereotypy in chronic lymphocytic leukemia: biological and clinical implications. Hematol Oncol Clin N Am.2021;35:687–702.
Alsadhan A, Chen J, Gaglione EM, Underbayev C, Tuma PL, Tian X, et al. CD49d expression identifies a biologically distinct subtype of chronic lymphocytic leukemia with inferior progression-free survival on BTK inhibitor therapy. Clin Cancer Res. 2023;29:3612–21.
Tissino E, Benedetti D, Herman SEM, Ten Hacken E, Ahn IE, Chaffee KG, et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J Exp Med. 2018;215:681–97.
Tissino E, Pozzo F, Benedetti D, Caldana C, Bittolo T, Rossi FM, et al. CD49d promotes disease progression in chronic lymphocytic leukemia: new insights from CD49d bimodal expression. Blood. 135:1244–54.
Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489:309–12.
Minici C, Gounari M, Ubelhart R, Scarfo L, Duhren-von Minden M, Schneider D, et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8:15746.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60.
Bomben R, Dal Bo M, Capello D, Forconi F, Maffei R, Laurenti L, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: results from an Italian multicentre study. Br J Haematol. 2009;144:492–506.
Bomben R, Rossi FM, Vit F, Bittolo T, D’Agaro T, Zucchetto A, et al. TP53 mutations with low variant allele frequency predict short survival in chronic lymphocytic leukemia. Clin Cancer Res. 2021;27:5566–75.
Pozzo F, Bittolo T, Arruga F, Bulian P, Macor P, Tissino E, et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia. 2016;30:182–9.
Pozzo F, Bittolo T, Vendramini E, Bomben R, Bulian P, Rossi FM, et al. NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia. 2017;31:2407–15.
Chigaev A, Blenc AM, Braaten JV, Kumaraswamy N, Kepley CL, Andrews RP, et al. Real time analysis of the affinity regulation of alpha 4-integrin: the physiologically activated receptor is intermediate in affinity between resting and Mn(2+) or antibody activation. J Biol Chem. 2001;276:48670–8.
Chigaev A, Waller A, Amit O, Halip L, Bologa CG, Sklar LA. Real-time analysis of conformation-sensitive antibody binding provides new insights into integrin conformational regulation. J Biol Chem. 2009;284:14337–46.
Köhler F, Hug E, Eschbach C, Meixlsperger S, Hobeika E, Kofer J, et al. Autoreactive B cell receptors mimic autonomous pre-B cell receptor signaling and induce proliferation of early B cells. Immunity. 2008;29:912–21.
Meixlsperger S, Köhler F, Wossning T, Reppel M, Müschen M, Jumaa H. Conventional light chains inhibit the autonomous signaling capacity of the B cell receptor. Immunity. 2007;26:323–33.
Benedetti D, Tissino E, Caldana C, Dal Bo M, Bomben R, Marconi D, et al. Persistent CD49d engagement in circulating CLL cells: a role for blood-borne ligands? Leukemia. 2016;30:513.
Härzschel A, Zucchetto A, Gattei V, Hartmann TN. VLA-4 expression and activation in B cell malignancies: functional and clinical aspects. Int J Mol Sci. 2020;21:2206.
Stamatopoulos K, Agathangelidis A, Rosenquist R, Ghia P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia. 2017;31:282–91.
Bomben R, Dal Bo M, Capello D, Benedetti D, Marconi D, Zucchetto A, et al. Comprehensive characterization of IGHV3-21-expressing B-cell chronic lymphocytic leukemia: an Italian multicenter study. Blood. 2007;109:2989–98.
Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111:1524–33.
Bystry V, Agathangelidis A, Bikos V, Sutton LA, Baliakas P, Hadzidimitriou A, et al. ARResT/AssignSubsets: a novel application for robust subclassification of chronic lymphocytic leukemia based on B cell receptor IG stereotypy. Bioinformatics. 2015;31:3844–6.
Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher-order connections between stereotyped subsets: implications for improved patient classification in CLL. Blood. 2021;137:1365–76.
Martínez-Viñambres E, García-Trujillo JA, Rodríguez-Martín E, Villar LM, Coll J, Roldán E. CD29 expressed on plasma cells is activated by divalent cations and soluble CD106 contained in the bone marrow plasma: refractory activation is associated with enhanced proliferation and exit of clonal plasma cells to circulation in multiple myeloma patients. Leukemia. 2012;26:1098–105.
Rose DM, Cardarelli PM, Cobb RR, Ginsberg MH. Soluble VCAM-1 binding to alpha4 integrins is cell-type specific and activation dependent and is disrupted during apoptosis in T cells. Blood. 2000;95:602–9.
Grove RA, Shackelford S, Sopper S, Pirruccello S, Horrigan J, Havrdova E, et al. Leukocyte counts in cerebrospinal fluid and blood following firategrast treatment in subjects with relapsing forms of multiple sclerosis. Eur J Neurol. 2013;20:1032–42.
Maity PC, Bilal M, Koning MT, Young M, van Bergen CAM, Renna V, et al. IGLV3-21*01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci USA. 2020;117:4320–7.
Nicolò A, Linder AT, Jumaa H, Maity PC. The determinants of B cell receptor signaling as prototype molecular biomarkers of leukemia. Front Oncol. 2021;11:771669.
Mazzarello AN, Gentner-Göbel E, Dühren-von Minden M, Tarasenko TN, Nicolò A, Ferrer G, et al. B cell receptor isotypes differentially associate with cell signaling, kinetics, and outcome in chronic lymphocytic leukemia. J Clin Investig. 2022;132:e149308.
Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–77.
Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating pro-survival signals derived from the B-cell receptor in chronic lymphocytic leukemia B-cells. Blood. 2007;110:341.
Strati P, Parikh SA, Chaffee KG, Achenbach SJ, Slager SL, Call TG, et al. CD49d associates with nodal presentation and subsequent development of lymphadenopathy in patients with chronic lymphocytic leukaemia. Br J Haematol. 2017;178:99–105.
Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129:1469–79.
Bonfiglio S, Sutton LA, Ljungström V, Capasso A, Pandzic T, Weström S, et al. BTK and PLCG2 remain unmutated in one-third of patients with CLL relapsing on ibrutinib. Blood Adv. 2023;7:2794–806.
Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. 2007;109:259–70.
Packham G, Krysov S, Allen A, Savelyeva N, Steele AJ, Forconi F, et al. The outcome of B-cell receptor signaling in chronic lymphocytic leukemia: proliferation or anergy. Haematologica. 2014;99:1138–48.
Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109:4424–31.
Duckworth A, Glenn M, Slupsky JR, Packham G, Kalakonda N. Variable induction of PRDM1 and differentiation in chronic lymphocytic leukemia is associated with anergy. Blood. 2014;123:3277–85.
Zucchetto A, Bomben R, Dal Bo M, Bulian P, Benedetti D, Nanni P, et al. CD49d in B-cell chronic lymphocytic leukemia: correlated expression with CD38 and prognostic relevance. Leukemia. 2006;20:523–5.
Pepper AGS, Zucchetto A, Norris K, Tissino E, Polesel J, Soe Z, et al. Combined analysis of IGHV mutations, telomere length and CD49d identifies long-term progression-free survivors in TP53 wild-type CLL treated with FCR-based therapies. Leukemia. 2022;36:271–4.
Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26.
Ganghammer S, Hutterer E, Hinterseer E, Brachtl G, Asslaber D, Krenn PW, et al. CXCL12-induced VLA-4 activation is impaired in trisomy 12 chronic lymphocytic leukemia cells: a role for CCL21. Oncotarget. 2015;6:12048–60.
Lafouresse F, Bellard E, Laurent C, Moussion C, Fournié JJ, Ysebaert L, et al. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood. 2015;126:1336–45.
Ugarte-Berzal E, Bailón E, Amigo-Jiménez I, Albar JP, García-Marco JA, García-Pardo A. A novel CD44-binding peptide from the pro-matrix metalloproteinase-9 hemopexin domain impairs adhesion and migration of chronic lymphocytic leukemia (CLL) cells. J Biol Chem. 2014;289:15340–9.
Acknowledgements
The present study is supported in part by PNRR-MAD-2022-12375673 (Next Generation EU, M6/C2_CALL 2022), Italian Ministry of Health, Rome, Italy; Associazione Italiana Ricerca Cancro (AIRC), Investigator Grant IG-21687; Associazione Italiana contro le Leucemie, linfomi e mielomi (AIL), Venezia Section, Italy; “5 × 1000 Intramural Program”, Centro di Riferimento Oncologico, Aviano, Italy; Associazione Italiana Ricerca Cancro (AIRC), AIRC 5 × 1000 (No. 21198). PCM is supported by Fritz-Thyssen-Stiftung (10.23.1.012MN). We thank all the patients for participating and donating samples to make this research possible.
Author information
Authors and Affiliations
Contributions
ET, AZ and VG designed the research concepts and wrote the manuscript; ET, AZ, AG, FP, TB, PN, IC, EZ, GC, GF, RM, AN, AH, AMZ, GI and MD performed research; ET, AZ, FP, AN and PCM analyzed data; FMR and RB provided biological data; JO, GDA, LL, FZ, AC, GAP, EAM, MG, GG, DR, GDP, RL and MIDP provided well-characterized biological samples, and revised the manuscript; HJ, PCM and TNH provided key reagents and scientific, technical or material support; VG and AZ supervised the study. All the authors agreed on the final form of the manuscript with the only exclusion of GDP (deceased).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tissino, E., Gaglio, A., Nicolò, A. et al. The VLA-4 integrin is constitutively active in circulating chronic lymphocytic leukemia cells via BCR autonomous signaling: a novel anchor-independent mechanism exploiting soluble blood-borne ligands. Leukemia (2024). https://doi.org/10.1038/s41375-024-02376-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41375-024-02376-7
- Springer Nature Limited