
Abstract
Prostacyclin (PGI2) plays key roles in shaping the immune microenvironment and modulating vasodilation, whereas its contribution to endometriosis (EMs) remains largely unclear. Our study suggested that prostacyclin synthase (PTGIS)-dependent PGI2 signaling was significantly activated in EMs, which was involved in the hypoxic microenvironment of ectopic lesions and deficient methylation status of the PTGIS promoter. Notably, in vitro assays, hypoxia promoted PTGIS expression through DNA methyltransferase 1 (DNMT1)-mediated DNA methylation deficiency in endometrial stromal cells (ESCs); PTGIS overexpression enhanced the adhesive ability of ESCs and led to elevated PGI2 production, and PGI2 triggered CD16− (encoded by FCGR3, Fc fragment of IgG receptor IIIa) natural killer (NK)-cell differentiation through PGI2 receptor (IP, PTGIR) in an ESC/NK-cell coculture system. Our rodent model experiment suggested that treatment with the PGI2 analog iloprost and adoptive transfer of fcgr3 knockout (fcgr3−/−) NK cells aggravated EMs progression and that genetic ablation of ptgis (ptgis−/−) in ectopic lesions and treatment with the PTGIR antagonist RO1138452 partially rescued this outcome. Thus, our findings identified the contribution of PGI2 to EMs progression via enhancement of the adhesive ability of ESCs and inhibition of the activity of NK cells. We hypothesized that PGI2 is a target for EMs intervention and provide a rationale for studying pharmacological PTGIR inhibition and PTGIS genetic depletion therapies as therapeutic strategies for EMs.
Similar content being viewed by others


Introduction
Endometriosis (EMs) is an enigmatic benign estrogen-dependent gynecological disease characterized by viable endometrial glands and stroma survived outside the uterine cavity; EMs is predominantly, but not exclusively, present in the ovaries and affects approximately 10% of women of reproductive age worldwide1,2,3. Accumulating evidence suggests that EMs is a multigenic disorder4,5, and intrinsic disorder in endometrial stromal cells (ESCs) and dysfunctional immunosuppression serve as two crucial contributors to its prevalence6,7,8; however, the underlying pathogenesis remains to be elucidated. Prostaglandins are the metabolic products of arachidonic acid metabolism, namely, PGD2, PGE2 and PGF2α, and PGE2 has been shown to promote inflammatory cell infiltration, cellular phenotype modulation and immunity dysregulation in EMs patients9,10,11. In contrast, other analogs of PGE2 have rarely been investigated in EMs, such as prostacyclin (PGI2). PGI2 is predominantly expressed in the vascular endothelium, and its biosynthesis is catalyzed by the rate-limiting enzyme prostacyclin synthase (PTGIS, PGIS or CYP8A1), which is a member of family 8 (CYP8) in the cytochrome P450 superfamily. Based on previous publications, the biological function of PGI2 was quite different from that of PGE2, and PGI2 was proposed to be a vasodilator and has been applied to treat pulmonary arterial hypertension (PAH) in the clinic12,13,14,15,16. In addition, PGI2 profoundly modulates immune responses mediated by regulatory T cells, natural killer (NK) cells, immature dendritic cells, B cells and CD4+ T-cell subsets in diseases17,18,19,20,21,22,23,24,25, and the concentration of PGI2 was reported to be increased in the peritoneal fluid of EMs patients26, whereas the role of PGI2 in EMs remains unknown.
CD3−CD56+ NK cells play pivotal roles in innate immunity, and NK cells with a CD16+ (encoded by FCGR3, Fc fragment of IgG receptor IIIa) phenotype are naturally cytotoxic and mainly exert killing effects, while CD16− NK cells were efficient cytokine producers endowed with immunoregulatory properties that can become cytotoxic upon activation27. In general, CD16 expression is downregulated following NK-cell activation; CD16 is not only an activating receptor of NK cells but also matters in disease progression. Remarkably, CD16− NK-cell infiltration has been reported to be profoundly associated with papillary thyroid cancer progression28, with these cells demonstrating an impaired capability to kill non-small cell lung cancer cells27 and displaying an enhanced antioxidative capacity29. Additionally, CD56dimCD16− NK-cell subsets are increased in the peripheral blood of advanced invasive breast cancer patients30. We reported an increased proportion of CD16− NK cells in the peritoneal fluid of EMs patients, which served as an active mediator for promoting EMs development; however, the underlying mechanism of this abnormal differentiation is not fully defined31. Bryan Simons reported that PGI2 was also endowed with the ability to modulate the number and properties of NK cells resident in the respiratory tract, relating to allergic asthma19, suggesting that PGI2 might correlate with the abnormal NK-cell differentiation in EMs. Therefore, our study focused on analyzing whether the augmented CD16− NK-cell population in EMs resulted from PTGIS/PGI2 signaling in ESCs, aiming to provide a theoretical foundation for developing new approaches to combat this disease.
Materials and methods
Cells and culture
A human endometrial stromal cell (HESC) line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and NK92 cells were a generous gift from Professor Qiang Fu at Binzhou Medical University. HESC and primary ESCs were cultured in DMEM/F12 (HyClone, USA) supplemented with 10% fetal bovine serum (FBS) (ScienCell, USA) and a 1% penicillin–streptomycin–amphotericin B solution (NCM, Suzhou). Human peripheral blood NK cells were cultured in RPMI 1640 medium (HyClone) supplemented with FBS and antibiotics. NK92 cells were cultured in serum-free medium for lymphocytes (DAKEWE, Shenzhen) supplemented with 20% FBS and 1% antibiotics. All cells were cultured in an incubator with 5% CO2 at 37 °C.
Clinical specimen collection and isolation of primary ESCs
This study was approved by the Ethics Committee of Tongji University School of Medicine affiliated Shanghai First Maternity and Infant Hospital. In total, 53 ovarian EMs patients and 47 tubal infertility, leiomyoma, or benign ovarian cyst patients provided informed consent were enrolled in the study, and none of the patients had used hormonal contraceptives in the past 6 months. Twenty-four cases of ovarian endometriotic cyst tissues (defined as ectopic endometrium), 20 cases of normal endometrium specimens and 56 cases of peritoneal fluid samples were collected during surgical procedures, and the menstrual cycle phase of each specimen was further verified by experienced histopathologists. The disease stage of EMs patients was defined according to the American Society of Reproductive Medicine (ASRM) revised classification system, and the clinical characteristics of the patients are summarized in Table 1 and Supplementary Table 1. The endometriotic lesions of EMs patients and normal endometrial tissues are collected into culture medium and immediately transferred to the laboratory on ice. Each collected biopsy was divided into 3 parts: one was frozen in liquid nitrogen, one was stored in 4% formalin (Biotech Well, Shanghai) for immunohistochemical analysis, and one was dissected, cut into pieces and digested with collagenase IV (Sigma, USA) at 37 °C for 30 min (min). The digested samples were filtered through a 70-μm strainer (FALCON, USA) and centrifuged at 1000 rpm for 10 min. Then, the cell pellets were resuspended and cultured in medium in an incubator. Primary ESCs were confirmed by detecting the expression levels of vimentin and cytokeratin 7. In addition, 29 endometriotic and 27 control peritoneal fluid samples were centrifuged at 3000 rpm for 20 min, and the supernatants were stored at −80 °C until further analyzed.
Animals and EMs models
All in vivo experiments in the study were approved by the Animal Care Committee of Tongji University. Wild-type (WT) female mice on the C57BL/6 J (C57) background (aged 6–8 weeks (w)) were purchased from Shanghai Jiesjie Laboratory Animal Co., Ltd. Ptgis−/− mice and fcgr3−/− mice on the C57 background were constructed, bred and fed in an SPF environment by Shanghai Model Organisms Center, Inc. The methods for establishment the syngeneic mouse EMs model were previously described and slightly improved31. Briefly, ~6 to 8 w-old female donor and recipient mice were intramuscularly injected with 3 μg/mouse 17 β-estradiol (Sigma) on Day 1 and Day 4. Then, the donor mice were euthanized, and the uterus was collected and dissected into 1-mm3 fragments on Day 7. The uterine fragments with endometrial tissue in the proliferative phase were resuspended in PBS and injected into the peritoneal cavity of recipient mice, and when the endometrial debris was implanted, the EMs models were successfully established. In some groups of EMs model mice, drugs or PBS were injected intraperitoneally daily for 10 days beginning on Day 10. On Day 21, the EMs model mice were sacrificed, and we observed, collected and reserved the endometriosis-like lesions implanted in the peritoneal cavity.
GEO database bioinformatic analysis
EMs-related Gene Expression Omnibus (GEO) datasets (GSE23339, GSE7305, GSE5108, GSE25628, GSE11691, GSE78851, GSE12768, GSE6364, GSE94414, GSE58178, GSE87809, GSE31515, GSE47360, GSE73950, GSE47361, and GSE87621), consisting of several types of EMs, were downloaded from the website (https://www.ncbi.nlm.nih.gov/gds/?term=endometriosis); these datasets were uploaded and generously shared by an increasing number of researchers in related fields. The samples in these datasets were mainly ectopic endometriotic lesion biopsies, normal endometrial tissues and isolated ESCs, and more detailed information is summarized in Table 2. We determined the original expression and methylation status of PTGIS in all samples according to the annotations of relative platforms when the datasets were complied with a standard. The data we used were mainly generated with two approaches: RNA-sequencing or gene microarray and DNA methylation sequencing. All data were standardized and further analyzed by statistical analysis.
Lentiviral transfection
The lentiviruses CON077, LV-HIF-1A-RNAi, LV-DNMT1-RNAi (hU6-MCS-Ubiquitin-EGFP-IRES-puromycin), CON238, LV-PTGIS and LV-DNMT1 (Ubi-MCS-3FLAG-SV40-EGFP-IRES-puromycin) were all constructed by GENECHEM (Shanghai). HESCs were treated with trypsin containing 0.25% EDTA (NCM), centrifuged and cultured in 6-well plates (Corning, USA). Then, a lentivirus and transfection enhancer reagent were added to the culture medium when the cells reached 50% confluence, and the medium was exchanged 12 h (h) after transfection. After transfection for 72 h, the HESCs were inspected by fluorescence microscopy, and the transfection efficiency was verified by qRT–PCR and western blotting assays. Then, the cells were treated with 2 μg/ml puromycin (MCE, China) for 7 days to select the successfully transfected cells.
Human peripheral blood and mouse spleen NK-cell isolation
Normal peripheral blood was extracted from a vein in the elbow of female volunteers. Then, peripheral blood mononuclear cells (PBMCs) were separated with Ficoll (GE Healthcare, USA); we reserved the layer containing these cells and washed it with sterile PBS 3 times. Peripheral NK cells (pNK cells) were isolated from the PBMCs with a human NK-cell isolation kit via negative selection (STEMCELL, Canada) and further cultured in RPMI 1640 (HyClone) medium supplemented with FBS and antibiotics. To isolate NK cells from the mouse spleen, ~6- to 8-w-old female mice on the C57 background were sacrificed, and their spleens were taken out. We cut the spleens into pieces, removed the debris with 40-μm filters (FALCON) and centrifuged the cell suspensions at 1000 rpm for 10 min. Then, red blood cell lysis buffer (BioLegend, China) was used to lyse the red blood cells. Then, all the cells were incubated with reagents from a mouse NK-cell isolation kit (Miltenyi, Germany) and passed through a magnetic column (Miltenyi), and the mouse NK cells were isolated. The purity of isolated human pNK cells and mouse spleen NK cells was evaluated by flow cytometric analysis, and cells were used in further studies when their purity was greater than 90%.
RNA extraction and quantitative Real-Time PCR (qRT–PCR)
Tissue and cell samples were treated with TRIzol (TAKARA, Japan) for 15 min on ice, and RNA was extracted with traditional extraction methods. After the RNA samples were quantified and qualified, they were used as templates for the reverse transcription of complementary DNA (cDNA), with a total of 1000 ng RNA used for each sample. The reaction system was processed at 37 °C for 15 min, 85 °C for 5 s (s) and 4 °C indefinitely. Then, the products were evaluated by real-time PCR; the reaction system was set and processed according to the manufacturer’s suggestions as follows: 95 °C for 30 sec for stage 1; 95 °C for 5 s and 60 °C for 30 s, repeated for 40 cycles for stage 2; and 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s for the melting curve stage. The primers used are shown in Table 3 and were constructed by Sangon Biotech (Shanghai), and the data were further analyzed to calculate relative gene expression values.
Immunohistochemical (IHC) staining and semiquantitative analysis
Collected normal and ectopic endometrial tissues were fixed with 4% formalin (Biotech Well) for 12 h at 4 °C, embedded in paraffin and cut into 5-µm-thick slices. Then, the specimens underwent dewaxing, dehydration and antigen retrieval, and they were further incubated with primary antibodies against PTGIS and HIF-1α (Abcam, UK) in a humidified box overnight at 4 °C. The following day, the specimens were washed with PBS 3 times and then incubated with an HRP-conjugated goat anti-rabbit antibody (Abcam) for 30 min at room temperature. Then, all slides were reacted with DAB substrate and hematoxylin dye reagents (Biotech Well). Rabbit-derived isotype-matched immunoglobulin G was incubated with slides used as negative controls. Detailed information on the antibodies and reagents is listed in Supplementary Table 2. Finally, the specimens were observed and imaged randomly under a microscope. The staining intensity of the slides was examined and verified independently by two observers, and scores were calculated as the percentage of positive immunoreactive cells. No staining was recorded as no staining; staining of less than 10% was defined as weak staining; staining of 11–50% was defined as moderate staining; and staining of more than 50% was defined as strong staining.
Western blotting
Tissues and cells were washed with precooled PBS and treated with RIPA buffer (Beyotime, Shanghai) on ice for 30 min. Total protein was collected from the cell lysates and centrifuged at 4 °C and 12000 rpm for 20 min, and the supernatants of all lysates were reserved. Then, we determined the protein concentration of all samples by BCA methods (Beyotime). Loading buffer was added to the protein samples, and they were heated at 4 °C for 10 min. Then, all samples were ready for use in further studies. After a SDS–PAGE gel (Beyotime) was prepared, 25 μg of protein sample was added per well for each sample. By electrophoresis, the proteins with different molecular weights were separated in the gel, and then they were transferred to a PVDF membrane (Millipore, USA). The membrane was incubated with a primary antibody at 4 °C overnight. On the following day, after being washed with TBST (Beyotime) 3 times, the PVDF membrane was incubated with an HRP-conjugated secondary antibody for 1 h at room temperature on an orbital shaker. Finally, the PVDF membrane was exposed to light for imaging with immobilon western chemiluminescence HRP substrate (Millipore), and the relative intensity of the protein band was determined with Image-Pro Plus software version 6.0 (Media Cybernetics, Rockville, MD, USA).
Flow cytometric analysis (FCM)
Cells were trypsinized with 0.25% non-EDTA trypsin and centrifuged at 1000 rpm for 10 min. Then, the cells were resuspended in precooled PBS, and a single-cell suspension was prepared for detecting surface and intracellular molecules. To determine the expression level of cell-surface molecules, cells were directly incubated with a fluorophore-conjugated antibody (BioLegend) for 30 min at room temperature protected from light, with negative control cells incubated with isotype reagents. To stain for intracellular molecules, cells underwent fixation and permeabilization processes and were then incubated with a fluorophore-conjugated antibody. Thirty minutes later, the cells were washed with PBS and centrifuged at 1000 rpm for 5 min. Then, the cell pellets were resuspended in 300 μl PBS and further detected on a flow cytometer machine, and the expression level of each molecule was calculated according to the mean fluorescence intensity.
Immunofluorescence assay
For evaluation by immunofluorescence staining, cells were digested, plated in dishes (3.5 cm in diameter) (NEST, USA) and cultured in a 5% CO2, 37 °C atmosphere for 24 h. Then, as the cells were attached to the bottom of the dishes, we removed the culture medium, washed the cells with PBS 3 times and fixed them with 4% paraformaldehyde (Biotech Well) for 10 min. After permeabilization and blocking processes, the cells were incubated with a diluted primary rabbit antibody (Abcam) at 4 °C overnight, with negative control cells incubated with PBS. On the following day, the antibody was recycled, and the cells were washed with PBS 3 times and then incubated with a fluorescent HRP-conjugated goat anti-rabbit antibody (Abcam) at room temperature for 1 h protected from light. One hour later, we removed the secondary antibody, washed the cells with PBS 3 times and then stained them with DAPI reagent (Biotech Well) for 5 min in the dark. Finally, the cells were inspected by confocal microscopy and imaged.
Enzyme-linked immunosorbent assay (ELISA)
The level of 6-keto prostaglandin F1α was determined by ELISA (Cayman, USA); 6-keto prostaglandin F1α is a stable metabolite of PGI2 and can be detected as an indicator of the PGI2 level. The supernatants of cells, tissue lysates and peritoneal fluid from volunteers were collected and centrifuged at 3000 rpm for 20 min. Then, we collected the supernatants for further detection. An ELISA kit was removed from the refrigerator and allowed to warm to room temperature. When all reagents were prepared, we added standard reagents, supernatant samples or PBS (used as a negative control) to the wells of a 96-well plate and incubated the plate for 18 h at 4 °C. The next day, the 96-well plate was removed from the refrigerator, and we removed the supernatants, washed the plate thoroughly with wash buffer 5 times and added reconstituted Ellman’s reagents. Then, the plate was developed on an orbital shaker protected from light for 90 min at room temperature according to the manufacturer’s instructions. Finally, the optical density in the wells of the plate was detected at a wavelength of 420 nm on a microplate reader, and we determined the PGI2 concentrations of all samples according to the standard curve.
DNA methylation sequencing
To determine the methylation status of a PTGIS gene reporter, we performed functional promoter sequence analysis. Then, we verified the DNA methylation status of the PTGIS gene promoter in two groups of isolated primary ESCs by performing genomic bisulfite sequencing and dissociation curve analysis. The procedure was performed as follows: we extracted DNA from 8 cases of nESCs and 8 cases of eESCs, and the DNA was treated with bisulfite. Then, the products further underwent PCR process, and the PCR primers are listed in Table 4. Samples were detected in triplicate, and we sequenced 8–10 individual clones from each reaction. Then, the methylation data were collected and analyzed.
Adhesion assay
Negative control CON238 HESCs and LV-PTGIS HESCs were digested with 0.25% EDTA-trypsin, and then the cells were resuspended and centrifuged at 1000 rpm for 10 min. The cell pellets were resuspended in culture medium. These two groups of HESCs were counted and plated in 24-well plates in triplicate (5 × 105 cells per well). Twenty-four hours later, when the cells had attached to the bottom of the plate, the HESCs were digested, centrifuged, resuspended in medium and stained with PKH26 (Sigma). Then, we plated the cells in the previous plate to coculture them with CON238 and LV-PTGIS HESCs. The plate was then incubated in a 37 °C incubator protected from light. Thirty minutes later, we removed the culture medium, resuspended the HESCs, and gently washed the cells with sterile PBS 3 times. Then, the plate was inspected under a fluorescence microscope. We randomly acquired 5 images for each well and calculated the adhesion index of CON238 and LV-PTGIS HESCs.
Mouse intraperitoneal NK-cell depletion and adoptive transfer of NK cells
We used an anti-NK1.1 antibody (BioLegend) to eliminate peritoneal NK cells in mouse EMs models. On Day 7 and Day 9, 200 μg anti-NK1.1 was intraperitoneally injected into EMs model mice to deplete NK cells, and negative control reagents were intraperitoneally injected into other EMs model mice. On Day 11, some of the anti-NK1.1- and negative control reagent-injected EMs model mice were sacrificed, and we injected 1 ml of PBS into the peritoneal cavity and collected the peritoneal lavage fluid. The cells in the peritoneal lavage fluid were analyzed by FCM to verify the NK-cell depletion efficiency. On the same day, donor WT and fcgr3−/− mice were sacrificed, and we collected the spleens from the peritoneal cavity, cut them into pieces with sterile scissors, and filtered them through 40-μm filters (FALCON). Subsequently, we lysed the red blood cells, and then mouse spleen NK cells were isolated with magnetic beads using a negative selection method (Miltenyi). Isolated NK cells (5 × 105) were adoptively transferred into the peritoneal cavity of each recipient EMs model mouse via intraperitoneal injection. Ten days after NK-cell adoptive transfer, the EMs model mice were all sacrificed to collect the peritoneal lavage fluid and endometriotic lesions.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM), and statistical analysis was performed using Student’s t test, one-way analysis of variance (one-way ANOVA) and the Mann–Whitney test with Prism version 6.0 GraphPad software (USA). A P value less than 0.05 was defined as significant.
Results
PTGIS and PGI2 are increased in ESCs from patients with EMs
PGI2 production is specifically catalyzed by PTGIS; thus, we first investigated the expression of PTGIS in EMs samples in EMs transcriptional profile-related GEO datasets (GSE23339, GSE7305, GSE5108, GSE25628, GSE11691, GSE78851, GSE12768, GSE6364, GSE94414, GSE58178, GSE87809, GSE31515 and GSE47360), and the results revealed substantially elevated PTGIS levels in endometriotic lesions and isolated ovarian endometrioma ESCs (Fig. 1a). To exclude any possible bias resulting from distinctions among tissue types, samples were broadly grouped into different subgroups according to EMs localization, such as ovarian endometriotic cysts, deep infiltrating EMs (DIE) and adenomyosis. Concordant with our finding, most types of EMs exhibited observably increased levels of PTGIS (Fig. 1b, c). To verify the transcriptional profile of PTGIS identified in the GEO datasets, we collected normal endometrial tissues (nEM), endometriotic lesions (eEM) and primary ESCs from volunteers. Assays were performed to determine the expression level of PTGIS, and we observed robustly elevated levels of PTGIS in endometriotic lesions and ovarian ESCs (Fig. 1d–g), consistent with the results for the GEO datasets. Notably, EMs specimens derived from eEM lysates, peritoneal fluid samples and primary ESCs supernatants all exhibited increased concentrations of PGI2 (Fig. 1h–j). To examine the catalytic efficiency of PTGIS in PGI2 production, we upregulated the expression of PTGIS in HESCs, and the results showed that elevated PTGIS enabled overproduction of PGI2 (Fig. 1k, Supplementary Fig. 1a, b).

a Information from GEO datasets. The expression of PTGIS was elevated in most endometriotic lesions. b Nearly all types of EMs specimens displayed increased levels of PTGIS. c Ectopic ESCs had higher levels of PTGIS than normal ESCs in GEO datasets. d The gene expression level of PTGIS was increased in endometriotic lesions (n = 20) compared to the normal endometrium (n = 24), and ectopic ESCs (n = 12) had higher gene levels of PTGIS than normal ESCs (n = 12). e, f The protein level of PTGIS was substantially increased in endometriotic lesions and eESCs. g IHC staining was performed with samples of normal endometrium, eutopic endometrium of EMs and ectopic lesions of EMs, and it showed that PTGIS expression was elevated in the eutopic and ectopic endometrium. An immunofluorescence assay visualized the augmented PTGIS expression in eESCs. h–j Tissue lysates of endometriotic lesions (n = 16), peritoneal fluid collected from EMs (n = 29) volunteers and supernatants of eESCs (n = 12) exhibited higher concentrations of PGI2. k Upregulation of PTGIS expression in HESCs encouraged overproduction of PGI2. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. NS no significant difference, GEO Gene Expression Omnibus, EMs endometriosis, nESC normal endometrial stromal cells, euESC eutopic endometrial stromal cells, ecESC ectopic endometrial stromal cells, NET normal endometrial tissues, EET ectopic endometrial tissues, eESCs ectopic endometrial stromal cells, IHC immunohistochemistry, DAPI nuclear stain. Scale bars: 100 μm and 30 μm for IHC and 100 μm for immunofluorescence.
Hypoxia upregulates PTGIS/PGI2 expression in ESCs by altering the methylation status of the PTGIS promoter
To uncover the mechanism underlying the elevated PTGIS/PGI2 in EMs, the microenvironment of disease-modified lesions was investigated. It is widely accepted that endometriotic lesions are in a state of hypoxia32,33,34, and we determined the hypoxia-inducible factor 1 alpha (HIF-1α) levels in all samples. Consistent with a previous study, HIF-1α was extensively augmented in EMs (Supplementary Fig. 1c–g). HESCs were treated with DMOG and COCL2 to simulate the hypoxic milieu in EMs, and the results showed that DMOG and COCL2 accelerated PTGIS/PGI2 production in a concentration-dependent manner; KC7F2 produced opposing effects (Fig. 2a, c–f). Moreover, downregulating the expression of HIF-1α in HESCs hindered PTGIS expression (Fig. 2b, Supplementary Fig. 1h), implicating hypoxia as a critical mediator in PTGIS/PGI2 expression promotion. More importantly, epigenetic modification, such as DNA methylation, has also been emphasized to regulate PTGIS expression35,36,37. Thus, we analyzed the methylation status of the PTGIS promoter in 3 existing EMs-related GEO datasets (GSE73950, GSE87621 and GSE47361). The methylation of the PTGIS promoter in EMs specimens was extensively inhibited (Supplementary Fig. 2a–c), which was consistent with our verification that ovarian ESCs exhibited a deficient methylation level in the PTGIS promoter compared to normal ESCs (Fig. 2g, h). To examine the mechanism underlying this phenomenon, we measured the expression levels of the 3 main DNA methyltransferases, DNMT1, DNMT3A and DNMT3B, in tissues and primary ESCs and found a significant reduction in the DNMT1 level but no significant differences in the DNMT3A and DNMT3B levels in EMs (Fig. 2i, j, Supplementary Fig. 1i), implying that the deficient methylation of the PTGIS promoter was favored by hampered DNMT1 expression. These results were further confirmed by knocking down DNMT1 in HESCs, which led to substantially increased levels of PTGIS/PGI2 (Fig. 2k–n, Supplementary Fig. 1j), indicating that the abnormal PTGIS/PGI2 levels in EMs were also modulated by the DNMT1-mediated methylation deficiency in the PTGIS promoter. As two regulatory pathways were identified in our study, we hypothesized that there might be some correlation between hypoxia and DNMTI, as it has been reported that hypoxia destabilizes DNMT1 expression in ESCs38. Thus, a hypoxic milieu was created in vitro by culturing PESCs in a 2% oxygen incubator or treating HESCs with DMOG and COCL2, and the results revealed that 2% oxygen substantially hampered the expression of DNMT1 in PESCs and that DMOG and COCL2 treatment inhibited DNMT1 expression in a concentration-dependent manner (Fig. 2o, p). Moreover, KC7F2 treatment promoted DNMT1 expression (Fig. 2p). Mechanistically, knocking down DNMT1 in HESCs rescued PTGIS expression, which was inhibited by HIF-1α deficiency, and overexpressing DNMT1 in HESCs partly reversed the high expression level of PTGIS induced by hypoxia (Fig. 2q), indicating that hypoxia promoted PTGIS/PGI2 production through the DNMT1-mediated deficient methylation status of the PTGIS promoter.

a When compared to control (Ctrl) treatment, DMOG (250 μM, 48 h) and COCL2 (200 μM, 48 h) treatment promoted PTGIS expression in HESCs, and KC7F2 (5 μM, 48 h) produced opposing effects. b Knocking down HIF-1α depressed the expression of PTGIS in HESCs. c, d DMOG (250 μM, 48 h) promoted and KC7F2 (5 μM, 48 h) hindered the production of PGI2 in PESCs (n = 10). e DMOG (100, 250 and 1000 μM) (48 h) and COCL2 (100, 200 and 500 μM) (48 h) treatment promoted PTGIS expression in a concentration-dependent manner, and KC7F2 (2.5, 5, and 10 μM) (48 h) treatment produced opposing effects. f DMOG (250 μM, 48 h) and COCL2 (200 μM, 48 h) treatment promoted PTGIS expression, and KC7F2 (5 μM, 48 h) produced opposing effects. g, h The methylation of the PTGIS promoter was decreased in eESCs (n = 8) compared to nESCs (n = 8). i The expression levels of DNMT1 and DNMT3B were reduced in endometriotic lesions, while the level of DNMT3A was not significant different. j The mRNA level of DNMT1 was decreased in eESCs, while the levels of DNMT3A and DNMT3B did not show significant differences. k–n Knocking down DNMT1 expression in HESCs promoted PTGIS expression and PGI2 production. o When compared to Ctrl treatment (normoxic condition), 2% oxygen (hypoxic condition) treatment obviously hindered DNMT1 expression in PESCs. p DMOG (100, 250 and 1000 μM) (48 h) and COCL2 (100, 200 and 500 μM) (48 h) treatment hindered DNMT1 expression in HESCs, and KC7F2 (2.5, 5 and 10 μM) (48 h) produced opposing effects in a concentration-dependent manner. q Knocking down the expression level of HIF-1α elevated the level of DNMT1 and hindered the expression of PTGIS, and downregulating DNMT1 partly reversed these effects. Overexpression of DNMT1 in HESCs partly reversed the high expression of PTGIS induced by hypoxia. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. NS: no significant difference, *P < 0.05, **P < 0.01. HIF-1α hypoxia-inducible factor 1 alpha, WB western blotting, HESCs human endometrial stromal cells, PESCs primary endometrial stromal cells. Scale bar: 100 μm, white dots: unmethylated regions, black dots: methylated regions. C: control group, refers to the normoxic condition; H: 2% oxygen group, refers to the hypoxic condition.
PTGIS/PGI2 signaling accelerates EMs progression in vivo
Since increased production of PTGIS/PGI2 was present in endometriotic lesions, we investigated the roles of PTGIS/PGI2 in EMs. Specifically, we carried out in vivo experiments, and well-established EMs models were separated into iloprost (a stable PGI2 analog) and vehicle (PBS) groups. Iloprost at the manufacturer’s recommended doses or PBS was injected intraperitoneally into EMs model mice, and the iloprost group exhibited an increased number and weight of endometriotic lesions (Fig. 3a–d), indicating that the increased PGI2 concentration in the mouse peritoneal cavity facilitated EMs progression. Moreover, to precisely decipher the function of the aberrant level of PTGIS in disease-modified endometriotic lesions, we constructed ptgis−/− mice, and the PGI2 levels in the peritoneal lavage fluid and plasma of male and female ptgis−/− mice were considerably reduced (Supplementary Fig. 2d, e). Ptgis−/− mice were used as donors for intraperitoneal injection of uterine fragments into recipient mice to establish EMs models. As shown, the EMs model mice that received ptgis−/− uterine fragments had dramatically attenuated weights of endometriosis-like lesions in the peritoneal cavity compared to the recipient mice in the WT group (Fig. 3e, f), demonstrating that the activated PTGIS/PGI2 signaling pathway facilitated EMs progression in vivo.

a Schematic of the EMs model establishment process. b Collected ectopic lesions in the abdominopelvic cavity of EMs model mice. c, d Iloprost (a stable PGI2 analog) treatment (20 mg/kg) (n = 23) increased the weight and number of ectopic lesions in EMs model mice compared to control mice (n = 22). e Collected ectopic lesions from 3 groups of EMs model mice. f The weights of ptgis−/− (n = 7) and ptgis−/+ (n = 9) uterine debris-originated ectopic lesions were decreased compared to those of lesions in the WT group (n = 10). Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. NS no significant difference, *P < 0.05, **P < 0.01. WT wild type, ptgis−/+ heterozygote, ptgis−/− homozygote.
Hypoxia promotes CD16− NK-cell differentiation by activating PTGIS/PGI2 signaling
To uncover the mechanism by which the activated PTGIS/PGI2 signaling pathway accelerates EMs progression, we considered two critical factors, namely, intrinsic disorders of ectopic ESCs and dysfunctional immunosuppression. RNA-sequencing results showed that PTGIS mainly induced substantially augmented expression of adhesion molecules in HESCs, with other pathways showing no significant differences (Supplementary Fig. 3a–d), and the adhesive ability was considerably enhanced in PTGIS-overexpressing HESCs (Supplementary Fig. 3e, f), suggesting that the PTGIS/PGI2 signaling pathway triggered EMs by strengthening the adhesive ability of ESCs. On the other hand, we investigated the roles of PTGIS/PGI2 in reprogramming the immune milieu in the peritoneal cavity of EMs patients, and then we analyzed the PTGIS levels in all peritoneal fluid cells collected from healthy and EMs volunteers. Among all peritoneal fluid cells, NK cells had the lowest expression level of PTGIS, while macrophages had the highest, with ESCs having a PTGIS expression level similar to that of as macrophages and much higher than that of NK cells (Supplementary Fig. 4a–e). Thus, we speculated that ectopic ESC-derived PGI2 might have considerable influence on the function of NK cells. Moreover, we determined the PGI2-specific membrane superficial receptor (PI, PTGIR) levels in NK cells, and the results showed that PTGIR was actually expressed on the surface membrane of NK cells (Fig. 4a, b). Additionally, treatment with the PTGIR antagonist RO1138452 favored enlargement of the CD16+ NK92 cell subpopulation and promoted CD107a, interferon gamma (IFN-ɣ) and granzyme B expression (Fig. 4c–g), indicating that blocking PTGIR was of great importance in activating NK cells and that ESC-derived PGI2 indeed repressed NK-cell activity.

a PTGIR was expressed on the surface membrane of NK cells. b The expression level of PTGIR in NK cells increased after coculture with HESCs. c–g Treatment with the PTGIR antagonist RO1138452 (50 μM) decreased the percentage of CD16− NK92 cells and promoted CD107a, IFN-ɣ and granzyme B expression. h The purity of pNK cells isolated from the peripheral blood of volunteers and the expression level of CD16 were determined by FCM. i, j The CD16− pNK-cell subpopulation increased and expression level of CD16 reduced after coculture with ectopic ESCs. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001. PTGIR PGI2 receptor, RO1138452 PTGIR antagonist, MFI mean fluorescence intensity, IFN-ɣ interferon gamma, NESC normal endometrial stromal cells, ECESC ectopic endometrial stromal cells, pNK peripheral NK cells, FCM flow cytometric analysis.
In a previous study, we reported that ESCs promoted CD16− pNK-cell differentiation when ESCs were cocultured with pNK cells, and ovarian ESCs exerted more obvious effects31, which was further validated in this study (Fig. 4h–j). Based on the results showing that the PTGIR antagonist RO1138452 favored the differentiation of CD16+ NK cells, we speculated that the enlarged subpopulation of CD16− NK cells observed after coculturing ESCs with NK cells was ascribed to the function of ESC-derived PGI2. Consistent with our speculation, the level of PTGIR increased when NK cells were cocultured with HESCs (Fig. 4b), and we found that DMOG-conditioned HESCs promoted the differentiation of CD16− NK cells when cocultured with pNK cells (Fig. 5a–c). PTGIS-overexpressing HESCs also had similar effects when they were cocultured with NK92 or pNK cells (Fig. 5d, e). Furthermore, the expression of NK-cell activators, CD107a and IFN-ɣ was decreased when PTGIS-overexpressing HESCs were cocultured with pNK cells (Fig. 5f–i), and RO1138452 reversed the promotive effects of HESCs on CD16− NK-cell differentiation (Fig. 5j–m), demonstrating that hypoxia-modified HESCs promoted CD16− NK-cell differentiation through induced PTGIS/PGI2 production via activating PTGIR. More importantly, the role of PTGIS/PGI2 in regulating NK-cell differentiation was also observed in vivo. Specifically, DMOG and COCL2 treatment accelerated EMs progression and promoted CD3−NK1.1+CD16− (CD16−) NK-cell differentiation in mouse models (Supplementary Fig. 5a–d, Fig. 6a–e). In addition, ptgis−/− uterine fragments that survived in the abdominal cavity of recipient mice attenuated the differentiation of CD16− NK cells, with no significant difference in NK-cell percentages (Fig. 6f–i), which was consistent with the results of the in vitro experiments.

a–c Hypoxia-conditioned ESCs accelerated CD16− NK-cell differentiation and hindered the expression of CD16 in NK cells. d, e Upregulating the expression of PTGIS in HESCs promoted CD16− NK92 cell differentiation. f–i Upregulating the expression of PTGIS in HESCs accelerated CD16− pNK-cell differentiation and inhibited CD107a and IFN-ɣ expression. j, k RO1138452 (50 μM) treatment rescued NK cells from the inhibitory effect of DMOG (250 µM) on CD16 expression. l, m Upregulation of PTGIS in HESCs inhibited the expression of CD16 in pNK cells, and RO1138452 (50 μM) partly rescued this effect. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.

a NK cells in the peritoneal lavage fluid of EMs model mice were analyzed by FCM. b–e DMOG (20 mg/kg and 40 mg/kg) and COCL2 (40 mg/kg) treatment promoted CD16− NK-cell differentiation in the peritoneal cavity of EMs model mice but had no influence on the NK-cell ratio. f–i Ptgis−/− ectopic lesions in EMs model mice hindered CD16− NK-cell differentiation and increased the CD16 expression level of NK cells in the peritoneal fluid of EMs model mice, but the NK-cell ratio was not significantly different among the three groups. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. NS no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. WT wild type, ptgis−/+ heterozygote, ptgis−/− homozygote.
CD16− NK cells accelerate EMs progression in vivo
Our study confirmed that ESCs induced CD16− NK-cell differentiation, but the function of this subpopulation of NK cells in vivo, especially in EMs, remains uncertain. Then, we constructed fcgr3−/− mice, and both WT and fcgr3−/− mice were used as recipients and treated with RO1138452 or PBS plus DMSO for 10 days. The number and weight of endometriotic lesions in the abdominal cavity of fcgr3−/− recipient mice were extensively increased, and RO1138452 partially rescued these effects (Figs. 7a–e, 8a–c), which indicated an immunosuppressive milieu in the abdominal cavity of fcgr3−/− mice. This phenomenon might partly be ascribed to decreased NK-cell cytotoxicity. To exclude any influence of other types of immune cells, we depleted NK cells in the abdominal cavity of EMs model mice by intraperitoneally injecting anti-NK1.1, and the depletion efficiency was verified (Supplementary Fig. 6b, c). We isolated spleen NK cells from WT and fcgr3−/− mice and divided the well-established peritoneal NK-cell-depleted EMs model mice into two groups. Then, we adoptively transferred isolated spleen NK cells isolated from WT or fcgr3−/− mice into the abdominal cavity of the two groups of EMs model mice. Ten days later, the number and weight of endometriotic lesions and the percentage of CD16− NK cells were increased in the EMs model mice adoptively transferred with fcgr3−/−- spleen NK cells (Fig. 7f–i, Supplementary Fig. 6a, d, e), suggesting that the CD16− NK-cell subpopulation cells substantially favored the deterioration of EMs.

a EMs model instruction process. b Ectopic lesions collected from groups of EMs model mice. c, d The weight of every ectopic lesion and total weight of ectopic lesions in fcgr3−/+ and fcgr3−/− EMs model mice were increased. e The number of ectopic lesions was increased in fcgr3−/+ and fcgr3−/− EMs model mice, but the difference was not significant. f Process of adoptively transferring spleen NK cells. g Ectopic lesions collected from the WT and fcgr3−/− groups of EMs model mice. h The total weight of ectopic lesions was elevated in the fcgr3−/− group (n = 12) compared to the WT group (n = 10). i Fcgr3−/− EMs model mice had an elevated number of ectopic lesions. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. *P < 0.05, **P < 0.01. WT wild type, fcgr3−/+ heterozygote, fcgr3−/− homozygote.

a Process of EMs model establishment and treatment. b Ectopic lesions collected from groups of EMs model mice. c RO1138452 (800 mg/kg) treatment attenuated the increase in the weight of ectopic lesions resulting from CD16 deficiency in NK cells in vivo. d Schematic presenting the molecular mechanism by which PTGIS/PGI2 promotes EMs progression. Statistical analysis was performed with Student’s t test, one-way analysis of variance (ANOVA) or the Mann–Whitney test. *P < 0.05, ***P < 0.001. WT wild type, fcgr3−/+ heterozygote, fcgr3−/− homozygote.
Discussion
Molecules involved in arachidonic acid metabolism, such as COX-2 and PGE2, have been clearly identified to be critical modulators in EMs, emphasizing that lipid mediators may potentially play some roles in this disease. Herein, we identified another arachidonic acid metabolite, PGI2, which was extensively augmented in EMs. Notably, our findings revealed the contribution of elevated PTGIS expression to PGI2 overproduction in endometriotic lesions, and the regulatory mechanism underlying the increased PTGIS levels attracted our attention. Although PTGIS has been reported to be predominantly located in ovarian and bladder tissues, GEO datasets provided an informative setting for analyzing PTGIS expression levels in all types of EMs, and we excluded the possible bias resulting from the tissue of origin. As several studies have reported, the expression of COX-2 was increased in endometriotic-like loci39,40, and as a downstream enzyme of COX-2, aberrant PTGIS expression might be promoted by COX-2. Nevertheless, research on PTGIS is limited at present, and gene variants41, targeted repression of miR-199a/b42 and miR-140-3p.143 and genetic modifications, such as DNA methylation, have been reported to contribute to PTGIS expression35,36,37. We identified a deficient methylation status for the PTGIS promoter in EMs, which was mediated by hypoxia/DNMT1; however, the mechanism by which hypoxia modulates DNMT1 expression remains unclear. Several papers have reported that hypoxia destabilizes DNMT1 transcription through HIF1α/2α44,45, the AU-rich element binding factor 1 (AUF1)/miR-148a axis38, the PI3K/Akt-DNMT1-p53 pathway46, and PAS domain-containing protein 1 (EPAS1) protein transactivation47, which could provide some clues. Additionally, other regulatory mechanisms independent of DNA methylation may also contribute to hypoxia-induced PTGIS expression. Notably, it has been reported that HIF-1α directly binds to the HRE sequence in the PTGIS promoter region37, but whether this mechanism is relevant in EMs requires further exploration.
Using RNA-sequencing analysis, PTGIS-overexpressing HESCs were shown to exhibit an enhanced adhesive ability, indicating that the elevated PTGIS level in ESCs contributed to the early implantation of endometrial debris. Furthermore, this reprogrammed cellular phenotype resulting from increased PTGIS levels or PGI2 overproduction is controversial and equivocal. Research on PGI2 has substantially concentrated on immunity regulation and vasodilation modulation, while the role of PGI2 in remodeling the cell phenotype has been relatively less studied. It has been reported that iloprost promotes myotube formation, migration, and fusion48 and that PTGIR and PPARs trigger white to brown adipocyte conversion49. However, an emerging role for PTGIS in modulating cell phenotypes has been proposed in recent years; specifically, it was reported that PTGIS hinders hepatic stellate cell activation, induces apoptosis-related protein expression and alleviates CCL4-mediated liver fibrosis35. Additionally, PTGIS was associated with liver hepatocellular carcinoma and with liver metastasis in colorectal cancer50,51,52. Knocking down the expression level of PTGIS was shown to promote the migration and proliferation of bladder cancer cells37, demonstrating a more complicated role for PTGIS. In addition, ptgis−/− mice displayed a phenotype of abnormalities in both the cardiovascular and renal systems (http://www.informatics.jax.org/marker/MGI:1097156), and the former was ascribed to a disorder in the vasodilatory function of PGI2, while the etiology of the latter remains a mystery, which suggests that PTGIS could perform additional unknown roles beyond our understanding at present. Regarding the mechanism underlying the enhanced adhesive ability of ESCs, it has been proposed that inhibition of PTGIS reduces stat3 activation in human breast cancer cells53, that PTGIS regulates the JAK/STAT signaling pathway in macrophages43 and that PTGIR regulates the PKA/CREB and PI3K-γ/PKC-ζ/TRB3/AKT pathways in diabetes54. Thus, the biological functions of PTGIS/PGI2 need to be further investigated, which might provide insight into the mechanism underlying the elevated adhesive molecule expression.
The crosstalk between ESCs and immune cells has been explored, but the linkage remains largely obscure. Our study identified that ESC-derived PGI2 contributed to CD16− NK-cell differentiation in a PTGIR-dependent manner and that the CD16− NK-cell subpopulation promoted EMs progression in vivo, which reinforced the widely accepted hypothesis that CD16− NK cells possess a strengthened regulatory function and weakened killing activity; thus, the approach of hindering CD16− NK-cell accumulation in the peritoneal cavity of EMs patients might be considered an immune-based therapy for combating EMs. Furthermore, our study revealed that the HESC–NK-cell direct coculture system promoted PTGIR expression in the NK cells, that PTGIS-overexpressing HESCs unexpectedly exerted a weaker effect than control HESCs and that upregulating the expression level of PTGIS hindered PTGIR expression in HESCs. We speculated that the overproduction and accumulation of PGI2 might negatively impact PTGIR expression. However, how PTGIR modulates CD16 expression is still unclear, and the existing informative research is profoundly limited. CD16 is an activating receptor of NK cells; thus, PTGIR-modified NK-cell inactivation could possibly contribute to the downregulation of CD16 expression. As reported by Bryan Simons, elevated CX3CL1 levels in the airways of ptgir−/− mice led to the overproduction of IFN-γ in lung NK cells19, which implied a potential role for CX3CL1 in regulating PTGIR-mediated CD16− NK-cell differentiation. Uncovering the detailed mechanism might provide a solid foundation for the application of the PTGIR antagonist RO1138452 in treating EMs in the clinic. Additionally, there are several limitations to our work. First, PTGIS expression was lowest in NK cells among all cells in the peritoneal fluid, and whether these results were equivalent to NK cells producing the lowest concentration of PGI2 warrants further investigation in the future. Furthermore, whether ESC-derived PGI2 influences the proliferation and apoptosis of NK cells remains unknown. In addition, the PTGIS/PGI2 signaling pathway may widely affect angiogenic and inflammatory processes, which are also essential factors in EMs development, and substantial work needs to be done to decipher more roles in the future. Last, the hypoxic milieu of endometriotic lesions also exerted a direct effect on NK cells, which should be taken into account alongside the effects of PGI2 derived from ESCs.
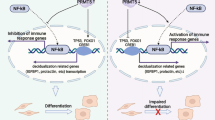
Overall, as shown in Fig. 8d, the hypoxic milieu of endometriotic foci destabilized the expression of DNMT1 and reduced the methylation of the PTGIS promoter, further promoting the transcription of PTGIS and catalyzing the biosynthesis of PGI2. On the one hand, increased levels of PTGIS/PGI2 demonstrated a hitherto unknown role in enhancing the adhesive ability of ESCs, which was critical for the survival of endometriotic debris in the early phase. On the other hand, the overproduction of secreted PGI2 promoted CD16− NK-cell differentiation through PTGIR, impairing the cytotoxic activity of NK cells, which contributed to immunosuppressive milieu formation in the peritoneal cavity of EMs patients. Together, these two critical factors accelerated EMs progression. In conclusion, we hope that our findings will provide innovative therapeutic approaches for EMs treatment and insight that helps uncover the underlying mechanism of EMs occurrence.
References
Bulun, S. E. et al. Endometriosis. Endocr. Rev. 40, 1048–1079 (2019).
Zondervan, K. T., Becker, C. M. & Missmer, S. A. Endometriosis. N. Engl. J. Med. 382, 1244–1256 (2020).
Peiris, A. N., Chaljub, E. & Medlock, D. Endometriosis. JAMA 320, 2608 (2018).
Koninckx, P. R. et al. Pathogenesis of endometriosis: the genetic/epigenetic theory. Fertil. Steril. 111, 327–340 (2019).
Zhang, T., De Carolis, C., Man, G. C. W. & Wang, C. C. The link between immunity, autoimmunity and endometriosis: a literature update. Autoimmun. Rev. 17, 945–955 (2018).
Vallvé-Juanico, J., Houshdaran, S. & Giudice, L. C. The endometrial immune environment of women with endometriosis. Hum. Reprod. Update 25, 564–591 (2019).
Jeljeli, M. et al. Macrophage immune memory controls endometriosis in mice and humans. Cell Rep. 33, 108325 (2020).
Alvarado-Díaz, C. P., Núñez, M. T., Devoto, L. & González-Ramos, R. Iron overload-modulated nuclear factor kappa-B activation in human endometrial stromal cells as a mechanism postulated in endometriosis pathogenesis. Fertil. Steril. 103, 439–447 (2015).
Rudzitis-Auth, J., Nickels, R. M., Menger, M. D. & Laschke, M. W. Inhibition of cyclooxygenase-2 suppresses the recruitment of endothelial progenitor cells in the microvasculature of endometriotic lesions. Am. J. Pathol. 188, 450–460 (2018).
Yamanaka, K. et al. Dienogest inhibits aromatase and cyclooxygenase-2 expression and prostaglandin E2 production in human endometriotic stromal cells in spheroid culture. Fertil. Steril. 97, 477–482 (2012).
Fagotti, A. et al. Analysis of cyclooxygenase-2 (COX-2) expression in different sites of endometriosis and correlation with clinico-pathological parameters. Hum. Reprod. 19, 393–397 (2004).
McLaughlin, V. V. et al. Results of an expert consensus survey on the treatment of pulmonary arterial hypertension with oral prostacyclin pathway agents. Chest 157, 955–965 (2020).
Carvalho, J. et al. Orphan G protein-coupled receptor GPRC5B controls smooth muscle contractility and differentiation by inhibiting prostacyclin receptor signaling. Circulation 141, 1168–1183 (2020).
Waxman, A. B. Oral prostacyclin therapy for pulmonary arterial hypertension: another step forward. Circulation 127, 563–565 (2013).
Pluchart, H., Khouri, C., Blaise, S., Roustit, M. & Cracowski, J. L. Targeting the prostacyclin pathway: beyond pulmonary arterial hypertension. Trends Pharmacol. Sci. 38, 512–523 (2017).
Hutchison, M. R. & White, P. C. Prostacyclin regulates bone growth via the Epac/Rap1 pathway. Endocrinology 156, 499–510 (2015).
Tamosiuniene, R. et al. Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ. Res. 122, 1689–1702 (2018).
Peshavariya, H. M. et al. Prostacyclin signaling boosts NADPH oxidase 4 in the endothelium promoting cytoprotection and angiogenesis. Antioxid. Redox Signal. 20, 2710–2725 (2014).
Simons, B. et al. PGI2 controls pulmonary NK cells that prevent airway sensitization to house dust mite allergen. J. Immunol. 198, 461–471 (2017).
Boswell, M. G., Zhou, W., Newcomb, D. C. & Peebles, R. S. Jr. PGI2 as a regulator of CD4+ subset differentiation and function. Prostaglandins Other Lipid Mediat. 96, 21–26 (2011).
Toki, S. et al. PGI2 signaling inhibits antigen uptake and increases migration of immature dendritic cells. J. Leukoc. Biol. 94, 77–88 (2013).
Wendler, S. et al. Immune modulation to enhance bone healing-a new concept to induce bone using prostacyclin to locally modulate immunity. Front. Immunol. 10, 713 (2019).
Zhou, W. et al. Prostaglandin I2 suppresses proinflammatory chemokine expression, CD4 T cell activation, and STAT6-independent allergic lung inflammation. J. Immunol. 197, 1577–1586 (2016).
Zhou, W. et al. The PGI2 analog cicaprost inhibits IL-33-induced Th2 responses, IL-2 production, and CD25 expression in mouse CD4+ T cells. J. Immunol. 201, 1936–1945 (2018).
Kim, J., Lee, S., Kim, Y. M., Jeoung, D. I. & Choe, J. Human follicular dendritic cells promote germinal center B cell survival by providing prostaglandins. Mol. Immunol. 55, 418–423 (2013).
Drake, T. S., O’Brien, W. F., Ramwell, P. W. & Metz, S. A. Peritoneal fluid thromboxane B2 and 6-keto-prostaglandin F1 alpha in endometriosis. Am. J. Obstet. Gynecol. 140, 401–404 (1981).
Carrega, P. et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 112, 863–875 (2008).
Gogali, F., Paterakis, G., Rassidakis, G. Z., Liakou, C. I. & Liapi, C. CD3(-)CD16(-)CD56(bright) immunoregulatory NK cells are increased in the tumor microenvironment and inversely correlate with advanced stages in patients with papillary thyroid cancer. Thyroid 23, 1561–1568 (2013).
Harlin, H. et al. The CD16- CD56(bright) NK cell subset is resistant to reactive oxygen species produced by activated granulocytes and has higher antioxidative capacity than the CD16+ CD56(dim) subset. J. Immunol. 179, 4513–4519 (2007).
Mamessier, E. et al. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J. Immunol. 190, 2424–2436 (2013).
Mei, J. et al. Suppression of autophagy and HCK signaling promotes PTGS2high FCGR3− NK cell differentiation triggered by ectopic endometrial stromal cells. Autophagy 14, 1376–1397 (2018).
Hsiao, K. Y., Chang, N., Lin, S. C., Li, Y. H. & Wu, M. H. Inhibition of dual specificity phosphatase-2 by hypoxia promotes interleukin-8-mediated angiogenesis in endometriosis. Hum. Reprod. 29, 2747–2755 (2014).
Li, W. N., Wu, M. H. & Tsai, S. J. HYPOXIA AND REPRODUCTIVE HEALTH: The role of hypoxia in the development and progression of endometriosis. Reproduction 161, F19–F31 (2021).
Xiong, Y. et al. Hypoxia-inducible factor 1α-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Hum. Reprod. 31, 1327–1338 (2016).
Pan, X. Y. et al. DNA Methylation of PTGIS Enhances Hepatic Stellate Cells Activation and Liver Fibrogenesis. Front. Pharmacol. 9, 553 (2018).
Frigola, J. et al. Hypermethylation of the prostacyclin synthase (PTGIS) promoter is a frequent event in colorectal cancer and associated with aneuploidy. Oncogene 24, 7320–7326 (2005).
Lu, M. et al. CIRBP is a novel oncogene in human bladder cancer inducing expression of HIF-1α. Cell Death Dis. 9, 1046 (2018).
Hsiao, K. Y. et al. Coordination of AUF1 and miR-148a destabilizes DNA methyltransferase 1 mRNA under hypoxia in endometriosis. Mol. Hum. Reprod. 21, 894–904 (2015).
Ota, H., Igarashi, S., Sasaki, M. & Tanaka, T. Distribution of cyclooxygenase-2 in eutopic and ectopic endometrium in endometriosis and adenomyosis. Hum. Reprod. 16, 561–566 (2001).
Hirata, T. et al. Interleukin-17F increases the secretion of interleukin-8 and the expression of cyclooxygenase 2 in endometriosis. Fertil. Steril. 96, 113–117 (2011).
Wang, X. J. et al. Association of rare PTGIS variants with susceptibility and pulmonary vascular response in patients with idiopathic pulmonary arterial hypertension. JAMA Cardiol. 5, 677–684 (2020).
Bai, Y. P. et al. Induction of microRNA-199 by nitric oxide in endothelial cells is required for nitrovasodilator resistance via targeting of prostaglandin I2 synthase. Circulation 138, 397–411 (2018).
Pan, X. Y. et al. Alternative activation of macrophages by prostacyclin synthase ameliorates alcohol induced liver injury. Lab. Invest. 101, 1210–1224 (2021).
Kitajima, S. et al. Hypoxia-inducible factor-2 alpha up-regulates CD70 under hypoxia and enhances anchorage-independent growth and aggressiveness in cancer cells. Oncotarget 9, 19123–19135 (2018).
Gao, X., Hicks, K. C., Neumann, P. & Patel, T. B. Hypoxia inducible factors regulate the transcription of the sprouty2 gene and expression of the sprouty2 protein. PLoS. One 12, e0171616 (2017).
Xu, R., Sun, Y., Chen, Z., Yao, Y. & Ma, G. Hypoxic preconditioning inhibits hypoxia-induced apoptosis of cardiac progenitor cells via the PI3K/Akt-DNMT1-p53 Pathway. Sci. Rep. 6, 30922 (2016).
Xu, X. H. et al. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 32, 6694–6705 (2018).
Murray, J., Whitson, R. H. & Itakura, K. Reduced prostaglandin I2 signaling in Arid5b−/− primary skeletal muscle cells attenuates myogenesis. FASEB J. 32, 1868–1879 (2018).
Ghandour, R. A. et al. IP-receptor and PPARs trigger the conversion of human white to brite adipocyte induced by carbaprostacyclin. Biochim. Biophys. Acta 1861, 285–293 (2016).
Cebola, I. et al. Epigenetics override pro-inflammatory PTGS transcriptomic signature towards selective hyperactivation of PGE2 in colorectal cancer. Clin. Epigenetics 7, 74 (2015).
Sun, L. C. et al. Overexpression of PTGIS could predict liver metastasis and is correlated with poor prognosis in colon cancer patients. Pathol. Oncol. Res. 18, 563–569 (2012).
Bai, K. H. et al. Identification of cancer stem cell characteristics in liver hepatocellular carcinoma by WGCNA analysis of transcriptome stemness index. Cancer Med. 9, 4290–4298 (2020).
Marotta, L. L. et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 121, 2723–2735 (2011).
Yan, S. et al. I prostanoid receptor-mediated inflammatory pathway promotes hepatic gluconeogenesis through activation of PKA and inhibition of AKT. Diabetes 63, 2911–2923 (2014).
Acknowledgements
We show great appreciation to Professor Qiang Fu at Binzhou Medical University for the kind donation of NK92 cells, and this work was supported by the Natural Science Foundation of China (NSFC82071615 and NSFC81771548, to Dong Zhao).
Author information
Authors and Affiliations
Contributions
Haiyan Peng conducted the experiments and conceived and wrote the original manuscript. Lichun Weng, Shating Lei, Shuhui Hou, and Shaoliang Yang supervised the experiments. Mingqing Li and Dong Zhao designed the experiments and reviewed the manuscript. All authors listed contributed to, read and approved the final manuscript for submission and publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, H., Weng, L., Lei, S. et al. Hypoxia-hindered methylation of PTGIS in endometrial stromal cells accelerates endometriosis progression by inducing CD16− NK-cell differentiation. Exp Mol Med 54, 890–905 (2022). https://doi.org/10.1038/s12276-022-00793-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00793-1
- Springer Nature Limited
This article is cited by
-
Tumor-suppressive functions of protein lysine methyltransferases
Experimental & Molecular Medicine (2023)