
Abstract
Proliferating cancer cells rely largely on glutamine for survival and proliferation. Glutamine serves as a carbon source for the synthesis of lipids and metabolites via the TCA cycle, as well as a source of nitrogen for amino acid and nucleotide synthesis. To date, many studies have explored the role of glutamine metabolism in cancer, thereby providing a scientific rationale for targeting glutamine metabolism for cancer treatment. In this review, we summarize the mechanism(s) involved at each step of glutamine metabolism, from glutamine transporters to redox homeostasis, and highlight areas that can be exploited for clinical cancer treatment. Furthermore, we discuss the mechanisms underlying cancer cell resistance to agents that target glutamine metabolism, as well as strategies for overcoming these mechanisms. Finally, we discuss the effects of glutamine blockade on the tumor microenvironment and explore strategies to maximize the utility of glutamine blockers as a cancer treatment.
Similar content being viewed by others
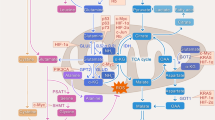

Introduction
Metabolic reprogramming, a hallmark of cancer cells, is a process by which cancer cells ensure a sufficient supply of proteins, nucleotides, and lipids to support rapid growth and proliferation1. The importance of cancer cell metabolism and the limitations of conventional cancer therapies (e.g., resistance to chemotherapy or radiotherapy) have prompted the development of strategies aimed at targeting this biological process2. Several drugs that do just that have been introduced and have shown promising results in animal studies; a few have entered clinical trials2. In particular, glutamine metabolism has attracted much attention as a therapeutic target because cancer cells are heavily reliant on this amino acid for growth and proliferation3.
Glutamine is a nonessential/conditionally essential amino acid that plays a pivotal role in clinical illness and stress conditions4. Glutamine in cancer cells plays critical and diverse roles by providing not only a source of nitrogen for amino acid and nucleotide biosynthesis but also a source of carbon to replenish the tricarboxylic acid (TCA) cycle and lipid biosynthesis pathways; thus, cancer cells are “addicted” to glutamine5. Glutamine metabolism and closely linked metabolic networks involving glutamine transporters, glutaminase, aminotransferase, and redox homeostasis are essential for cancer cell survival6. Targeting each step of glutamine metabolism has shown promising results in cancer treatment, prompting the discovery of druggable targets and the development of anticancer drug candidates3. In addition, given that immune checkpoint inhibitors are now widely used to treat cancer, the role of glutamine blockade within the tumor microenvironment (TME) has gained much attention7.
This review summarizes each step of glutamine metabolism in cancer cells and highlights opportunities for clinical intervention. Furthermore, we discuss resistance mechanisms and the role of glutamine blockade in the TME.
The role of glutamine in cancer cell growth
Rapidly proliferating cancer cells take up glutamine from plasma via various amino acid transporters, and then it is converted to glutamate in the mitochondria by the two forms of glutaminase: kidney-type glutaminase (GLS)1 and liver-type GLS28. Notably, GLS-mediated deamination of glutamine to glutamate is the first and rate-limiting step of glutaminolysis, making it an attractive druggable target9. GLS1 is overexpressed in various cancer cells, and this phenotype is associated with a higher disease stage and a poor prognosis10. Mechanistically, the expression of GLS1 is regulated indirectly by Myc (via repression of miR-23a and miR-23b) and mTORC111,12. Unlike GLS1, GLS2 suppresses the proliferation and migration of cancer cells13. In hepatocellular carcinoma (HCC), GLS2 inhibited proliferation in vitro and lung metastasis in a xenograft mouse model by inhibiting the small GTPase Rac113. However, several studies have shown that GLS2 is highly expressed in triple-negative basal-like breast cancer (TNBC) and metastatic lung cancer and that it confers radioresistance in advanced human cervical cancer cells, suggesting that GLS2 may reduce reactive oxygen species (ROS) levels by increasing the level of cellular reduced glutathione (GSH), NADH, or NADPH14,15,16. The confounding results regarding the function of GLS2 in cancer metabolism suggest that it may act in a context-specific manner17.
The TCA cycle is an essential hub for several metabolic pathways and for the interconversion of metabolites, which are renewed constantly in rapidly proliferating cancer cells18. Thus, replenishment of metabolic intermediates via the TCA cycle is vital to cancer cells, making them reliant on glutamine, a phenomenon called anaplerosis8. During anaplerosis, mitochondrial glutamate dehydrogenase 1 (GLUD1) plays a key role by catalyzing the conversion of glutamate to alpha-ketoglutarate (α-KG) and releasing ammonia, which regulates autophagy and neutralizes the intracellular pH in cancer cells19,20. α-KG is generated for the TCA cycle and is used for oxidative phosphorylation (OXPHOS)21. In addition, glutamine-derived α-KG is oxidized to succinate and fumarate, which maintain the TCA cycle in cancer cells by providing ATP, NADH, and FADH2 and by acting as oncometabolites22. Indeed, GLUD1 is overexpressed in various cancer cells, promoting epithelial-mesenchymal transition and drug resistance23. Mechanistically, Myc drives glutaminolysis by upregulating GLUD1 and induces a concurrent increase in the expression of GLS and SLC1A524.
Amino acids are required by cancer cells for proliferation under genotoxic, oxidative, and nutritional stress conditions; these amino acids serve as building blocks for protein synthesis and act as substrates for glucose, lipid, and nucleic acid synthesis25,26. In particular, glutamine plays a vital role in this process not only by providing a carbon source to the TCA cycle but also by acting as a nitrogen source for the biosynthesis of alanine, aspartate, and serine (Fig. 1)8. Therefore, the role of aminotransferases such as glutamate pyruvate transaminase (GPT) and glutamate oxaloacetate transaminase (GOT) in glutamine metabolism in cancer cells has been studied extensively8. Regarding GPTs, cytosolic GPT1 and mitochondrial GPT2 play major roles in energy metabolism in cancer cells by providing alanine for protein synthesis and by replenishing TCA cycle intermediates27. Indeed, GPT2 is a significant contributor to tumorigenesis in breast cancer, glioblastoma, and KRAS-driven colorectal cancer (CRC) cells28,29,30,31. Because cancer cells do not take up aspartate very well, GOT fuels tumorigenesis by providing cytosolic aspartate, which is used as a precursor for protein and nucleotide synthesis and for redox homeostasis32. Cytosolic GOT1 and mitochondrial GOT2, which together comprise the malate-aspartate shuttle, interconvert oxaloacetate and aspartate using glutamate or α-KG as substrates33. Indeed, both GOT1 and GOT2 are overexpressed in KRAS-driven pancreatic ductal adenocarcinoma (PDAC) cells34,35. Glutamine is also required for de novo synthesis of asparagine via asparagine synthetase (ASNS), which is induced by either the amino acid response or the unfolded protein response pathways36,37. Asparagine activates mTORC1 and contributes to the biosynthesis of purines and pyrimidines, as well as to the exchange of extracellular amino acids such as histidine, aspartate, and serine37. Indeed, the role of ASNS in tumorigenesis and metastasis has been reported, and it is associated with poor survival in various types of breast cancer, non-small cell lung cancer (NSCLC), and sarcoma38,39,40.

Glutamine transporters (SLC1A5, SLC38A1/SLC38A2, and SLC6A14) expressed on the cell membrane transport glutamine into the cytosol. Next, the SLC1A5 variant transports glutamine to the mitochondrial matrix, where it is converted to glutamate by GLS; this is the rate-limiting step of glutaminolysis. Glutamine-derived glutamate is catalyzed into α-KG by GLUD1, GOT2, and GPT2 to release ammonia, aspartate, and alanine, respectively. Glutamine-derived glutamate in the mitochondria is also transported to the cytosol by SLC25A18/SLC25A22. GOT1, which is part of the malate-aspartate shuttle, contributes to the maintenance of redox homeostasis by converting OAA to aspartate, and GPT1 converts pyruvate to alanine. SLC7A11 transports cysteine to the cytosol in exchange for glutamate. Glutamine-derived glutamate and cysteine are ligated by GCLM/GCLC, which is in turn utilized by GSS to form GSH, which scavenges cellular ROS. Inhibitors of each step of glutamine metabolism are shown in white boxes. GLS, glutaminase; α-KG, α-ketoglutarate; GLUD1, glutamate dehydrogenase 1; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; GCLM, glutamate-cysteine ligase modifier subunit; GCLC, glutamate-cysteine ligase catalytic subunit; GSS, glutathione synthetase; GSH, reduced glutathione; ROS, reactive oxygen species; ASNS, asparagine synthetase; PSAT1, phosphoserine aminotransferase 1.
Targeting glutaminase and transaminase as a treatment for cancer
GLS, which is highly expressed in cancer cells and plays a role in cancer progression, has been investigated extensively as a druggable target41. Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl (BPTES), a potent orally available GLS1 inhibitor that spares GLS2, shows promising antitumor effects against human lymphoma B cells in vitro and in a xenograft mouse model42; it also suppresses the growth of platinum-resistant CRC and ovarian cancer cells, suggesting that combined treatments based on conventional drugs and glutamine-modulating compounds will yield clinically relevant results43,44. Recently, another selective inhibitor of GLS1, CB-839 (telaglenastat), showed no significant side effects in preclinical trials and is currently undergoing full clinical trials45. Previous studies showed that it did not significantly suppress the growth of KRAS-derived PDAC cells in vitro or in vivo because these cells mounted an adaptive metabolomic response, suggesting the importance of combined therapy for avoiding metabolic adaptation in response to GLS inhibition45. Thus, clinical trials are currently testing the following drugs in combination with CB-839: nivolumab as a treatment for melanoma, renal cell carcinoma (RCC), and NSCLC (clinicaltrials.gov ID: NCT02771626); everolimus for RCC (clinicaltrials.gov ID: NCT03163667); palbociclib for KRAS-derived PDAC, NSCLC and CRC (clinicaltrials.gov ID: NCT03965845); and cabozantinib for advanced RCC (clinicaltrials.gov ID: NCT03428217)46. Furthermore, CB-839 increased the radiosensitivity of head and neck squamous carcinoma (HNSCC) and NSCLC cells both in vitro and in a xenograft mouse model by abolishing GSH synthesis47,48, making it useful for concurrent chemotherapy and radiotherapy in a clinical setting.
Previous studies have shown that targeting GLUD1 inhibits the proliferation and migration of cancer cells, suggesting that GLUD1 is a druggable target for cancer therapy23. Epigallocatechin gallate (EGCG), an inhibitor of GLUD1 and 2, suppresses the proliferation of neuroblastoma, glioma, and CRC cells49. Recently, the purpurin analog R162 (an inhibitor of GLUD1) also showed promising results with respect to attenuating the proliferation of breast, NSCLC, and glioma cells in vitro and in patient-derived xenograft mouse models23. In addition, cotreatment of docetaxel-resistant NSCLC with docetaxel plus R162 inhibited cancer cell growth and metastasis both in vitro and in xenograft mouse models, again suggesting that combination therapy with anticancer drugs plus a GLUD1 inhibitor is an effective cancer treatment23.
Role of glutamine metabolism in redox homeostasis
ROS levels are elevated persistently in proliferating cancer cells, and ROS damage DNA and cellular components; therefore, redox homeostasis plays a pivotal role in protecting cancer cells against them. Notably, GSH acts as a critical antioxidant that protects cancer cells from any form of programmed cell death (i.e., autophagy, apoptosis, necroptosis, and ferroptosis)50,51,52. Given that glutathione is a tripeptide composed of glutamate, glycine, and cysteine, glutamine-derived glutamate and cysteine need to be ligated by glutamate-cysteine ligase (GCL), which itself comprises two separately encoded proteins: a catalytic subunit (GCLC) and a modifier subunit (GCLM)53,54. Next, glutathione synthetase (GSS) adds glycine to the ligated glutamate-cysteine (Fig. 1)53,54. While glutamate and glycine are abundant in cells, cysteine is the least abundant amino acid; therefore, it must be transported into the cells by SLC7A11 (xCT) in exchange for glutamate, which implies that SLC7A11-mediated GSH biosynthesis largely relies on glutamine metabolism55. Intriguingly, GOT1 and malic enzyme 1 (ME1) are also associated with redox homeostasis; in PDAC, GOT1 and ME1 maintain the NADPH/NADP+ ratio via the malate-aspartate shuttle by maintaining GSH levels56. In addition, we previously showed that upon inhibition of glutamine, cancer cells reduce the amount of GSH by exporting oxidized glutathione (GSSG) out of the cell via GSSG transporters and multiple-drug resistance-associated proteins57 and by extracellular degradation of GSSG58. Given that glutamine metabolism increases the amount of GSH in cells by maintaining the NADPH/NADP+ ratio and by preventing export and extracellular degradation of GSSG, glutamine is the primary amino acid that controls cellular GSH homeostasis.
Targeting redox homeostasis for cancer treatment
Approximately one-third of glutamine taken up by human fibroblast cells is exchanged for cysteine by SLC7A1159,60. This suggests that SLC7A11 not only plays a critical role in protein and GSH synthesis through cysteine uptake but also dictates glutamine dependence61. Therefore, targeting SLC7A11 is a promising therapeutic option, and its efficacy can be increased by combining it with drugs that target glutaminolysis60. Indeed, sulfasalazine (which inhibits SLC7A11) effectively suppresses the proliferation of glutamine-depleted TNBC in vitro and in vivo60. Moreover, glutamine-dependent PDAC is sensitive to the SLC7A11 inhibitor erastin, which induces ferroptosis62,63. However, although erastin shows antitumor effects, it has not entered clinical trials because it is poorly soluble in water, and its metabolism in vivo is unpredictable; therefore, imidazole ketone erastin (IKE) and piperazine erastin were developed (which are more soluble in water), and both show strong antitumor effects against diffuse large B-cell lymphoma (DLBCL) and fibrosarcoma64,65. Intriguingly, and as mentioned above, PDAC cells are dependent on GOT1 and the malate-aspartate shuttle; GOT1 knockout combined with cysteine depletion by erastin or IKE showed potent antitumor effects against these cells by reducing GSH and increasing ferroptosis66. In addition, sorafenib, a kinase inhibitor approved for the treatment of RCC and HCC, inhibits SLC7A11 to suppress the growth of these tumors via the induction of ferroptosis67,68.
Targeting glutamine transporters as a treatment for cancer
Cancer cells require an abundant supply of glutamine from the extracellular milieu; therefore, upregulation of glutamine transporters SLC1A5, SLC38A1, SLC38A2, and SLC6A14 at the cell membrane is required (Fig. 1)69. Indeed, high expression of these transporters contributes to cancer cell growth and is a marker of clinically poor outcomes for patients with NSCLC, prostate cancer, breast cancer, and acute myeloid leukemia70,71,72,73. Thus, cancer treatment strategies have focused on pharmacological inhibition of these transporters.
SLC1A5
SLC1A5 (ASCT2) is an obligatory sodium-dependent transporter of neutral amino acids, which are exchanged for asparagine, threonine, or serine74. SLC1A5 has high affinity for glutamine, particularly in an acidic environment75, and is thus more effective at transporting glutamine into cancer cells that thrive in acidic environments76. Indeed, SLC1A5 is highly expressed in various solid cancers73,76,77, such as squamous lung cancer, in which it is responsible for approximately 50% of glutamine uptake73. The expression of SLC1A5 is regulated by various transcriptional regulators, including ATF4 and Myc78,79. In TNBC, high expression of ATF4 and Myc is associated with overexpression of SLC1A5 and indicates poor survival outcomes77. In addition, Myc-dependent expression of ATF4 in DLBCL cells, human colon adenocarcinoma cells, and mouse embryonic fibroblasts drives the expression of SLC1A5 during metabolic adaptation to stress conditions73,80,81. A recent study showed that HIF-2α-mediated overexpression of SLC1A5 variants in mitochondria plays an essential role in glutamine metabolism in pancreatic cancer cells by inducing chemotherapy resistance82.
Therefore, SLC1A5 is a promising druggable target83. Benzylserine and benzylcysteine were the first molecules found to inhibit SLC1A5 in breast and gastric cancer cells, but they are nonspecific84,85. L-γ-glutamyl-p-nitroanilide (GPNA) suppresses the growth of TNBC, different types of lung cancer, and neuroblastoma cells73,77,81. In addition, combined treatment with GPNA and a monoclonal antibody (cetuximab) targeting EGFR effectively suppressed the growth of gastric cancer cells in vitro and in vivo86. However, amino acid analogs are unsuitable for clinical use due to their low affinity, lack of specificity, and toxicity87. V-9302 (2-amino-4-bis (aryloxy benzyl) aminobutanoic acid) was originally discovered as an SLC1A5 inhibitor; it showed a 100-fold increase in potency over GPNA and attenuated the growth of cancer cells, including HCC, CRC, lung cancer, and breast cancer cells83. Recent studies have shown that synthetic monoclonal antibodies specific for SLC1A5 (i.e., KM4008, KM4012, and KM4018) are an effective therapeutic option for suppressing glutamine-dependent growth of CRC cells, but their efficacy in other cancer cells remains unclear88. Despite the significance of SLC1A5 in some cancer cells, there are few specific and effective SLC1A5-inhibiting drugs89.
SLC38A1 and SLC38A2
SLC38A1 (SNAT1) and SLC38A2 (SNAT2) are sodium-dependent neutral amino acid transporters that drive glutamine influx into cells90. SLC38A1 is overexpressed in melanoma, breast, gastric, osteosarcoma, and endometrial cancer cells, showing a close association with proliferation and migration91,92,93. SLC38A2 is highly expressed in prostate cancer, HCC, and TNBC cells, thereby contributing to tumorigenesis94,95. Interestingly, silencing of SLC1A5 does not suppress the proliferation of epithelial cervical cancer and osteosarcoma cells; rather, it induces an amino acid starvation response by upregulating the expression of SLC38A1, suggesting that SLC38A1 is a major importer of glutamine into these cells90. In addition, amino acid starvation upregulates SLC38A2 via activation of GCN2 and ATF4, which help to maintain the intracellular glutamine pool36,90. Therefore, strategies designed to target glutamine metabolism should consider the combined blockade of these transporters.
N-methyl-aminoisobutyric acid (MeAIB) has been investigated in studies attempting to elucidate the function of SLC38A1 and/or SLC38A2 in various cells; indeed, MeAIB exerts an antitumor effect against various cancer cells90,96. Recently, it was proposed that the aforementioned drug V-9302 targets SLC38A2 and SLC7A5 rather than SLC1A587. A previous study supported this, showing that treatment of SLC1A5-knockdown HNSCC cell lines with V-9302 led to marked inhibition of glutamine metabolism, thereby suppressing growth and proliferation both in vitro and in vivo97. Therefore, combination therapy with V-9302 and SLC1A5-specific inhibitors may be a promising therapeutic option for some cancers97.
SLC6A14
SLC6A14, also known as amino acid transporter B0,+ (ATB0,+), maintains a unidirectional influx of glutamine, coupled with 2 Na+ and 1 Cl-, along a transmembrane gradient98. Given the functional role of SLC6A14 in extending the range of amino acid uptake (including glutamine and leucine, both of which are activators of mTORC1), as well as providing substrates for SLC1A5 and SLC7A5, the molecule has attracted much attention99,100. Indeed, SLC6A14 is overexpressed in colon, cervical, ER-positive breast, and pancreatic cancer cells and is associated with their proliferation101,102,103,104. High expression of SLC6A14 in PDAC and CRC cells is closely associated with metastasis and a poor outcome101,105,106. Mechanistically, SLC6A14 expression is regulated by the Wnt signaling pathway, and genetic or pharmacological inhibition of the transporter and its downstream effectors suppresses the growth of CRC cells both in vitro and in vivo102.
Given that tryptophan is a substrate for SLC6A14, the inhibitor α-methyltryptophan (α-MT) suppresses the growth of SLC6A14-positive breast cancer, PDAC, and CRC cells but not SLC6A14-negative cells107,108,109. Combined treatment of pancreatic cancer cells with gemcitabine and α-MT significantly inhibited proliferation and migration110. Although the role of SLC6A14 in cancer cells is becoming clearer, few compounds targeting SLC6A14 have been developed; thus, an effective drug targeting this transporter needs to be developed.
Mechanisms that induce resistance to glutamine-targeting therapies
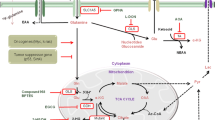
Although targeting glutamine metabolism is a promising therapeutic approach, few drugs have been developed. Tumor metabolism is affected by a multitude of microenvironmental factors, including nutrient availability. There are several mechanisms by which cancer cells escape the effects of inhibitors of glutamine metabolism; these include increased metabolic flexibility, uptake of extracellular amino acids via compensatory transporters and macropinocytosis, and expression of nutrient stress-response proteins (Fig. 2).

a Glutamine starvation induces metabolic flexibility, in which the influx of glucose-derived pyruvate via MPC and fatty acid-derived acyl-CoA via CPT1 into the mitochondria drives TCA cycle activity. b Under conditions of glutamine deprivation, the tumor suppressor protein p53 induces the expression of the SLC1A3 and SLC7A2 transporters. Aspartate uptake through SLC1A3 transporters increases the amount of malate, which is a TCA cycle intermediate, leading to an increase in oxidative phosphorylation and glutamine synthesis. Aspartate is used for nucleotide synthesis. Arginine uptake through SLC7A3 transporters restores mTORC1 expression, which is suppressed by glutamine depletion. The high level of intracellular asparagine increases the expression of GLUL proteins, thereby increasing glutamine and protein synthesis. c Under conditions of nutrient stress, macropinocytosis internalizes extracellular macromolecules to supply amino acids. Membrane ruffling aids in the uptake of extracellular macromolecules, such as serum albumin, via the formation of macropinosomes. After fusion between macropinosomes and lysosomes, albumin is degraded to supply amino acids to the cytosol and the mitochondrial TCA cycle. d Glutamine deprivation increases the expression of p53 and its target genes (Sestrin2, Gadd45a, and Cdkn1) and increases the phosphorylation of C/EBPβ and its target gene (Sestrin2), all of which maintain energy and redox balance and increase cancer cell survival. MPC, mitochondrial pyruvate carrier; CPT1, carnitine palmitoyltransferase I; TCA, tricarboxylic acid cycle; Asp, aspartate; Arg, arginine; Asn, asparagine; Gln, glutamine; ROS, reactive oxygen species; GLUL, glutamate-ammonia ligase; C/EBPβ, CCAAT/enhancer binding protein β.
Metabolic flexibility
Although glutamine is the primary carbon source for the TCA cycle in some cancer cells, replenishment of TCA cycle intermediates using alternative anaplerotic substrates reduces bioenergetic stress, thereby enabling resistance to inhibition of glutamine metabolism. There are two main anaplerotic flux pathways that feed the citric acid cycle: glutamine flux via glutaminase and glucose flux via pyruvate carboxylase110. Upon interruption of glutamine metabolism, glutamine-addicted tumor cells employ compensatory anaplerotic mechanisms via pyruvate carboxylase, which generates the oxaloacetate required to maintain TCA cycle flux; thus, the levels of pyruvate carboxylase can greatly affect the sensitivity of tumor cells to inhibition of glutamine metabolism111. In addition, deletion of GLS1 genes from Myc-driven liver tumors upregulates several metabolic compensatory pathways, including glycolysis and aminotransferases111. Thus, combined inhibition of glycolytic genes encoding hexokinase II or aminotransferases increases the efficacy of the GLS1 inhibitor CB-839111. In contrast, CB-839 showed no antitumor activity in PDAC mouse models due to the use of alternative metabolic pathways (e.g., fatty acid and lipid metabolism) by these cancer cells45. Integrated metabolomics and proteomics platforms revealed a marked increase in fatty acid oxidation-related metabolites, as well as proteome changes, in PDAC treated with GLS1 inhibitors, suggesting that treatments should target multiple metabolic pathways to overcome metabolic plasticity112,113.
Extracellular amino acid uptake via compensatory transporters
Amino acids, including aspartate, arginine, and asparagine, are associated with resistance to glutamine depletion. The tumor suppressor protein p53 increases the expression of the aspartate/glutamate transporter SLC1A3 and the arginine transporter SLC7A3 upon glutamine depletion, leading to resistance to treatments that deplete extracellular glutamine114,115. Increases in the levels of intracellular aspartate via SLC1A3 contribute to nucleotide synthesis and maintain the electron transport chain and TCA cycle114. Although the uptake of arginine by SLC7A3 transporters does not maintain TCA cycle flux under conditions of glutamine depletion, arginine activates mTORC1 and contributes to metabolic adaptation and tumor growth115. Uptake of extracellular asparagine prevents the death of glioblastoma cells in response to glutamine depletion by blocking the apoptotic function of a glutamine-induced endoplasmic reticulum stress marker protein, ATF4, and by increasing glutamate-ammonia ligase (GLUL)-mediated glutamine and protein synthesis116,117. Thus, blocking amino acid transporters or depleting amino acids such as L-asparaginase may be effective therapeutic strategies to overcome resistance to glutamine withdrawal.
Macropinocytosis
Macropinocytosis, a nutrient-scavenging pathway, is a compensatory route that supplies amino acids to nutrient-starved cancer cells harboring oncogenic mutations in KRAS or PTEN118,119,120. Experiments using isotope-labeled extracellular proteins show that when supplied with extracellular serum albumin, Ras-transformed cells, which rely on glutamine metabolism to support growth, utilize macropinocytosis to maintain proliferation under glutamine-limiting conditions120. More recent studies have shown that regional depletion of glutamine from PDAC tumors stimulates macropinocytosis by activating EGFR/PAK signaling and supplying glutamine via degradation of extracellular proteins in lysosomes121. Macropinocytosis also facilitates the survival of hypoxic HCC cells. Thus, HCC cells can internalize extracellular proteins by increasing the expression of a membrane ruffling protein called EH domain-containing protein 2, leading to resistance to glutamine deprivation under hypoxic conditions122. Although targeting macropinocytosis could be a key strategy for overcoming resistance to glutamine uptake blockade, further studies are necessary to examine whether macropinocytosis can overcome tumor cell resistance to glutamine antimetabolites or GLS inhibitors that target enzymes involved directly in glutamine metabolism.
Nutrient stress-response proteins
Limiting glutamine utilization regulates nutrient stress-response proteins and transcription factors. Upon glutamine deprivation, lung cancer cells increase the phosphorylated CCAAT/enhancer binding protein β (p-C/EBPβ)-dependent metabolic protein called Sestrin2 to maintain ATP levels and prevent excessive production of ROS through differential regulation of mTORC1 and mTORC2123. Sestrin2-mediated suppression of mTORC1 and mTORC2 activation reprograms lipid metabolism to limit ATP and NADPH consumption, thereby enabling cancer cells to survive under glutamine-depleted conditions. Other studies have shown that ROS production in response to glutamine deprivation increases the expression of p53-dependent genes (Gadd45a, Cdkn1, and Sestrin2) via B55α or IKKβ124,125. Upregulation of Gadd45a and Cdkn1 induces cell cycle arrest in response to glutamine deprivation, which alleviates oxidative stress and reduces energy consumption126,127. Cotargeting proteins involved in glutamine metabolism and the stress response under conditions of glutamine depletion would therefore be a promising therapeutic strategy for overcoming adaptive/resistance mechanisms in cancer cells.
Effects of targeting glutamine metabolism in the TME
The TME is a complex milieu that surrounds tumor cells, often providing immunosuppressive cover that facilitates immune invasion. Specifically, competition for nutrients or cell-intrinsic programming between cancer cells and immune cells induces nutrient deficiency and metabolic reprogramming of immune cells, leading to modulation of antitumor immunity128,129. Given that activation and differentiation of immune cells are coupled to metabolic reprogramming, regulating the metabolic activity of immune cells should be considered in the development of potential strategies that target glutamine metabolism130,131.
Glutamine metabolism in immune cells
Accumulating evidence shows that glutamine is an immunomodulatory nutrient in immune cells. Naïve T cells are metabolically quiescent, undergoing basal levels of glycolysis and glutaminolysis sufficient to maintain minimal biosynthesis; however, T-cell receptor (TCR)-stimulated activation increases the expression of the Myc transcription factor, glutamine transporters (SLC38A1, SLC38A2), and glutaminolysis-related enzymes (GLS, GLUD1, GOT, GPT) to meet bioenergetic and biosynthetic requirements, resulting in T-cell proliferation130,131,132,133. TCR-induced activation of mTORC1 and metabolic signaling pathways requires SLC1A5-dependent uptake of glutamine; indeed, an SLC1A5-deficient mouse model shows decreased induction of T helper 1 (Th1) and Th17 cells but not Th2, regulatory T (Treg), or CD8(+) T cells, leading to a decrease in proinflammatory T-cell responses134. Recent studies have shown that glutamine-derived α-KG regulates the differentiation of CD4(+) T cells into Th1-type effector T cells or Treg cells via DNA methylation and lipid homeostasis135,136. Mechanistically, α-KG decreases Treg differentiation by inhibiting FOXP3 and upregulating inflammatory cytokines such as IFN-γ, Tbet, and Rorc, suggesting that Th1-type effector T cells are more dependent on glutaminolysis than Treg cells136. Moreover, effector T cells are capable of adapting their metabolism in response to nutrient limitation. Activated T cells rely on glutamine-dependent OXPHOS to maintain energetic homeostasis under energy-related stress (e.g., low glucose levels)137.
The antitumor functions of natural killer (NK) cells are upregulated by the expression of c-Myc proteins, which are required for IL-2/IL-12-induced NK cell metabolism and function138. Although amino acids are essential for the function of NK cells, their main role in NK cells is the maintenance of signaling (e.g., via c-Myc or mTOR)139. Unlike other lymphocyte subsets, glutaminolysis and the TCA cycle do not sustain OXPHOS in activated NK cells. Glutamine withdrawal, but not inhibition of glutaminolysis, results in loss of c-Myc protein, reduced cell growth, and impaired NK cell responses138. Consistent with this, receptor-simulated production of IFN-γ by NK cells is not impaired under glutamine-limited conditions140.
In macrophages, glutamine metabolism is a critical metabolic pathway for differentiation. Macrophages undergo metabolic switching during differentiation into inflammatory (M1) or anti-inflammatory (M2) phenotypes. Tumor-associated macrophages (TAMs) can exhibit either an antitumor M1-like phenotype or a protumor M2-like phenotype. Glutamine starvation inhibits M2 polarization but not M1 polarization by suppressing UDP-GlcNAc biosynthesis and N-glycosylation of M2-related proteins such as Relmα, CD206, and CD301141. Consistent with this, glutaminolysis-derived α-KG promotes M2 activation by increasing fatty acid oxidation and Jmjd3-dependent epigenetic reprogramming of M2-related genes142. In contrast to the inhibition of glutaminolysis, pharmacological or genetic targeting of GLUL in macrophages reprograms M2-polarized macrophages to an M1-polarized phenotype143. Mechanistically, macrophage-specific inhibition of GLUL leads to accumulation of succinate and HIF-1α via glutamine-dependent γ-aminobutyric acid (GABA) shunting (thereby inhibiting vessel sprouting and metastasis) and via stimulation of T effector cells; however, IL-10-induced expression of GLUL promotes vessel sprouting, immunosuppression, and metastasis143.
Given the importance of glutamine metabolism to immune cells, including activated lymphocytes, it is crucial to determine whether blockade of glutamine metabolism in tumor cells hampers anticancer immune responses; the answer may be key to the success of therapeutic strategies targeting glutamine metabolism.
Glutamine blockade in the TME
The metabolism of cancer cells and immune cells in the TME is regulated by cell-intrinsic programs through mTORC1 signaling128. PET tracers showed that cancer cells rely heavily on glutamine uptake via mTORC1 signaling, while myeloid cells in the TME are more dependent on glucose, as are T cells and cancer cells (but to a lesser extent)128. Although administration of V-9302 to decrease glutamine availability increases glucose uptake by cancer cells and immune cells in allograft models, the growth of tumors harboring tumor-infiltrating Tregs, CD8(+) T cells, and NK cells is suppressed128. Given that cancer cells are much more dependent on glutamine than immune cells and that V-9302 does not impair CD8(+) T-cell viability and activation83, pharmacological inhibitors of SLC1A5 might have high therapeutic potential.
Accumulating evidence shows that inhibitors of glutamine metabolism, such as V-9302, JHU-083, and CB-839, elicit stronger antitumor effects when used in combination with immune checkpoint inhibitors144,145,146 (Fig. 3). In a previous study, we showed that V-9302 induces the expression of PD-L1 by tumor cells and augments immune evasion in synergistic murine models58. Mechanistically, glutamine limitation decreases GSH levels and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) glutathionylation, resulting in reduced SERCA activity58. Upregulation of cytosolic Ca2+ activates calcium/calmodulin-dependent protein kinase II (CAMKII), leading to aberrant NF-κB signaling and downstream expression of PD-L158. Therefore, agents that target glutamine utilization may, when used in combination with an anti-PD-L1 antibody, boost antitumor immunity58. Similar results were reported for several tumors147,148,149,150. Glutamine starvation increases the expression of PD-L1 in RCC and bladder cancer cells via the EGFR/ERK/c-Jun signaling pathway147,148. Furthermore, bladder tumors in mice supplemented with glutamine showed lower PD-L1 levels than control tumors148. In natural killer T-cell lymphoma (NKTCL), blocking SLC1A1-mediated glutamine addiction in tumor cells induced PD-L1 expression and inhibited CD8(+) T-cell activity149. As a therapeutic option, combined treatment with asparaginase and an anti-PD-1 antibody could be useful because glutamine-addicted cells are sensitive to asparaginase149.

a Glutamine deprivation and transporter inhibition decrease glutamine metabolism, thereby boosting EGFR/ERK/c-Jun signaling and calcium/NF-kB signaling, leading to upregulation of PD-L1. PD-L1 suppresses antitumor immune responses by blocking T-cell activation in the tumor microenvironment. b Treatment with glutamine analogs, including DON and JHU-083, decreases glucose and glutamine metabolism, leading to inhibition of tumor growth via a decrease in hypoxia, acidosis, and nutrient depletion in the tumor microenvironment. Furthermore, DON decreases the recruitment of MDSCs by suppressing the secretion of CSF3 by tumor cells and blocking the production of the immunosuppressive metabolite kynurenine; this inhibits the synthesis of the hyaluronan-rich ECM, resulting in the activation and infiltration of T cells. PD-L1, programmed death-ligand 1; CSF3, colony stimulating factor 3; MDSC, myeloid-derived suppressor cell; ECM, extracellular matrix.
In contrast to PD-L1-mediated T-cell dysfunction induced by glutamine limitation, DON and its prodrug JHU-083 skew CD8(+) T cells toward a highly activated, persistent, and proliferative phenotype, thereby facilitating immunogenic cancer cell death58,149,150 (Fig. 3). DON treatment activates AMPK in cancer cells by increasing the AMP/ATP ratio and decreasing the expression of c-Myc proteins, thereby inhibiting glycolytic metabolism and suppressing cancer cell growth in vitro145. However, CD8(+) T cells in vitro overcome metabolic stress through glucose-dependent anaplerosis and acetate catabolism. Another study showed that in JHU-083-treated cancer cell allograft models, an increase in nutrient levels and oxygen and a decrease in the acidity of the TME resulted in T-cell-mediated tumor suppression145, whereas another study demonstrated the effects of JHU-083 on myeloid-derived suppressor cells (MDSCs) and TAMs150. JHU-083 markedly suppresses the recruitment of MDSCs by decreasing tumor-derived CSF3 levels via degradation of C/EBPβ, which in turn increases the numbers of proinflammatory TAMs and enhances antigen presentation to CD8(+) T cells, resulting in stronger T-cell responses150. In addition, JHU-083 reduces the expression of IDO (an enzyme that mediates tryptophan metabolism) by inhibiting the phosphorylation of STAT1 and STAT3 in tumor cells, MDSCs, and TAMs, thereby decreasing the kynurenine/tryptophan ratio and enhancing the functions of antitumor T cells150. Treatment of PDAC with DON decreases uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) levels, which affects synthesis of the hyaluronan-rich extracellular matrix (ECM); this suggests that glutamine inhibitors such as DON could deplete the ECM and allow infiltration of tumors by CD8(+) T cells151.
Two compounds (V-9302 and JHU-083) that target SLC1A5 and glutamine metabolism in allograft models elicited different T cell-mediated immune responses; however, because these compounds do not reduce the activation or viability of CD8(+) T cells83,145, combined immunotherapy significantly improves their antitumor effects.
Conclusion
Glutamine metabolism plays a central role in regulating uncontrolled tumor growth by modulating bioenergetic and redox homeostasis and by serving as a precursor for the synthesis of biomass. Although targeting glutamine metabolism is a promising strategy for cancer therapy, there are many hurdles to be overcome before we develop a clinically effective drug. Metabolic flexibility or adaptation by cancer cells, as well as reduced antitumor immunity, may be unwanted consequences of inhibiting glutamine metabolism. A comprehensive understanding of the TME is of the utmost importance because it provides valuable insights into pathways that could be targeted by novel metabolic therapies for advanced or drug-resistant cancers.
References
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012).
Stine, Z. E., Schug, Z. T., Salvino, J. M. & Dang, C. V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 21, 141–162 (2022).
Wang, Z. et al. Targeting glutaminolysis: new perspectives to understand cancer development and novel strategies for potential target therapies. Front. Oncol. 10, 589508 (2020).
Cruzat, V., Macedo Rogero, M., Noel Keane, K., Curi, R. & Newsholme, P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients 10, 1564 (2018).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016).
Matés, J. M., Di Paola, F. J., Campos-Sandoval, J. A., Mazurek, S. & Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 98, 34–43 (2020).
Ma, G. et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun. Signal. 20, 114 (2022).
Choi, Y. K. & Park, K. G. Targeting glutamine metabolism for cancer treatment. Biomol. Ther. (Seoul) 26, 19–28 (2018).
Grinde, M. T. et al. Glutamine to proline conversion is associated with response to glutaminase inhibition in breast cancer. Breast Cancer Res. 21, 61 (2019).
Xiang, L. et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 10, 40 (2019).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Csibi, A. et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 24, 2274–2280 (2014).
Zhang, C. et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 5, e10727 (2016).
Saha, S. K. et al. Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J. Clin. Med. 8, 355 (2019).
Dias, M. M. et al. GLS2 is protumorigenic in breast cancers. Oncogene 39, 690–702 (2020).
Xiang, L. et al. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim. Biophys. Acta—Mol. Cell Res. 1833, 2996–3005 (2013).
López de la Oliva, A. R. et al. Nuclear translocation of glutaminase GLS2 in human cancer cells associates with proliferation arrest and differentiation. Sci. Rep. 10, 2259 (2020).
Cardaci, S. & Ciriolo, M. R. TCA cycle defects and cancer: when metabolism tunes redox state. Int. J. Cell Biol. 2012, 161837 (2012).
Jin, L. et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27, 257–270 (2015).
Yelamanchi, S. D. et al. A pathway map of glutamate metabolism. J. Cell Commun. Signal 10, 69–75 (2016).
Vanhove, K. et al. Glutamine addiction and therapeutic strategies in lung cancer. Int. J. Mol. Sci. 20, 252 (2019).
Yang, M., Soga, T. & Pollard, P. J. Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest. 123, 3652–3658 (2013).
Wang, Q. et al. Therapeutic targeting of glutamate dehydrogenase 1 that links metabolic reprogramming and Snail-mediated epithelial–mesenchymal transition in drug-resistant lung cancer. Pharmacol. Res. 185, 106490 (2022).
Tambay, V., Raymond, V. A. & Bilodeau, M. MYC rules: leading glutamine metabolism toward a distinct cancer cell phenotype. Cancers (Basel) 13, 4484 (2021).
Wei, Z., Liu, X., Cheng, C., Yu, W. & Yi, P. Metabolism of amino acids in cancer. Front. Cell Dev. Biol. 8, 603837 (2021).
Yuan, H. X., Xiong, Y. & Guan, K. L. Nutrient sensing, metabolism, and cell growth control. Mol. Cell 49, 379–387 (2013).
Ahn, C. S. & Metallo, C. M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 3, 1 (2015).
Korangath, P. et al. Targeting glutamine metabolism in breast cancer with aminooxyacetatetargeting glutamine metabolism in breast cancer. Clin. Cancer Res. 21, 3263–3273 (2015).
Zhang, B., Chen, Y., Bao, L. & Luo, W. GPT2 is induced by hypoxia-inducible factor (HIF)-2 and promotes glioblastoma growth. Cells 11, 2597 (2022).
Kim, M., Gwak, J., Hwang, S., Yang, S. & Jeong, S. M. Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 38, 4729–4738 (2019).
Shen, Y., Huang, Q., Zhang, Y., Hsueh, C.-Y. & Zhou, L. A novel signature derived from metabolism-related genes GPT and SMS to predict prognosis of laryngeal squamous cell carcinoma. Cancer Cell Int. 22, 226 (2022).
Gorgoglione, R. et al. Glutamine-derived aspartate biosynthesis in cancer cells: role of mitochondrial transporters and new therapeutic perspectives. Cancers (Basel) 14, 245 (2022).
Yang, H. et al. SIRT3-dependent GOT2 acetylation status affects the malate–aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 34, 1110–1125 (2015).
Sun, W. et al. Aspulvinone O, a natural inhibitor of GOT1 suppresses pancreatic ductal adenocarcinoma cells growth by interfering glutamine metabolism. Cell Commun. Signal 17, 111 (2019).
Kerk, S. A. et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. eLife 11, e73245 (2022).
Kilberg, M. S., Shan, J. & Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 20, 436–443 (2009).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 (2016).
Knott, S. R. V. et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381 (2018).
Cai, D.-J. et al. Asparagine synthetase regulates lung-cancer metastasis by stabilizing the β-catenin complex and modulating mitochondrial response. Cell Death Dis. 13, 566 (2022).
Hettmer, S. et al. Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. Elife 4, e09436 (2015).
Yang, W. H., Qiu, Y., Stamatatos, O., Janowitz, T. & Lukey, M. J. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer 7, 790–804 (2021).
Shukla, K. et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem. 55, 10551–10563 (2012).
Hudson, C. D. et al. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 7, 41637–41649 (2016).
Chen, P. et al. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol. Ther. Oncolytics 20, 228–239 (2021).
Biancur, D. E. et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965 (2017).
Varghese, S. et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell–mediated immunotherapies. Mol. Cancer Ther. 20, 500–511 (2021).
Wicker, C. A. et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 502, 180–188 (2021).
Boysen, G. et al. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Biol. 95, 436–442 (2019).
Peeters, T. H. et al. Isocitrate dehydrogenase 1-mutated cancers are sensitive to the green tea polyphenol epigallocatechin-3-gallate. Cancer Metab. 7, 4 (2019).
Bansal, A. & Simon, M. C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 217, 2291–2298 (2018).
Huang, R. et al. Dual role of reactive oxygen species and their application in cancer therapy. J. Cancer 12, 5543–5561 (2021).
Lv, H. et al. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell Longev. 2019, 3150145 (2019).
Anderson, M. E. Glutathione: an overview of biosynthesis and modulation. Chem. Biol. Interact. 111-112, 1–14 (1998).
Kang, Y. P. et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 33, 174–189.e7 (2021).
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620 (2021).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
Keppler, D. Export pumps for glutathione S-conjugates. Free Radic. Biol. Med. 27, 985–991 (1999).
Byun, J. K. et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol. Cell 80, 592–606.e8 (2020).
Bannai, S. & Ishii, T. A novel function of glutamine in cell culture: Utilization of glutamine for the uptake of cystine in human fibroblasts. J. Cell. Physiol. 137, 360–366 (1988).
Timmerman, L. A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013).
Muir, A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713 (2017).
Jyotsana, N., Ta, K. T. & DelGiorno, K. E. The role of cystine/glutamate antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 12, 858462 (2022).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Yan, R. et al. The structure of erastin-bound xCT–4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 32, 687–690 (2022).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e9 (2019).
Kremer, D. M. et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat. Commun. 12, 4860 (2021).
Zhang, L. et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol. Sin. 44, 622–634 (2023).
Zhang, Y. et al. MITD1 deficiency suppresses clear cell renal cell carcinoma growth and migration by inducing ferroptosis through the TAZ/SLC7A11 pathway. Oxid. Med. Cell. Longev. 2022, 7560569 (2022).
Bhutia, Y. D. & Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 1863, 2531–2539 (2016).
Wang, Q. et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 236, 278–289 (2015).
Willems, L. et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 122, 3521–3532 (2013).
Cha, Y. J., Kim, E.-S. & Koo, J. S. Amino acid transporters and glutamine metabolism in breast cancer. Int. J. Mol. Sci. 19, 907 (2018).
Hassanein, M. et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 19, 560–570 (2013).
Scalise, M. et al. Cysteine 467 of the ASCT2 amino acid transporter is a molecular determinant of the antiport mechanism. Int. J. Mol. Sci. 23, 1127 (2022).
Sastrasinh, M. & Sastrasinh, S. Effect of acute pH change on mitochondrial glutamine transport. Am. J. Physiol. 259, F863–F866 (1990).
Li, X. et al. Role of glutamine and its metabolite ammonia in crosstalk of cancer-associated fibroblasts and cancer cells. Cancer Cell Int. 21, 479 (2021).
van Geldermalsen, M. et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 35, 3201–3208 (2016).
Kahya, U., Köseer, A. S. & Dubrovska, A. Amino Acid Transporters on the Guard of Cell Genome and Epigenome. Cancers (Basel) 13, 125 (2021).
Zhou, X. et al. Activated amino acid response pathway generates apatinib resistance by reprograming glutamine metabolism in non-small-cell lung cancer. Cell Death Dis. 13, 636 (2022).
Tameire, F. et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 21, 889–899 (2019).
Ren, P. et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 235, 90–100 (2015).
Yoo, H. C. et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab 31, 267–283.e12 (2020).
Schulte, M. L. et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 24, 194–202 (2018).
Sun, H. J., Meng, L. Y., Shen, Y., Zhu, Y. Z. & Liu, H. R. S-benzyl-cysteine-mediated cell cycle arrest and apoptosis involving activation of mitochondrial-dependent caspase cascade through the p53 pathway in human gastric cancer SGC-7901 cells. Asian Pac. J. Cancer Prev. 14, 6379–6384 (2013).
van Geldermalsen, M. et al. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 18, 689 (2018).
Ma, H. et al. Inhibition of glutamine uptake improves the efficacy of cetuximab on gastric cancer. Integr. Cancer Ther. 20, 15347354211045349 (2021).
Bröer, A., Fairweather, S. & Bröer, S. Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Front. Pharmacol. 9, 785 (2018).
Suzuki, M., Toki, H., Furuya, A. & Ando, H. Establishment of monoclonal antibodies against cell surface domains of ASCT2/SLC1A5 and their inhibition of glutamine-dependent tumor cell growth. Biochem. Biophys. Res. Commun. 482, 651–657 (2017).
Nachef, M., Ali, A. K., Almutairi, S. M. & Lee, S.-H. Targeting SLC1A5 and SLC3A2/SLC7A5 as a potential strategy to strengthen anti-tumor immunity in the tumor microenvironment. Front. Immunol. 12, 624324 (2021).
Bröer, A., Rahimi, F. & Bröer, S. Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J. Biol. Chem. 291, 13194–13205 (2016).
Böhme-Schäfer, I., Lörentz, S. & Bosserhoff, A. K. Role of amino acid transporter SNAT1/SLC38A1 in human melanoma. Cancers (Basel) 14, 2151 (2022).
Wang, K. et al. Activation of SNAT1/SLC38A1 in human breast cancer: correlation with p-Akt overexpression. BMC Cancer 13, 343 (2013).
Zhou, F.-f et al. SLC38A1 promotes proliferation and migration of human colorectal cancer cells. J. Huazhong Univ. Sci. Technolog. Med. Sci. 37, 30–36 (2017).
Okudaira, H. et al. Putative transport mechanism and intracellular fate of trans-1-amino-3-18F-fluorocyclobutanecarboxylic acid in human prostate cancer. J. Nucl. Med. 52, 822–829 (2011).
Morotti, M. et al. Increased expression of glutamine transporter SNAT2/SLC38A2 promotes glutamine dependence and oxidative stress resistance, and is associated with worse prognosis in triple-negative breast cancer. Br. J. Cancer 124, 494–505 (2021).
Tang, Y. et al. The regulatory role of meaib in protein metabolism and the mTOR signaling pathway in porcine enterocytes. Int. J. Mol. Sci. 19, 714 (2018).
Zhang, Z. et al. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 122, 82–93 (2020).
Sloan, J. L. & Mager, S. Cloning and functional expression of a human Na(+) and Cl(-)-dependent neutral and cationic amino acid transporter B(0+). J. Biol. Chem. 274, 23740–23745 (1999).
Dejure, F. R., Butzer, J., Lindemann, R. K. & Mardin, B. R. Exploiting the metabolic dependencies of the broad amino acid transporter SLC6A14. Oncotarget 11, 4490–4503 (2020).
Sniegowski, T., Korac, K., Bhutia, Y. D. & Ganapathy, V. SLC6A14 and SLC38A5 drive the glutaminolysis and serine-glycine-one-carbon pathways in cancer. Pharmaceuticals (Basel) 14, 216 (2021).
Gupta, N. et al. Upregulation of the amino acid transporter ATB0,+ (SLC6A14) in colorectal cancer and metastasis in humans. Biochim. Biophys. Acta—Mol. Basis Dis. 1741, 215–223 (2005).
Sikder, M. O. F. et al. SLC6A14, a Na+/Cl−-coupled amino acid transporter, functions as a tumor promoter in colon and is a target for Wnt signaling. Biochem. J 477, 1409–1425 (2020).
Babu, E. et al. Deletion of the amino acid transporter Slc6a14 suppresses tumour growth in spontaneous mouse models of breast cancer. Biochem. J 469, 17–23 (2015).
Kou, L. et al. Endocytosis of ATB0,+(SLC6A14)-targeted liposomes for drug delivery and its therapeutic application for pancreatic cancer. Expert Opin. Drug Deliv. 17, 395–405 (2020).
Nałęcz, K. A. Amino acid transporter SLC6A14 (ATB0,+)—a target in combined anti-cancer therapy. Front. Cell Dev. Biol. 8, 594464 (2020).
Penheiter, A. R. et al. Transcriptomic and immunohistochemical profiling of SLC6A14 in pancreatic ductal adenocarcinoma. Biomed. Res. Int. 2015, 593572 (2015).
Lu, Y. et al. Blockade of the amino acid transporter SLC6A14 suppresses tumor growth in colorectal Cancer. BMC Cancer 22, 833 (2022).
Coothankandaswamy, V. et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 173, 3292–3306 (2016).
Karunakaran, S. et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 286, 31830–31838 (2011).
Cai, A. et al. Synergism between SLC6A14 blockade and gemcitabine in pancreactic cancer: a 1H-NMR-based metabolomic study in pancreatic cancer cells. Biochem. J. 477, 1923–1937 (2020).
Cheng, T. et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl Acad. Sci. USA 108, 8674–8679 (2011).
Mendez-Lucas, A. et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2, 335–350 (2020).
Zhang, G. et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 19, 4983–4993 (2013).
Tajan, M. et al. A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 28, 721–736.e6 (2018).
Lowman, X. H. et al. p53 promotes cancer cell adaptation to glutamine deprivation by upregulating Slc7a3 to increase arginine uptake. Cell Rep. 26, 3051–3060.e4 (2019).
Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 (2014).
Pavlova, N. N. et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428–438.e5 (2018).
Byun, J. K. et al. Macropinocytosis is an alternative pathway of cysteine acquisition and mitigates sorafenib-induced ferroptosis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 41, 98 (2022).
Jeong, S. et al. Transcription factor Eb is required for macropinocytosis-mediated growth recovery of nutrient-deprived kras-mutant cells. Nutrients 10, 1638 (2018).
Commisso, C. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013).
Lee, S. W. et al. EGFR-PAK signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392.e5 (2019).
Zhang, M. S. et al. Hypoxia-induced macropinocytosis represents a metabolic route for liver cancer. Nat. Commun. 13, 954 (2022).
Byun, J. K. et al. A positive feedback loop between Sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells. Cell Rep. 20, 586–599 (2017).
Reid, M. A. et al. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 50, 200–211 (2013).
Ishak Gabra, M. B. et al. IKKbeta activates p53 to promote cancer cell adaptation to glutamine deprivation. Oncogenesis 7, 93 (2018).
Abcouwer, S. F., Schwarz, C. & Meguid, R. A. Glutamine deprivation induces the expression of GADD45 and GADD153 primarily by mRNA stabilization. J. Biol. Chem. 274, 28645–28651 (1999).
Tran, T. Q. et al. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 36, 1991–2001 (2017).
Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
Zhang, L. & Romero, P. Metabolic control of CD8(+) T cell fate decisions and antitumor immunity. Trends Mol. Med. 24, 30–48 (2018).
Pearce, E. L., Poffenberger, M. C., Chang, C. H. & Jones, R. G. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).
Carr, E. L. et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol 185, 1037–1044 (2010).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Nakaya, M. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014).
Klysz, D. et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97 (2015).
Matias, M. I. et al. Regulatory T cell differentiation is controlled by alphaKG-induced alterations in mitochondrial metabolism and lipid homeostasis. Cell Rep. 37, 109911 (2021).
Blagih, J. et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 (2015).
Loftus, R. M. et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 9, 2341 (2018).
Terrén, I., Orrantia, A., Vitallé, J., Zenarruzabeitia, O. & Borrego, F. NK cell metabolism and tumor microenvironment. Front. Immunol. 10, 2278 (2019).
Keppel, M. P., Saucier, N., Mah, A. Y., Vogel, T. P. & Cooper, M. A. Activation-specific metabolic requirements for NK Cell IFN-γ production. J. Immunol. 194, 1954–1962 (2015).
Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015).
Liu, P. S. et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994 (2017).
Palmieri, E. M. et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 20, 1654–1666 (2017).
Tang, Y. et al. Simultaneous glutamine metabolism and PD-L1 inhibition to enhance suppression of triple-negative breast cancer. J. Nanobiotechnol. 20, 216 (2022).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019).
Varghese, S. et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell-mediated immunotherapies. Mol. Cancer Ther. 20, 500–511 (2021).
Ma, G. et al. Glutamine deprivation induces PD-L1 expression via activation of EGFR/ERK/c-Jun signaling in renal cancer. Mol. Cancer Res. 18, 324–339 (2020).
Wang, L. et al. Immunosuppression induced by glutamine deprivation occurs via activating PD-L1 transcription in bladder cancer. Front. Mol. Biosci. 8, 687305 (2021).
Xiong, J. et al. SLC1A1 mediated glutamine addiction and contributed to natural killer T-cell lymphoma progression with immunotherapeutic potential. EBioMedicine 72, 103614 (2021).
Oh, M. H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 130, 3865–3884 (2020).
Sharma, N. S. et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Invest. 130, 451–465 (2020).
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants NRF-2021R1A2C3005603, 2020R1A5A2017323, NRF-2022R1A2C1008591, and NRF-2021R1C1C2003405 funded by the Ministry of Science and ICT.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, J., Byun, JK., Choi, YK. et al. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med 55, 706–715 (2023). https://doi.org/10.1038/s12276-023-00971-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00971-9
- Springer Nature Limited
This article is cited by
-
IGF2BP3 promotes glutamine metabolism of endometriosis by interacting with UCA1 to enhances the mRNA stability of GLS1
Molecular Medicine (2024)
-
Glutamine-derived aspartate is required for eIF5A hypusination-mediated translation of HIF-1α to induce the polarization of tumor-associated macrophages
Experimental & Molecular Medicine (2024)
-
Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies
Signal Transduction and Targeted Therapy (2024)
-
Targeting the glutamine metabolism to suppress cell proliferation in mesenchymal docetaxel-resistant prostate cancer
Oncogene (2024)
-
Metabolic engineering for optimized CAR-T cell therapy
Nature Metabolism (2024)