
Abstract
Background
Threshold of Toxicological Concern (TTC) approaches are used for chemical safety assessment and risk-based priority setting for data poor chemicals. TTCs are derived from in vivo No Observed Effect Level (NOEL) datasets involving an external administered dose from a single exposure route, e.g., oral intake rate. Thus, a route-specific TTC can only be compared to a route-specific exposure estimate and such TTCs cannot be used for other exposure scenarios such as aggregate exposures.
Objective
Develop and apply a method for deriving internal TTCs (iTTCs) that can be used in chemical assessments for multiple route-specific exposures (e.g., oral, inhalation or dermal) or aggregate exposures.
Methods
Chemical-specific toxicokinetics (TK) data and models are applied to calculate internal concentrations (whole-body and blood) from the reported administered oral dose NOELs used to derive the Munro TTCs. The new iTTCs are calculated from the 5th percentile of cumulative distributions of internal NOELs and the commonly applied uncertainty factor of 100 to extrapolate animal testing data for applications in human health assessment.
Results
The new iTTCs for whole-body and blood are 0.5 nmol/kg and 0.1 nmol/L, respectively. Because the iTTCs are expressed on a molar basis they are readily converted to chemical mass iTTCs using the molar mass of the chemical of interest. For example, the median molar mass in the dataset is 220 g/mol corresponding to an iTTC of 22 ng/L-blood (22 pg/mL-blood). The iTTCs are considered broadly applicable for many organic chemicals except those that are genotoxic or acetylcholinesterase inhibitors. The new iTTCs can be compared with measured or estimated whole-body or blood exposure concentrations for chemical safety screening and priority-setting.
Significance
Existing Threshold of Toxicological Concern (TTC) approaches are limited in their applications for route-specific exposure scenarios only and are not suitable for chemical risk and safety assessments under conditions of aggregate exposure. New internal Threshold of Toxicological Concern (iTTC) values are developed to address data gaps in chemical safety estimation for multi-route and aggregate exposures.
Similar content being viewed by others


Introduction
Hazard and exposure data required for the human health assessment of thousands of chemicals are limited. Risk-based chemical priority setting methods are being developed and applied to identify those chemicals that pose the greatest health concern to focus resources and assessment efforts [1,2,3,4,5]. New Approach Methods (NAMs) are being developed and applied to reduce animal testing and address hazard and exposure data gaps, including the US Environmental Protection Agency’s ToxCast [6] and ExpoCast [7,8,9] programs and the Exposure And Safety Estimation (EAS-E) Suite platform (www.eas-e-suite.com). The Threshold of Toxicological Concern (TTC) is a well-established approach [10,11,12,13,14] used to address hazard data gaps by regulatory agencies such as Health Canada [15], the European Food Safety Agency (EFSA) [16], and the US Food and Drug Administration [17]. In the absence of chemical-specific toxicity data, the TTC approach provides an estimate for a level of exposure in which no appreciable human health risk is expected [18]. A review by EFSA and the World Health Organization (WHO) [19] concluded that the TTC is a fit-for-purpose approach with broad applicability for risk estimation. Subsequently, Patlewicz et al. [20] selected a daily oral exposure-based TTC and high throughput exposure predictions from ExpoCast for approximately 8000 chemicals to demonstrate a risk-based priority setting method.
Humans can be exposed simultaneously from multiple exposure pathways, i.e., aggregate exposure, and this limits the application of route-specific TTCs such as a daily oral exposure-based TTC. For example, Health Canada discusses the challenge in applying an oral TTC value for screening risk evaluations to chemicals for which exposure occurs by other routes, such as dermal or inhalation [15]. Current TTCs derived from in vivo experimental data are exposure-route specific. For example, oral (ingestion) based TTCs were first developed 15–20 years ago [11, 12, 14]. Following the general approach for ingestion based TTCs, inhalation TTCs, e.g., [21], and dermal TTCs, e.g., [22], have also been developed. Partosch et al. [23] proposed a route-to-route extrapolation method by revising the TTC to account for oral bioavailability; however, this proposed method does not adequately address the situation because it ignores other toxicokinetic (TK) processes [24, 25]. The concept of an internal TTC (iTTC) has been proposed, e.g., [26,27,28] to address current limitations of the TTC so the approach can be applied more broadly including comparisons with aggregate exposure estimates. Essentially iTTCs can be determined from the same data used to derive external TTCs by applying TK data and models to convert external exposure doses to internal exposure doses, e.g., concentrations in blood or plasma [27, 28]. However, as highlighted in proposed iTTC workflows, e.g., [27,28,29] one of the principal limitations in applying TK models in this manner is the paucity of chemical-specific TK data, e.g., biotransformation rates, for all chemicals in the TTC databases. The ExpoCast program and other research have advanced TK modeling for data poor chemicals using in silico and in vitro NAMs for high throughput toxicokinetic (HTTK) and exposure models, e.g., [8, 9, 30,31,32,33]. Advancements include the development and validation of quantitative structure-activity relationships (QSARs) for predicting total (terminal) elimination half-lives and whole-body biotransformation half-lives in humans (HLT and HLB, respectively) [34, 35] for parameterizing TK models for applications in regulatory decision-making [36].
This study applies available TK data and models to derive in vivo iTTCs for whole-body and blood using No Observed Effect Levels (NOELs) reported for oral exposures. This approach is demonstrated as a case study with the NOEL dataset used to derive the Munro oral exposure TTCs [11]. A tiered approach for applying TK data to estimate steady-state internal concentrations preferentially uses measured in vivo TK data when available and model estimates for TK parameters when measured in vivo data are lacking. A one-compartment physiologically-based toxicokinetic (1-CoPBTK) model for mammals parameterized with available in vitro, in vivo and in silico data is used to estimate chemical-specific TK parameters in the absence of measured in vivo TK data. Cumulative distributions of the new internal exposure concentrations corresponding to the Munro NOELs are used to select the 5th percentile values from which new iTTCs for whole-body and blood are calculated with commonly applied uncertainty factors. Recommendations for improving the approach and to expand and better define the chemical applicability domain of the new iTTCs are provided.
Methods
General toxicokinetic model
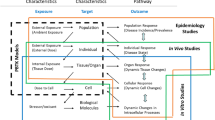
Figure 1 provides a conceptual overview of the general methods developed and applied to calculate internal concentrations from exposure doses and to derive new whole-body and blood iTTCs. The fundamental relationship between external exposures (used for TTCs) and internal exposures (used for iTTCs) is determined by chemical- and organism-specific toxicokinetics. An internal whole-body exposure (dose) can be calculated from an external oral ingestion rate (dose) as:
where CWB is the steady-state whole-body concentration (mg/kg-body), OD is the oral dose (mg/kg-body/d), AE is the chemical absorption efficiency (unitless) from the gastrointestinal tract (GIT), and kT is the first-order whole-body total (terminal) elimination rate constant (1/d). In the current application of the models, OD is replaced with reported NOEL (mg/kg-body/d). The AE parameter is different from oral bioavailability (F; often expressed as a percentage) because F includes first pass effects in the liver and AE quantifies chemical absorption from the GIT lumen into blood (portal vein) [37]. The kT parameter is the sum of individual chemical elimination process rate constants as:
where kR, kF, kU and kB are rate constants for respiratory elimination, fecal egestion, urinary excretion (renal clearance), and biotransformation (metabolism), respectively. These rate constants constitute the primary chemical elimination processes in an organism. For persistent chemicals that are very slowly eliminated and very slowly biotransformed, organism growth can influence the concentration, therefore, kG (rate constant to account for growth) can be included as a “pseudo” elimination rate process in the steady-state calculation. kT can be measured in vivo or calculated using PBTK models. First order rate constants operating at an organism level can be converted to whole-body half-lives and vice versa, e.g., HLT = ln2/kT and HLB = ln2/kB. Steady-state blood concentrations (CB; mg/L-blood) can be calculated from CWB using the steady-state volume of distribution referenced to blood (VD; L-blood/kg-body) as:

External dose NOELs were converted using a general toxicokinetic model to generate the cumulative distribution of internal NOELs, and then lower 5th percentile internal NOEL value was divided by a 100-fold adjustment factor to calculate the iTTC.
Munro TTC dataset
The NOELs from the original Munro database derived from administered oral doses were used in Eq. 1, i.e., OD = NOEL, to calculate internal dose CWB and CB. The original Munro oral TTC values were derived from 613 organic chemicals associated with 2941 NOELs from non-cancer endpoints in toxicity studies conducted in rats (n = 489), mice (n = 90), hamsters (n = 31), and rabbits (n = 3). As detailed in Munro et al. [11] the 613 chemicals were assigned to three Cramer classes based on the Cramer classification scheme [38] (i.e., Class I – low; n = 137, Class II – medium; n = 28, Class III – high level of concern; n = 448). A TTC for each Cramer class used by Munro et al. [11] was determined using the 5th percentile NOEL and dividing by an uncertainty factor of 100 to obtain intake rate based TTCs of 30, 9.0 and 1.5 µg/kg-bw/d for Cramer class I, II and III, respectively for a 60 kg adult. The “original” Munro dataset [11] as reported by Bassan et al. [39] which includes Simplified Molecular Input Line Entry System (SMILES) notations [40] was used. The Munro et al. NOEL toxicity data summarized by Bassan et al. [39] were not critically evaluated; however, a cursory analysis of the SMILES associated with the database was conducted. The exposure dose was determined as chemical quantity (e.g., mmol-chemical/kg/d), not mass (e.g., mg-chemical/kg/d) because it is the quantity of chemical, not its mass, at a site of toxic action that may elicit a biological response. Further comments on the Munro data are in the Supplementary Information (SI). Chemicals identified as genotoxic or acetylcholinesterase inhibitors as used in other TTCs were not included here.
Model parameterization
Figure 2 shows tiers of TK data used to parameterize kT in Eq. 1 to estimate internal doses (CWB and CB) from the NOELs reported in the Munro TTC dataset [11, 39]. In Tier 1 measured in vivo kT data are used to parameterize Eq. 1. For chemicals without in vivo kT data, Tier 2 uses the 1-CoPBTK model parameterized with in vitro and in silico (QSAR) estimates for kB to calculate kT that is then used in Eq. 1. The 1-CoPBTK model is generic in the sense that it can be readily parameterized for (i) many mammalian species requiring only minimal organism specific parameters to predict physiological processes, and (ii) many organic chemicals requiring only minimal chemical-specific parameters, e.g., kB and distribution ratios. The 1Co-PBTK model is part of a general modeling framework that has been applied and evaluated for various mammals over the past 20 years, e.g., [33, 34, 41,42,43,44]. Details of the 1-CoPBTK model are presented in the SI. The 1Co-PBTK models are coded in the EAS-E Suite HTTK module (www.eas-e-suite.com) for HTTK applications for an adult male human and rat.

Blood concentrations (CB or Cblood) are then calculated from whole body concentrations (CWB) as shown in Eq. (3). Absorption efficiency (AE) is used in the parameterization of the default and alternative #1 models while oral bioavailability (F) is used in the alternative model #2 calculations.
A literature search for measured in vivo mammalian TK data for the chemicals in the Munro database [11] was conducted. Rat in vivo kT data were selected preferentially for toxicity studies for rats, and mouse in vivo kT were selected preferentially for toxicity studies using mice; however, if there were no rat data for a chemical but mouse data were available then the in vivo mouse data were used and vice versa using allometric scaling. In vivo human kT,H values were scaled to test animal specific kT,A values using the following allometric relationship:
where MH is the human body mass (70 kg) assumed for the empirical kT,H data and MA is the experimental animal body mass (assumed 0.025 kg for mouse, 0.25 kg for rat and hamster, and 5 kg for rabbit studies).
In absence of any empirical in vivo kT data, the Tier 2 methods for parameterizing Eq. (1) were applied. In Tier 2 test animal specific kT,A values were calculated using the 1-CoPBTK model with kB,A estimates from (i) hepatocyte assays and common in vitro-in vivo extrapolation (IVIVE) models and (ii) QSARs for predicting HLB,H (in humans). Details of the IVIVE calculations for scaling in vitro biotransformation rates to kB are provided in the SI. The five HLB,H QSARs [34, 35] used here were developed from in vivo human data as described by Arnot and colleagues [34]. Predicted HLB,H values were converted to kB,H as kB,H = ln2/HLB,H and kB,H values were then scaled to toxicity test animal body mass values (kB,A) using allometric scaling Eq. 4 replacing kT with kB. The QSARs have been developed and validated following OECD guidance for QSAR applications in regulatory decision-making [36]. The QSAR predictions include Applicability Domain (AD) information as detailed in the SI. When in vitro biotransformation rates were available from rodents or humans the IVIVE estimates of kB were combined with “in” domain HLB-QSAR predictions to calculate a geometric mean for kB that was then used to parameterize the test animal 1-CoPBTK models. If in vitro biotransformation rates were not available, the geometric mean of the “in” domain QSAR predictions for kB were calculated to parameterize the 1-CoPBTK models.
Alternative TK model assumptions
Equation (1) requires an estimate for oral AE. An AE model that is primarily a function of the octanol-water partition coefficient (KOW) and chemical residence time in the GIT [45] was used in the default calculations (see SI for details). One principal limitation when applying the general TK model described in Eq. 1 is the absence of a first-pass effect in the liver. The parameter AE in Eq. (1) considers absorption from the GIT into the portal vein and does not explicitly account for the potential reduction in systemic blood concentrations (and whole-body concentrations) that may occur if there is significant biotransformation in GIT tissues or the liver. While AE is conservative for exposure assessment it may be inappropriate when estimating internal hazard values from external doses. Empirical estimates of bioavailability (F) from the literature from studies with mammals (humans, rats) were obtained to address the potential error of ignoring first-pass effects in the default model parameterization. In the absence of empirical estimates of F, predictions from the ACD Labs model (Release 2019.2.1, Build 3285. 16 Jan 2020) were obtained. The general model was then parameterized with F instead of AE for a second set of calculations and this is referred to as alternative model #1. For the TK model calculations, a minimum value of 1% was set for AE and F when estimates were < 1%.
The HTTK model used to estimate kT for the test animals includes two options for predicting renal clearance. The first option used in the default model assumes urine excretion is a function of equilibrium partitioning between the body and urine as is commonly used in 1-CoPBTK models used for environmental exposure estimation [33, 34, 41, 44]. A second option assumes the glomerular filtration rate methods that are more commonly used in IVIVE TK models [31, 32]. The glomerular filtration prediction method results in faster rates of renal clearance compared to the partitioning model for more water-soluble chemicals, i.e., log KOW < 3. When urinary excretion of chemicals is faster, kT becomes faster resulting in lower predicted internal concentrations. To address some of the uncertainty in renal elimination rates an additional set of simulations using the glomerular filtration calculation method was combined with the assumption of using F instead of AE and this approach is referred to as alternative model #2.
Hepatic blood flow limitations can occur for chemicals that are rapidly biotransformed in the liver and biotransformed slowly, or not at all, in other compartments of the body. Under these conditions the net effect of error is that kT values calculated from a 1-CoPBTK model can be faster than kT values calculated from a multicompartment PBTK model parameterized with hepatic clearance estimates only [44]. Many chemicals are biotransformed in other compartments of the body and for such chemicals the potential hepatic blood-flow limitations could be irrelevant. It is not currently possible to explicitly address the potential errors that may exist in the 1-CoPBTK model kT calculations for these conditions because the uncertainty in potential extra-hepatic clearance is not known for most chemicals in the Munro database. Fortunately, in the absence of empirical kT data, the approach here uses whole-body estimates of biotransformation (kB) that account for hepatic as well as extra-hepatic biotransformation [34].
Physical-chemical properties
Physical-chemical properties required to parameterize the generic 1-CoPBTK models include molar mass, pKa (for ionizable organics), KOW and octanol-air partition coefficients (KOA). Chemical data are summarized in the SI.
Results
Model input parameters
Munro dataset
The NOELs in the Munro TTC dataset span about 6 orders of magnitude ranging from 0.005 (Haloxyfop-methyl) to 7,203 (Calcium cyclamate) mg/kg/d with a median value of 30 mg/kg/d. When the NOELs were converted to a molar basis for the discrete organic moiety, the external NOELs span about 7 orders of magnitude ranging from 1.3 × 10−5 (dieldrin) to 114 mmol/kg/d (ethanol) with a median of 0.14 mmol/kg/d. When expressed as external molar concentrations some of the chemicals with the lowest NOEL doses (~ 10−5 mmol/kg/d) are dieldrin, haloxyfop-methyl, abamectin B1, zeranol. Chemicals with some of the highest external NOEL doses (~ 10–100 mmol/kg/d) include alcohols, e.g., ethanol, methanol, glycerol, propylene glycol.
The Munro TTC dataset comprises a diverse range of chemical classes and properties. The molar mass of the administered chemicals (including salt formulations) ranges from 30 to 1,135 g/mol with a median of 223. The molar mass of the discrete organic molecules in the dataset ranges from 23 to 1,135 g/mol with a median of 220. For the discrete organic chemical structures, the measured log KOW values (n = 402/613) range from −5.4 to 10.0 and the geometric means of the predicted log KOW values range from −6.0 (β-cyclodextrin) to 16.2 (stearyl tartrate) for the remaining chemicals. Due to a general lack of measured KOA data, the KOA values required to parameterize the 1-CoPBTK model were predicted from QSARs and polyparameter linear free energy relationship (ppLFERs). The geometric means of the predicted log KOA values range from 1.2 to 25. One hundred ninety-six of the chemicals were classified as mono-protic acids with pKas ranging from −3.6 to 11.3, with a median of 4.6. Fifty-eight of the chemicals were classified as mono-protic bases with pKas ranging from 4.0 to 13.6, with a median of 7.6.
Summary of in vitro, in vivo and in silico TK data
In vivo rat and mouse HLT data were found for 38 and 4 chemicals in the Munro database, respectively. In vivo HLT data from human studies were found for 63 chemicals. Combined there were empirical in vivo HLT estimates for 91 of the 613 chemicals. In vitro rat and human hepatocyte biotransformation rates were obtained for 18 and 102 chemicals, respectively. The in vitro biotransformation rates were critically evaluated for data quality using methods and criteria described elsewhere [46]. Combined there are in vitro measurements for 111 of the chemicals for which there were no in vivo HLT measurements. Whole-body biotransformation half-lives (HLB) predicted by QSARs [34, 35] that were within their defined applicability domains were used for 522 of the 613 Munro TTC database chemicals.
The measured in vivo HLT data (at 0.25 kg-bw) range from 0.02 to 4700 hours with a median of 1.6 h. The in vitro and in silico HLT data (at 0.25 kg-bw) range from 0.02 to 8,100 hours with a median of 1.1 h. Combining the in vivo with the in vitro and in silico estimates, the HLT values used to estimate internal exposures range from 0.02 to 4700 hours (spanning about 6 orders of magnitude) with a median of 1.1 h. The distribution of estimated HLT is very similar to the distribution of empirical HLT. Chemicals with the shortest HLT (ca. a few minutes) include formaldehyde, formic acid, and vinyl chloride. Chemicals with the longest HLT (ca. 60 to 200 d) include Persistent Organic Pollutants (POPs) listed under the Stockholm Convention like PCBs, mirex, polybrominated diphenyl ethers (PBDEs), p,p-DDT, lindane, dieldrin, and hexachlorobenzene.
Empirical estimates of oral bioavailability (F) from experimental studies with mammals were found for 53 chemicals (see TK databases in EAS-E Suite; www.eas-e-suite.com). The empirical estimates for F range from 5 to 100% (median = 74%) and are in reasonable agreement with the default AE model calculations and the ACD Labs F calculations. The net result of applying the alternative model #1 is a lower value in the numerator (Eq. 1) and hence a lower predicted internal concentration for chemicals when F is lower than AE. Most potential errors in estimating AE and F are considered relatively minor in the current context because if for example the true value is 50% instead of 100% then the predicted steady-state internal exposure concentrations are overestimated by only a factor of 2.
Model calculations
Default model
The calculated whole-body concentrations for the database range from 9.9×10−4 to 1.3×104 µmol/kg (spanning over 7 orders of magnitude) with a median of 9.7 µmol/kg. The calculated molar blood concentrations for the database range from 3.2×10−4 to 2.1×103 µmol/L-blood (spanning almost 7 orders of magnitude) with a median of 3.9 µmol/L-blood. When expressed as internal molar concentrations (whole-body or blood), many of the more potent chemicals in the dataset are known bioactive chemicals and biocides, e.g., glufosinate, abamectin B1, zeranol, haloxyfop-methyl, tetrakis(hydroxymethyl)phosphonium, and paraquat. Some examples of chemicals with the highest internal molar doses (indicating relative lower potency) are those often included in food, e.g., quercetin, various simple alcohols, some dyes (food additives), and relatively unreactive chemicals like DDT, PBDEs, and PCBs.
External and internal chemical concentration comparisons
Figure 3 compares administered external doses (µmol/kg/d) and internal whole-body concentration doses (µmol/kg) corresponding to the reported NOELs for the parent (discrete organic moiety) chemicals (n = 613). The internal concentrations include explicit consideration for chemical-specific toxicokinetics (i.e., absorption, elimination, and biotransformation of parent chemical) and thus the relative potencies are more directly comparable between chemicals in the internal dose dataset. While there is a positive correlation between the two datasets, the external dose only explains about 56% of the chemical potency, the rest can be explained by toxicokinetics. Many chemicals that appear to be relatively “more toxic” because of lower oral intake NOELs are actually relatively “less toxic” than other chemicals with respect to internal dose. In some cases, the apparent toxicity and relative potency as indicated by the two approaches differ by several orders of magnitude reflecting the influence of HLT, and to some degree AE, on internal exposure.

The dashed diagonal line represents the 1:1 line.
Alternative models
The model that considers some uncertainty in possible first-pass effects (alternative model #1) calculated whole-body molar concentrations ranging from 7.4×10−4 to 3.9×103 µmol/kg with a median of 6.2 µmol/kg. The molar blood concentrations range from 3.1×10−4 to 2.0×103 µmol/L-blood with a median of 2.3 µmol/L-blood. As expected, the internal concentrations for some chemicals are lower and the median concentrations are slightly less than twofold lower compared to the default model. The model that includes the potential for first-pass effects and higher rates of urinary excretion for lower KOW chemicals (alternative model #2) calculated whole-body molar concentrations ranging from 6.3×10−4 to 3.8×103 µmol/kg with a median of 5.5 µmol/kg. The calculated molar blood concentrations from 3.1×10−4 to 2.0×103 µmol/L-blood with a median of 2.0 µmol/L-blood. Here again for some chemicals the internal concentrations decrease but the distributions remain largely similar compared to the default and alternative #1 model calculations.
Deriving new TTCs and iTTCs
In this case study the database was not split into the three Cramer Classes associated with Munro TTCs so that the new iTTCs could be applied generally to organic chemicals that are not identified as being genotoxic or acetylcholinesterase inhibitors, i.e., certain carbamates and organophosphates, thus minimizing the need for decision-trees for an initial screening. The fifth percentile of the cumulative distribution of all NOELs in the Munro database expressed on a chemical mass oral intake rate basis is 280 µg/kg/d (not shown). Applying an assumed uncertainty factor of 100 consistent with previous methods for deriving TTCs from this dataset, the TTC is 2.8 µg/kg/d. This TTC for the entire database is included for context when comparing against other TTCs and iTTCs calculated in this study, and not as another TTC for consideration for regulatory applications. For example, the TTC based on the total Munro dataset is more conservative than the Cramer Class I and II TTCs and within a factor of 2 of the Cramer Class III TTC (1.5 µg/kg/d). The fifth percentile of the cumulative distribution of all NOELs expressed on a molar basis for the discrete organic molecules is 0.83 µmol/kg/d (not shown). Applying an uncertainty factor of 100 to this value results in a TTC of 0.0083 µmol/kg/d. Recall the range of molar mass for administered chemicals in the dataset is large, but the median is 220 g/mol approximating the difference between the two TTCs. The actual factor difference between the mg and mmol oral intake TTCs is 337 which is a result of treating the discrete organic molecules separately from the administered salt formulations (see SI for details).
Table 1 summarizes the new iTTCs. Figure 4 shows the cumulative distributions of the blood concentrations for the discrete organic molecules (n = 613) calculated from the Munro TTC NOELs using the three different model assumptions. There is general consistency between the 3 distributions and the alternative models #1 and #2 show slightly lower blood concentrations largely reflecting the differences in assumptions relating to reduced uptake and enhanced renal clearance. The 5th percentiles of steady-state blood concentrations are 22, 8.5 and 8.3 nmol/L for the default, alternative #1 and alternative #2 model calculations, respectively. Following the general operating procedures for applying safety (uncertainty) factors for calculating TTCs from NOELs an uncertainty factor of 100 was applied to account for interspecies differences (factor of 10) and human variability (factor of 10) [47,48,49]. The new blood iTTCs are 0.22, 0.085 and 0.083 nmol/L for the default, alternative #1 and alternative #2 model calculations, respectively. Given the consensus in the iTTCs from the three models and the underlying uncertainty in the hazard and TK data a blood iTTC of 0.1 nmol/L (or pmol/mL-blood) is selected for organic chemicals that are not considered genotoxic and are not acetylcholinesterase inhibitors. This iTTC is therefore rather broadly applicable for a range of organic chemicals. Furthermore, because the new iTTC is expressed on a molar basis it can be readily converted to an iTTC expressed on a chemical mass basis by multiplying 0.1 nmol/L by the molar mass of the chemical of interest, for example when comparing to human exposure blood concentrations expressed in units of ng/L or pg/mL. As a first approximation, using the median value for molar mass in the dataset (220 g/mol) corresponds to a blood iTTC of 22 ng/L-blood (or 22 pg/mL-blood).

The figure on the right provides a better view of the lower 20% of the data in the cumulative distribution.
Figure 5 shows the cumulative distributions of the whole-body concentrations for the discrete organic molecules (n = 613) calculated from the NOELs using the three different model assumptions. As with blood, there is general consistency between the 3 distributions and the alternative models #1 and #2 show slightly lower concentrations. The 5th percentiles of steady-state whole-body concentrations are 0.074, 0.029 and 0.023 µmol/kg for the default, alternative #1 and alternative #2 model calculations, respectively. Applying an uncertainty factor of 100 results in new whole-body iTTCs of 0.74, 0.29 and 0.23 nmol/kg, respectively. Given the consensus in the iTTCs from the three models a whole-body iTTC of 0.5 nmol/kg is selected for organic chemicals that are not genotoxic and are not acetylcholinesterase inhibitors. This whole-body iTTC is therefore rather broadly applicable for a range of organic chemicals. As with the new blood iTTC, the whole-body iTTC can be expressed on a chemical mass basis by multiplying 0.5 nmol/kg by the molar mass (g/mol) of the chemical of interest. For example, using the median value for molar mass in the dataset (220 g/mol) corresponds to a whole-body iTTC of 110 ng/kg-bw. While the blood iTTC will likely be more applicable to human health studies, the whole-body iTTC may be helpful for applications to non-human receptors or when exposure estimates can only be obtained at a whole-body level.

The figure on the right provides a better view of the lower 20% of the data in the cumulative distribution.
Examining the steady-state assumption in the Munro TTC database
It has been recommended by experts at EFSA [49] that the TTC approach is not applicable “for chemicals that are known or predicted to bioaccumulate.” However, there was no practical or quantitative guidance provided for determining what constitutes a “bioaccumulative chemical” for exclusion. Leeman et al. [50] highlight that the Munro TTC database includes many POPs and “bioaccumulative chemicals” including PCBs (Arochlor 1254), PBDEs, chlordane, p,p-DDT, mirex and hexachlorobenzene [11]. Kroes et al. [14] had previously indicated that bioaccumulation is not accounted for in the TTC approach. We analyzed the experimental NOEL data to determine if the exposure durations in the sub-chronic and chronic experiments were sufficiently long for the organism to approach steady-state. The time to approach 95% of steady-state can be estimated as 4 × HLT. Using the in vivo, or default model estimates for HLT when necessary, and the reported experimental exposure durations, all but three chemicals are expected to approach 95% of steady-state within the experimental test duration (SI Section S5). Thus, from a hazard threshold perspective, the Munro TTCs can be considered “valid” with respect to the steady-state assumption and the exclusion of the TTC approach for bioaccumulative chemicals seems unwarranted.
Discussion
The Munro dataset comprises a diverse range of chemical classes, structures, physical-chemical properties, and biological half-lives. The new in vivo iTTCs for whole-body and blood concentrations from the Munro NOEL dataset can be considered for thousands of data poor chemicals in absence of chemical-specific hazard data. The iTTCs can be compared against measured blood concentrations and against predicted whole-body or blood concentrations from multi-route and aggregate exposure scenarios for risk (or safety) based screening and priority setting assessment objectives. For example, while existing oral-dose based TTCs have been compared with high-throughput external exposure estimates for risk-based priority setting [20], the new iTTC can be used with internal aggregate exposure estimates using tools like the PROduction-To-EXposure High Throughput (PROTEX-HT) model [33]. Blackburn et al., [25] have previously proposed an in vitro derived iTTC of 1 µmol/L for cosmetics; however, this proposed value was associated with several caveats and exclusions making it difficult to directly compare with the new in vivo blood iTTC derived herein, i.e., 0.10 nmol/L-blood. We are not aware of any other reported iTTCs for blood and whole-body level.
Ignoring TK obscures the true potency and relative hazard of chemicals when they are expressed in terms of external doses, e.g., Fig. 3. While whole-body and blood concentrations are still only surrogates for concentrations at target sites corresponding to molecular initiating events and biological perturbations, the internal exposures are much more representative than the external administered doses. Possible exceptions are for chemicals that exert effects upon epithelial tissues at the site of contact or portal of entry for which the administered concentrations and applied doses may be better surrogates than systemic blood concentrations.
This study highlights opportunities for more broadly converting external exposure doses to internal exposure doses to better compare and rank chemicals for relative potency in combination with other lines of evidence to address uncertainty in hazard characterization. Internal concentrations corresponding to oral NOELs and the iTTC can be compared against other existing and emerging data sources; however, it is imperative that the values used in the comparisons are in equal units and dimensions, e.g., [4]. The new internal concentrations can be compared with in vitro bioactivity data, e.g., from ToxCast, provided in vitro toxicokinetics are also considered, e.g., [51] and with in vivo tissue and body residues corresponding to effect responses, e.g., critical body residues [52]. Future comparisons of internal doses corresponding with biological effect, perturbations and no effects obtained from in vivo, in vitro and in silico methods will help strengthen weight of evidence approaches when establishing chemical-specific hazard thresholds and provide opportunities for further advancing Adverse Outcome Pathways (AOPs) by quantitatively linking internal exposure to key events and key event relationships.
Recommendations for future iTTCs
In future work, the new tiered TK methods can also be applied to other TTC datasets (e.g., acetylcholinesterase inhibitors, inhalation, and dermal data) to expand and better define the applicability domain of the new iTTCs. Multi-compartment PBTK models can also be considered when they can be parameterized with required TK data [29]. Future work to derive iTTCs should include a more intensive examination of in vitro, in vivo and in silico estimates for TK data used to parameterize the models and the development of validated QSARs for predicting in vitro or in vivo biotransformation rates and whole-body clearance in rodent species would be helpful. To address uncertainty in biotransformation rate data and to reduce unnecessary animal testing, long-term integrated testing strategies combining in vitro, in vivo and in silico methods for estimating biotransformation rates are strongly encouraged.
The emerging iTTC approach can also be aligned with general recommendations for improving the TTC approach, e.g., [53] and other on-going efforts to develop iTTCs [29]. There may be value in developing Cramer Class iTTCs in future work to better align with existing decision-trees, e.g., [14, 15]; however, we recommend a thorough evaluation of the in vivo NOEL data beforehand. Moreover, while the Cramer Class TTCs and other TTCs have proven useful, there are unique opportunities with iTTCs that could be explored for alternative classifications. Approaches to derive iTTCs for chemicals grouped by Mode of Action (MOA) or chemical class, may prove fruitful. The iTTC approach also fosters the capacity for improved inter-species comparisons and potential species sensitivities where uncertainties in inter-species toxicokinetics are greatly reduced. Finally, as discussed by Dankovic et al. [47] general approaches for applying uncertainty factors i.e., 100x, that seek to account for TK and toxicodynamic differences may or may not be sufficient to account for differences between animals and humans and thus examination of the application of uncertainty factors for extrapolating hazard data is warranted as more explicit TK modeling efforts are developed and applied.
Data availability
The supplemental material includes additional details regarding the toxicity data, the models, and the chemical information as well as summary model output results used in this study.
Change history
13 July 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41370-023-00582-6
References
U.S. EPA. Assessing and Managing Chemicals under TSCA; The Frank R. Lautenberg Chemical Safety for the 21st Century Act. Washington, DC. 2017.
U.S. EPA. A Working Approach for Identifying Potential Candidate Chemicals for Prioritization. Washington, DC. 2018.
Health Canada. Overview of the Chemicals Management Plan. Ottawa, ON. 2017.
National Academies of Sciences Engineering, and Medicine. Using 21st Century Science to Improve Risk-Related Evaluations. The National Academies Press: Washington, DC. 2017.
Kavlock RJ, Bahadori T, Barton-Maclaren TS, Gwinn MR, Rasenberg M, Thomas RS. Accelerating the pace of chemical risk assessment. Chem Res Toxicol. 2018;31:287–90.
Dix DJ, Houck KA, Martin M, Richard AM, Setzer RW, Kavlock RJ. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci. 2007;95:5–12.
Cohen Hubal EA, Richard A, Aylward L, Edwards S, Gallagher J, Goldsmith M-R, et al. Advancing exposure characterization for chemical evaluation and risk assessment. J Toxicol Environ Health Part B. 2010;13:299–313.
Wambaugh JF, Setzer RW, Reif DM, Gangwal S, Mitchell-Blackwood J, Arnot JA, et al. High-throughput models for exposure-based chemical prioritization in the ExpoCast project. Environ Sci Technol. 2013;47:8479–88.
Ring CL, Arnot JA, Bennett DH, Egeghy PP, Fantke P, Huang L, et al. Consensus modeling of median chemical intake for the U.S. population based on predictions of exposure pathways. Environ Sci Technol. 2019;53:719–32.
Frawley JP. Scientific evidence and common sense as a basis for food-packaging regulations. Food Cosmet Toxicol. 1967;5:293–308.
Munro IC, Ford RA, Kennepohl E, Sprenger JG. Correlation of structural class with no-observed-effect levels: A proposal for establishing a threshold of concern. Food Chem Toxicol. 1996;34:829–67.
Kroes R, Galli CL, Munro I, Schilter B, Tran L-A, Walker R, et al. Threshold of toxicological concern for chemical substances present in the diet: a practical tool for assessing the need for toxicity testing. Food Chem Toxicol. 2000;38:255–312.
Kroes R. The threshold of toxicological concern concept in risk assessment. Toxicol Sci. 2005;86:226–30.
Kroes R, Renwick AG, Cheeseman M, Kleiner J, Mangelsdorf I, Piersma A, et al. Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Chem Toxicol. 2004;42:65–83.
Health Canada. Threshold of Toxicological Concern (TTC)-based Approach for Certain Substances. Ottawa, ON. 2016.
More SJ, Bampidis V, Benford D, Bragard C, Halldorsson TI, Hernández-Jerez AF, et al. Guidance on the use of the threshold of toxicological concern approach in food safety assessment. EFSA J. 2019;17:e05708.
U.S. Food and Drug Administration. Food Additives; Threshold of regulation for substances used in food-contact articles. In, 1995. pp 21 CFR Parts 25, 25, 170, 171, and 174. Docket Nos. 177P-0122 and 0192N-0181.
Munro IC, Renwick AG, Danielewska-Nikiel B. The threshold of toxicological concern (TTC) in risk assessment. Toxicol Lett. 2008;180:151–6.
EFSA (European Food Safety Authority) and WHO (World Health Organization). Review of the Threshold of Toxicological Concern (TTC) approach and development of new TTC decision tree. 2016.
Patlewicz G, Wambaugh JF, Felter SP, Simon TW, Becker RA. Utilizing threshold of toxicological concern (TTC) with high throughput exposure predictions (HTE) as a risk-based prioritization approach for thousands of chemicals. Comput Toxicol. 2018;7:58–67.
Nelms MD, Patlewicz G. Derivation of new threshold of toxicological concern values for exposure via inhalation for environmentally-relevant chemicals. Front Toxicol. 2020;2:580347.
Safford RJ. The dermal sensitisation threshold - a TTC approach for allergic contact dermatitis. Regul Toxicol Pharm. 2008;51:195–200.
Partosch F, Mielke H, Stahlmann R, Kleuser B, Barlow S, Gundert-Remy U. Internal threshold of toxicological concern values: enabling route-to-route extrapolation. Arch Toxicol. 2015;89:941–8.
Chebekoue SF, Krishnan K. Derivation of occupational thresholds of toxicological concern for systemically acting noncarcinogenic organic chemicals. Toxicol Sci. 2017;160:47–56.
Blackburn KL, Carr G, Rose JL, Selman BG. An interim internal threshold of toxicologic concern (iTTC) for chemicals in consumer products, with support from an automated assessment of ToxCast™ dose response data. Regul Toxicol Pharm. 2020;114:104656.
Bessems JGM Opinion on the usefulness of in vitro data for human risk assessment. Suggestions for better use of non-testing approaches National Institute for Public Health and the Environment (RIVM), 2009. Report no.: 320016002/2009.
Coecke S, Pelkonen O, Leite SB, Bernauer U, Bessems JGM, Bois FY, et al. Toxicokinetics as a key to the integrated toxicity risk assessment based primarily on non-animal approaches. Toxicol Vitr. 2013;27:1570–7.
Ellison CA, Blackburn KL, Carmichael PL, Clewell HJ, Cronin MTD, Desprez B, et al. Challenges in working towards an internal threshold of toxicological concern (iTTC) for use in the safety assessment of cosmetics: Discussions from the Cosmetics Europe iTTC Working Group workshop. Regul Toxicol Pharm. 2019;103:63–72.
Ellison CA, Api AM, Becker RA, Efremenko AY, Gadhia S, Hack CE, et al. Internal threshold of toxicological concern (iTTC): Where we are today and what is possible in the near future. Front Toxicol. 2021;2:621541.
Wetmore BA, Wambaugh JF, Allen B, Ferguson SS, Sochaski MA, Setzer RW, et al. Incorporating high-throughput exposure predictions with dosimetry-adjusted in vitro bioactivity to inform chemical toxicity testing. Toxicol Sci. 2015;148:121–36.
Pearce RG, Setzer RW, Strope CL, Wambaugh JF, Sipes NS. httk: R Package for high-throughput toxicokinetics. J Stat Softw. 2017;79:1–26.
Wambaugh JF, Hughes MF, Ring CL, MacMillan DK, Ford J, Fennell TR, et al. Evaluating in vitro-in vivo extrapolation of toxicokinetics. Toxicol Sci. 2018;163:152–69.
Li L, Sangion A, Wania F, Armitage JM, Toose L, Hughes L, et al. Development and evaluation of a holistic and mechanistic modeling framework for chemical emissions, fate, exposure, and risk. Environ Health Persp. 2021;129:127006.
Arnot JA, Brown TN, Wania F. Estimating screening-level organic chemical half-lives in humans. Environl Sci Technol. 2014;48:723–30.
Papa E, Sangion A, Arnot JA, Gramatica P. Development of human biotransformation QSARs and application for PBT assessment refinement. Food Chem Toxicol. 2018;112:535–43.
OECD. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationship [(Q)SAR] models. Organisation for Economic Cooperation and Development, Environment Directorate: Paris, 2007.
OECD. Test No. 417: Toxicokinetics. Organisation for Economic Cooperation and Development, Environment Directorate: Paris, 2010.
Cramer GM, Ford RA, Hall RL. Estimation of toxic hazard—A decision tree approach. Food Cosmet Toxicol. 1976;16:255–76.
Bassan A, Fioravanzo E, Pavan M, Stocchero M. Applicability of physicochemical data, QSARs and read-across in threshold of toxicological concern assessment. EFSA Supporting Publ. 2011;8:159E.
Weininger D. SMILES, a chemical language and information-system.1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36.
Kelly BC, Ikonomou MG, Blair JD, Morin AE, Gobas FAPC. Food web-specific biomagnification of persistent organic pollutants. Science 2007;317:236–9.
Arnot JA, Brown TN, Wania F, Breivik K, McLachlan MS. Prioritizing chemicals and data requirements for screening-level exposure and risk assessment. Environ Health Persp. 2012;120:1565–70.
Lee Y-S, Lo JC, Otton SV, Moore MM, Kennedy CJ, Gobas FAPC. In vitro to in vivo extrapolation of biotransformation rates for assessing bioaccumulation of hydrophobic organic chemicals in mammals. Environ Toxicol Chem. 2017;36:1934–46.
Armitage JM, Hughes L, Sangion A, Arnot JA. Development and intercomparison of single and multicompartment physiologically-based toxicokinetic models: Implications for model selection and tiered modeling frameworks. Environ Int. 2021;154:106557.
Arnot JA, Mackay D. The influence of chemical degradation during dietary exposures to fish on biomagnification factors and bioaccumulation factors. Environ Sci Process Impacts. 2018;20:86–97.
Arnot JA, Toose L, Armitage JM, Embry M, Sangion A, Hughes L A weight of evidence approach for bioaccumulation assessment. Integr Environ Assess Manage. 2022; https://doi.org/10.1002/ieam.4583.
Dankovic DA, Naumann BD, Maier A, Dourson ML, Levy LS. The scientific basis of uncertainty factors used in setting occupational exposure limits. J Occup Environ Hyg. 2015;12:S55–S68.
U.S. EPA. A Review of the Reference Dose and Reference Concentration Processes. U.S. Environmental Protection Agency, Risk Assessment Forum. Washington, DC. 2002.
Antunovic B, Barlow S, Chesson A, Flynn A, Hardy A, Jeger MJ, et al. Scientific Opinion on Exploring options for providing advice about possible human health risks based on the concept of threshold of toxicological concern (TTC). EFSA Journal. 2012;10:2750.
Leeman WR, Rouhani-Rankouhi T, Vogels J, Krul L. Relevance of bioaccumulating substances in the TTC concept. Regul Toxicol Pharm. 2016;77:42–48.
Armitage JM, Wania F, Arnot JA. Application of mass balance models and the chemical activity concept to facilitate the use of in vitro toxicity data for risk assessment. Environ Sci Technol. 2014;48:9770–9.
Meador JP, Adams WJ, Escher BI, McCarty LS, McElroy AE, Sappington KG. The tissue residue approach for toxicity assessment: Findings and critical reviews from a Society of Environmental Toxicology and Chemistry Pellston Workshop. Integr Environ Assess Manag. 2011;7:2–6.
Nelms MD, Pradeep P, Patlewicz G. Evaluating potential refinements to existing threshold of toxicological concern (TTC) values for environmentally-relevant compounds. Regul Toxicol Pharm. 2019;109:104505.
Acknowledgements
JAA, LT, JMA, AS, AL, TNB, and LL acknowledge funding from the American Chemistry Council Long-Range Research Initiative. Because this publication has not been formally reviewed by the American Chemistry Council, views expressed in this document are solely those of the authors.
Author information
Authors and Affiliations
Contributions
JAA concept development, model development, model parameterization and applications, data analysis, writing. LT, JMA, PBTK model development, model coding, writing. AS, AL, TNB, LL literature searches and data suitability analysis of in vitro and in vivo TK data, QSAR applications. RAB concept development, writing. All authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
JAA, LT, JMA, AS, AL, TNB and LL acknowledge funding from the American Chemistry Council Long-Range Research Initiative (ACC-LRI). ARC Arnot Research and Consulting receives research funding from Health Canada, Environment and Climate Change Canada, and various chemical companies and industry groups. RB is a Senior Toxicologist and Director of the ACC-LRI at American Chemistry Council.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In the original publication the units in the a paragraph were 22 nmol/L-blood (or 22 pmol/mL-blood), but they should have been 22 ng/L-blood (or 22 pg/mL-blood). Additionally, the axes in Figure 3 of the original publication should have been presented on a molar basis (µmol) rather than a mass basis (µg).
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arnot, J.A., Toose, L., Armitage, J.M. et al. Developing an internal threshold of toxicological concern (iTTC). J Expo Sci Environ Epidemiol 32, 877–884 (2022). https://doi.org/10.1038/s41370-022-00494-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41370-022-00494-x
- Springer Nature America, Inc.