
Abstract
Targeting BTK has profoundly changed the face of CLL treatment over the past decade. Iterative advances in the cat and mouse game of resistance and redesign have moved BTK inhibitors from covalent to non-covalent and now targeted protein degraders. However, contrary to the presumption that protein degraders may be impervious to mutations in BTK, we now present clinical evidence that a mutation in the kinase domain of BTK, namely A428D, can confer disease resistance to a BTK degrader currently in clinical trials, that is BGB-16673. Modeling of a BTK A428D mutation places a negatively charged aspartic acid in place of the hydrophobic side chain of alanine within the binding pocket of another BTK-degrader in clinical development, namely NX-2127, suggesting that CLL cells with BTK A428D also may be resistant to NX-2127, as they already are known to be with either non-covalent or covalent inhibitors of BTK. Consequently, the two BTK degraders furthest advanced in clinical trials potentially may select for CLL cells with BTK A428D that are resistant to all approved BTKi’s.
Similar content being viewed by others

To the Editor:
The enzyme Bruton tyrosine kinase (BTK) is involved in B-cell signaling that promotes the migration, proliferation, and survival of neoplastic B-cells of patients with chronic lymphocytic leukemia (CLL) [1]. Consequently, drugs that block the action of BTK are highly effective and fundamentally have changed the standard of care for patients with CLL since the therapeutic introduction of the first BTK inhibitor (BTKi), ibrutinib, in 2013. Ibrutinib, and second-generation BTKi’s, such as acalabrutinib and zanubrutinib, form a covalent bond with BTK within its kinase domain at residue C481, causing permanent inactivation of BTK and requiring the leukemia cell to synthesize new enzyme to restore B-cell signaling [2]. A nonsynonymous mutation in BTK at C481, such as C481S, can block the capacity of these drugs to form a covalent bond with BTK, thus conferring clinical resistance to ‘covalent’ BTKi’s [3]. This stimulated development of ‘non-covalent’ BTKi’s, such as pirtobrutinib or nemtabrutinib, which may be effective in treating patients with CLL cells that harbor the BTK C481S mutation [4]. However, patients subsequently were noted to develop resistance to therapy with pirtobrutinib through acquisition of other nonsynonymous BTK mutations, e.g. T474I or L528W [5]. Furthermore, other BTK mutations that confer clinical resistance to covalent and/or noncovalent BTKi’s now have been identified, including V416L, A428D, and M437R [6,7,8]. This has stimulated interest in development of drugs that can target BTK for proteasomal degradation, the so-called “BTK-degraders” [9], which are presumed able to mitigate the risk of drug resistance due to mutations in BTK [10].
Here we describe a patient who successively acquired resistance to each generation of BTKi, including a BTK-degrader, BGB-16673, which has demonstrated clinical activity in early phase I clinical trials [11]. He was 66 years old when diagnosed in 2014 with CLL that expressed an unmutated IGHV with 100% homology to IGHV1-69 and that reportedly had trisomy 12 as the sole cytogenetic abnormality detected by Fluorescence in situ Hybridization (FISH). Due to rapid disease progression with a lymphocyte-doubling time of ≈4 months, he was treated with bendamustine and rituximab in 2015, achieving a clinical complete response (CR) by iwCLL criteria [12]. Due to relapsed progressive CLL in 2017, he presented to us for therapy with obinutuzumab and venetoclax (after debulking with high dose methylprednisolone), again achieving a clinical CR. He remained on therapy with venetoclax until 2019 when he developed rapidly progressive lymph node enlargement, for which he initiated ibrutinib, resulting in complete resolution of his bulky lymphadenopathy. In 2021 he experienced symptomatic cardiac arrythmias, thought possibly ibrutinib-related; consequently his therapy was switched to acalabrutinib. He maintained a good clinical response until 14 months later, when he again developed progressive lymphadenopathy. Next generation sequencing (NGS) of his marrow aspirate, of which 20% were CLL cells (07/07/2022, Table 1), revealed a mutation in BTK c1442G>C at a variant allelic frequency (VAF) of 3.1%, resulting in BTK p.C481S. Also noted was a mutation in TP53 c.730G>A at a VAF of 10%, resulting in TP53 p.G244S and a complex karyotype with del(17p) (Table 1). He initiated therapy with an inhibitor of the δ/γ isoforms of phosphoinositide-3-kinase, duvelisib, which again resulted in complete resolution of his lymphadenopathy. However, because of adverse effects of therapy, he discontinued duvelisib and initiated therapy with pirtobrutinib. His disease was well controlled on pirtobrutinib for 7 months, after which time he again developed rapidly progressive lymphadenopathy. NGS of his marrow aspirate, of which 42% were CLL cells (09/12/2023, Table 1), revealed a distinctive subclone of CLL harboring a mutation in BTK c.1421C>T at a VAF of 19%. This mutation resulted in BTK p.T474I, which prior studies found confers resistance to pirtobrutinib [6]. Also detected was the previously identified mutation in TP53 c.730G>A at a VAF of 35% and similar complex karyotype, but not the mutation in BTK c1442G>C (Table 1). After this analysis, he discontinued pirtobrutinib and enrolled into a clinical study, in which he was treated with the BTK-degrader, BGB-16673. After initially experiencing a marked reduction in bulky lymphadenopathy, he came off this study after 4 months of therapy because of rapidly progressive disease. Subsequent to his coming off-study, NGS of his marrow aspirate, of which 67% were CLL cells (02/15/2024, Table 1), revealed yet another distinctive subclone of CLL cells with a new mutation in BTK c.1283C>A at a VAF of 28%, resulting in BTK p.A428D, but without detectable BTK c1442G>C or BTK c.1421C>T, while maintaining the same mutation in TP53, namely c.730G>A, at a VAF of 62% and same complex karyotype. These findings support a model of selective emergence of distinct subclones during his successive therapy with each type of BTKi from cells harboring the same mutation in TP53, namely c.730G>A and complex karyotype as noted in earlier samples. Remarkably there did not appear to be substantial evolution in the karyotypic complexity noted in the cytogenetics or FISH analyses performed on samples collected on 07/22/2019, 07/07/2022, 09/12/2023, and 02/15/2024, which respectively were obtained before and after treatment with obinutuzumab, venetoclax, and successive BTKi’s (Table 1 and Supplementary Fig. 1 and Supplementary Table 1). In any event, this case highlights the challenges of targeting BTK in some patients with CLL.
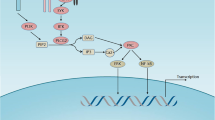
The BTK A428D mutation is located in the ATP-binding interface of BTK and significantly reduces its capacity to undergo autophosphorylation at Y223 or phosphorylation of its downstream substrate, phosphatidylinositol-specific phospholipase Cγ2 (PLCγ2) [7, 8]. However, upon B-cell receptor ligation the mutant BTK A428D, along with other catalytically inactive BTK mutants, e.g. BTK V416L or L528W, still can enable release of inositol-1-phosphate and provide for an enhanced Ca2+ flux in response to anti-µ stimulation in cells that have such mutant BTK when compared with cells with wildtype BTK [6, 8]. As such, these BTK mutant proteins can enable downstream signaling leading to activation of AKT, ERK, and NF-κB upon B-cell receptor ligation that is not inhibited by pirtobrutinib [6, 8, 9, 13]. Testament to this is the finding of the BTK A428D mutation in leukemia cells of patients who develop therapy resistance to pirtobrutinib [6]. Moreover, this mutation also may impair the sensitivity to other non-covalent inhibitors in clinical development, including ARQ-531, fenebrutinib, and vecabrutinib [6], as well as confer resistance to covalent BTKi’s [7]. Nevertheless, the BTK A428D mutation was not detected in the CLL cells of this patient until he developed resistance to the BTK degrader, BGB-16673.
This may be due to the selective pressure applied by BTK-degrader therapy. In work presented at the 2023 European Hematology Association Meeting, Feng and colleagues used BGB-16673 to treat various TMD8-derived lymphoma cell lines, each bearing either wildtype BTK or any one of the reported mutated BTK’s that are associated with BTKi resistance (e.g. V416L, A428D, M437R, T474I, T474L, M477I, C481S, C481F, C481Y, or L528W) [14]. They reported that BGB-16673 had a nanomolar IC50 and effected BTK protein degradation in each of these TMD8 lymphoma cell lines, except for those with the BTK A428D mutation, suggesting that BGB-16673 interacts with BTK at or around A428. It is noteworthy that the crystal structure of the BTK interacting “hook” of another BTK-degrader in clinical development, namely NX-2127, reveals a close drug interface with the BTK A428 residue [9, 15]. Modeling of a BTK A428D mutation places a negatively charged aspartic acid in place of the hydrophobic side chain of alanine within the binding pocket of NX-2127 to BTK (Fig. 1). This suggests that CLL cells with BTK A428D also may be resistant to NX-2127, as they already are known to be with either non-covalent or covalent BTKi’s [6]. Consequently, the two BTK degraders furthest advanced in clinical trials potentially may select for CLL cells with BTK A428D that are resistant to all approved BTKi’s.

A Crystal structure of wildtype BTK with residues that were found mutated highlighted in yellow. B BTK structure highlighting in orange the three modeled resistance mutation variants clustered in the BTK kinase domain. C The hook region (in pink) of the BTK degrader NX-2127 bound to wildtype BTK in proximity to the BTK A428 residue (in yellow). D Modeled BTK resistance mutation A428D with the aspartic acid side chain (in orange) inside the binding pocket for NX-2127.
In summary, targeting BTK has profoundly changed the face of CLL treatment over the past decade. Iterative advances in the cat and mouse game of resistance and redesign have moved BTK inhibitors from covalent to non-covalent and now targeted protein degraders. However, contrary to the presumption that protein degraders may be impervious to mutations in BTK, we now present clinical evidence that a mutation in the kinase domain of BTK can confer disease resistance to a BTK degrader currently in clinical trials. Moreover, we anticipate that patients with CLL cells that harbor the A428D mutation in BTK may prove refractory to therapy with either BGB-16673 or NX-2127.
References
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–32.
Estupinán HY, Berglöf A, Zain R, Smith CIE. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol. 2021;9:630942.
Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9:13023–35.
Mato AR, Woyach JA, Brown JR, Ghia P, Patel K, Eyre TA, et al. Pirtobrutinib after a covalent BTK inhibitor in chronic lymphocytic leukemia. N Engl J Med. 2023;389:33–44.
Naeem A, Utro F, Wang Q, Cha J, Vihinen M, Martindale S, et al. Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance. Blood Adv. 2023;7:1929–43.
Wang E, Mi X, Thompson MC, Montoya S, Notti RQ, Afaghani J, et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med. 2022;386:735–43.
Chirino A, Montoya S, Safronenka A, Taylor J. Resisting the resistance: navigating BTK mutations in chronic lymphocytic leukemia (CLL). Genes. 2023;14:2182.
Qi J, Endres S, Yosifov DY, Tausch E, Dheenadayalan RP, Gao X, et al. Acquired BTK mutations associated with resistance to noncovalent BTK inhibitors. Blood Adv. 2023;7:5698–702.
Montoya S, Bourcier J, Noviski M, Lu H, Thompson MC, Chirino A, et al. Kinase-impaired BTK mutations are susceptible to clinical-stage BTK and IKZF1/3 degrader NX-2127. Science. 2024;383:eadi5798.
Villanueva MT. BTK degraders tackle drug resistance. Nat Rev Drug Discov. 2024;23:173.
Seymour JF, Cheah CY, Parrondo R, Thompson MC, Stevens DA, Lasica M, et al. First results from A Phase 1, first-in-human study of the Bruton’s tyrosine kinase (BTK) Degrader Bgb-16673 In Patients (Pts) With Relapsed Or Refractory (R/R) B-cell malignancies (BGB-16673-101). ASH; 2023; San Diego, CA: Blood; 2023. p. 4401.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60.
Dhami K, Chakraborty A, Gururaja TL, Cheung LW, Sun C, DeAnda F, et al. Kinase-deficient BTK mutants confer ibrutinib resistance through activation of the kinase HCK. Sci Signal. 2022;15:eabg5216.
Feng X, Wang Y, Long T, Bai L, Yang X, Yang A, et al. P1239: Bruton Tyrosine Kinase (Btk) Protein Degrader Bgb-16673 Is less apt to cause, and able to overcome variable BTK resistance mutations compared to other BTK inhibitors (BTKi). Hemasphere. 2023;7:e368855c.
Robbins DW, Noviski MA, Tan YS, Konst ZA, Kelly A, Auger P, et al. Discovery and Preclinical Pharmacology of NX-2127, an orally bioavailable degrader of Bruton’s tyrosine kinase with immunomodulatory activity for the treatment of patients with B cell malignancies. J Med Chem. 2024;67:2321–36.
Acknowledgements
This work was supported in part by the University of California San Diego Foundation Blood Cancer Research Fund (BCRF), and in part by the National Institutes of Health, National Cancer Institute (R01-CA236361) (TJK).
Author information
Authors and Affiliations
Contributions
TJK and RLW conceptualized the study and wrote the paper, RLW performed the analysis required to prepare Fig. 1, MYC and TJK provided patient samples. TJK supervised the study and acquired funding. All authors reviewed and provided edits and/or helpful comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Wong, R.L., Choi, M.Y., Wang, HY. et al. Mutation in Bruton Tyrosine Kinase (BTK) A428D confers resistance To BTK-degrader therapy in chronic lymphocytic leukemia. Leukemia 38, 1818–1821 (2024). https://doi.org/10.1038/s41375-024-02317-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-024-02317-4
- Springer Nature Limited