
Abstract
Capicua (CIC) is an evolutionarily conserved transcription factor. CIC contains a high-mobility group (HMG) box that recognizes specific DNA sequences to regulate the expression of various target genes. CIC was originally identified in Drosophila melanogaster as a transcriptional repressor that suppresses the receptor tyrosine kinase signaling pathway. This molecule controls normal organ growth and tissue patterning as well as embryogenesis in Drosophila. Recent studies have also demonstrated its extensive functions in mammals. For example, CIC regulates several developmental and physiological processes, including lung development, abdominal wall closure during embryogenesis, brain development and function, neural stem cell homeostasis, T cell differentiation, and enterohepatic circulation of bile acids. CIC is also associated with the progression of various types of cancer and neurodegeneration in spinocerebellar ataxia type-1, systemic autoimmunity, and liver injury. In this review, I provide a broad overview of our current understanding of the regulation and functions of CIC in mammals and discuss future research directions.
Similar content being viewed by others
Introduction
In 2000, the capicua (cic) gene was first identified in Drosophila melanogaster as a transcriptional repressor involved in the regulation of embryogenesis1. Casanova and colleagues performed a P-element screen to identify genes required for anteroposterior patterning in Drosophila1. These researchers found that a mutant embryonic phenotype characterized by a lack of abdominal segmentation but maintenance of head and tail structures was caused by a mutation in capicua (thus explaining the gene name, derived from the Catalan term meaning “head-and-tail”)1. Cic is required for organ growth and tissue patterning as well as anteroposterior and dorsoventral formation during embryogenesis in Drosophila1,2,3,4,5,6,7,8,9. Cic represses the expression of genes downstream of receptor tyrosine kinases (RTKs), including Torso and epidermal growth factor receptor (EGFR)1,8. Therefore, Cic functions as a negative regulator of the RTK signaling pathway. Moreover, RTK signaling activation promotes the degradation and/or cytoplasmic translocation of Cic via phosphorylation, thereby inducing the expression of Cic target genes downstream of RTK pathways2,8,10.
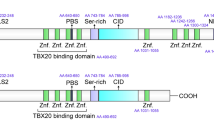
CIC is evolutionarily conserved from Caenorhabditis elegans to humans1,11,12. CIC exists as two isoforms, the short form (CIC-S) and the long form (CIC-L), which differ at their N-termini (Fig. 1a). CIC harbors two conserved domains, the high mobility group (HMG)-box and C1 domain (Fig. 1a), which cooperatively recognize specific octameric DNA sequences13. In mammals, CIC interacts with ataxin-1 (ATXN1), of which the polyglutamine (polyQ)-expanded form causes spinocerebellar ataxia type-1 (SCA1), a neurodegenerative disease14. CIC contributes to the pathogenesis of SCA1 in mice via interactions with mutant ATXN115,16. A fusion between CIC and a transcription activator domain of double homeobox 4 (DUX4) (CIC–DUX4 fusion protein) was identified in Ewing-like sarcoma cells17. CIC–DUX4 fusion proteins activate the expression of ETV1, ETV4, and ETV5, which encode oncogenic transcription factors18, thereby promoting cancer progression17. Many studies have verified that CIC functions as a tumor suppressor in various types of cancer19,20,21,22,23,24,25,26,27,28. Endogenous functions of CIC have been elucidated by examinations of the phenotypes of Cic mutant mice. CIC deficiency results in defects in lung development, bile acid homeostasis, abdominal wall closure during embryogenesis, neuronal cell differentiation, brain development, and T cell subset differentiation25,26,27,29,30,31,32,33,34. In this review, I focus on the roles of CIC in mammals; in particular, I summarize recent studies of (1) its functions in diseases, including neurological diseases and cancer, (2) its functions in development, and (3) its underlying regulatory mechanisms in mammalian cells.

a Schematic illustration of human CIC-S and CIC-L. CIC-L has a unique long N-terminal region compared with CIC-S. The amino acid regions of CIC responsible for the interaction with ATXN1/ATXN1L, 14-3-3, and ERK, the HMG box, nuclear localization signal (NLS), c-Src-mediated phosphorylation site, and C1 domain, are depicted. Numbers indicate amino acid positions. EBS: ERK binding site. b Regulatory mechanisms for CIC activity and stability. The left panel shows the RTK-ERK activation-mediated degradation and/or cytoplasmic translocation of CIC in mammalian cells. It is unclear whether CIC is degraded in the cytoplasm of mammalian cells. The right panel depicts the ATXN1/ATXN1L-mediated protection of mammalian CIC from proteasomal degradation. The molecular machinery mediating the degradation of CIC in the absence of ATXN1 and ATXN1L is unknown.
CIC functions in diseases
Spinocerebellar ataxia type-1 (SCA1)
SCA1 is one of nine polyQ disorders35,36. Expansion of the CAG repeat in ATXN1 results in a long polyQ tract-containing mutant ATXN1, which is associated with cerebellar neurodegeneration primarily due to Purkinje cell death35. Phosphorylation at the S776 residue of ATXN1 is critical for the neurotoxicity of the polyQ-expanded ATXN137,38. CIC binds with a high affinity to ATXN1 in human cells14. The CIC–ATXN1 complex is approximately 1.8 MDa in size, irrespective of the polyQ expansion in ATXN114. The S776A mutation reduces the incorporation of ATXN1 into large CIC–ATXN1 complexes, implying that the interaction with CIC contributes to the neurotoxicity of the polyQ-expanded ATXN114. Fryer et al. experimentally proved that CIC facilitates the pathogenesis of SCA1 using a Cic-deficient SCA1 mouse model (Atxn1154Q; Cic-L+/−) generated by crossing 154Q knock-in SCA1 (Atxn1154Q) mice with Cic hypomorphic (Cic-L−/−) mice15. A partial loss of CIC expression substantially attenuated the pathological and behavioral abnormalities of the Atxn1154Q mice15. Furthermore, the expression levels of some CIC target genes were downregulated in the cerebellum of the Atxn1154Q mice and were significantly rescued in the cerebellum of the Atxn1154Q; Cic-L+/− mice15. These findings suggest that the polyQ-expanded ATXN1 could enhance the transcriptional repressor activity of CIC for a subset of target genes, thereby contributing to the progression of SCA1. Disruption of the interaction between the polyQ-expanded ATXN1 and CIC inhibited the SCA1 disease phenotypes in mice, suggesting that SCA1 is caused by neurotoxicity driven by a gain-of-function of the polyQ-expanded ATXN1–CIC complex16.
Cancer
The first evidence for an association between CIC and cancer progression was the identification of the fusion between CIC and DUX4 as a result of a recurrent chromosomal translocation t(4;19)(q35;q13) in Ewing-like sarcomas17. The CIC–DUX4 chimaeras are composed of the majority of the CIC protein, except for a small portion of the C-terminus, and the C-terminal region of DUX4 involved in transcriptional activation17. The CIC–DUX4 fusion protein acquires transforming activity against NIH3T3 fibroblasts, indicating that CIC–DUX4 acts as a dominant oncogene17,39. The chimeric proteins transcriptionally activate the expression of CIC target genes, including PEA3 group genes that encode the oncogenic transcription factors ETV1, ETV4, and ETV517,18. Several other studies have identified various additional chromosomal translocations generating CIC–DUX4 chimeric transcripts in round cell sarcoma as well as Ewing sarcoma40,41,42,43,44. A xenograft mouse model subcutaneously injected with embryonic mesenchymal cells expressing CIC–DUX4 developed small round cell sarcoma45. Another study using a xenograft mouse model orthotopically injected with NIH3T3 mouse fibroblasts expressing CIC–DUX4 showed that the CIC–DUX4 proteins promote tumor growth and metastasis via the upregulation of CCNE1 and ETV4, respectively, suggesting that these proteins drive tumorigenesis and metastasis in sarcomas via distinct regulatory programs39.
CIC mutations occur most frequently in oligodendroglioma. Based on high-throughput DNA sequencing analyses, CIC was shown to harbor point mutations in 50–70% of oligodendrogliomas carrying the codeletion of chromosomes 1p and 19q23,24,46. The role of CIC point mutations in oligodendroglioma development and progression has not been experimentally verified. However, CIC deficiency promoted gliomagenesis in a xenograft mouse model orthotopically injected with PDGFB-expressing neural stem cells (NSCs)27. The glial cell-specific deletion of CIC did not induce tumor formation in the mouse brain, suggesting that defects in CIC itself may not be sufficient to initiate oligodendroglioma25. Many somatic mutations in CIC, including truncations, insertions, and deletions, have been identified in advanced-stage human lung adenocarcinoma specimens22. Okimoto et al. showed that the inactivation of CIC by point mutations promotes lung cancer metastasis via derepression of ETV4, which induces the expression of MMP2422. The CIC-ETV4-MMP24 metastatic axis is also involved in gastric adenocarcinoma22. Genetic ablation of CIC in adult mice caused T cell acute lymphoblastic leukemia/lymphoma (T-ALL)25,26, suggesting that CIC mutations could be considered driver mutations for T-ALL in humans. T-ALL also developed in hematopoietic lineage cell-specific Cic null mice26. However, T cell-specific Cic null mice did not show T-ALL phenotypes up to 14 months of age33, suggesting that the loss of CIC in T cells may be insufficient to cause T-ALL, and CIC deficiency in other types of immune cells may contribute to disease onset in mice. Decreased CIC expression at the protein level is frequently observed in various types of cancer19,20,21,28,46. Moreover, the CIC protein levels are often not correlated with the mRNA levels within the same cancer samples, suggesting that CIC exhibits robust post-transcriptional regulation in cancer cells19,21. Nuclear expression of CIC decreases gradually as prostate cancer (PC) becomes more aggressive20. CIC levels are also substantially downregulated in hepatocellular carcinoma (HCC), glioblastoma (GBM), and colorectal cancer (CRC)19,21,28. The decreased expression of CIC leads to the derepression of PEA3 group genes, thereby promoting cell growth and invasion in PC, HCC, GBM, and CRC cell lines19,20,21,28. Notably, the major PEA3 group members (e.g., ETV1, ETV4, or ETV5) regulated by CIC differ among cancer cell types; the expression of ETV5 and ETV4 is most highly and significantly upregulated by CIC deficiency in PC and HCC cell lines, respectively20,21. CIC is also involved in the control of cancer stem cell properties. CIC deficiency promotes the self-renewal capacity and increases the expression of cancer stem cell markers, including EpCAM+/CD44hi/CD24lo and ALDH47,48, via derepression of ETV4, ETV5, and SOX2 in breast cancer cell lines49. Consistent with this result, CIC levels were decreased in breast cancer patient samples with a CD44 high and CD24 low phenotype49. These data suggest that CIC suppresses breast cancer formation by restricting cancer stemness and identify CIC as a potential regulator of stem cell maintenance.
Functions of CIC in development
Lung development
Defective lung alveolarization has been observed in Cic-L−/− mice, in which CIC-L expression is completely abolished and CIC-S expression is substantially reduced but incompletely blocked15,31. Cic-L−/− mice exhibited perinatal lethality; approximately 83% of Cic-L−/− mice died before postnatal day 14 (P14; unpublished data), and the survivors were smaller than the wild-type (WT) littermates31. Cic-L−/− survivors had lung alveolarization defects causing air space enlargement accompanied by MMP9 overexpression in the lungs at P2031. Another germline Cic mutant (Cic△2–6/△2–6) mouse with deletions in Cic exons 2–6 (i.e., the HMG box-encoding exons), which expresses mutant CIC-L and CIC-S isoforms that lack the HMG box in the whole body, also exhibited defects in the terminal differentiation of the respiratory epithelium at embryonic day 18.5 (E18.5), potentially leading to delayed or altered alveolar maturation during postnatal development25. The CIC levels were relatively high in the lungs of E18.5 embryos31.
Abdominal wall closure
Characterization of Cic△2–6/△2–6 mice revealed that CIC is required for late embryonic development. Homozygous Cic△2–6/△2–6 embryos were present in Mendelian ratios at E18.5 but died immediately after birth25. Approximately 70% of the E18.5 Cic△2–6/△2–6 embryos had an omphalocele, a mild type of abdominal wall closure defect25. In this case, the gut protrudes into the umbilical ring in the late embryonic stage. Therefore, one explanation for the early death of Cic mutant mice is that a part of the internal organs, such as the intestines, is cannibalized when the mother removes the placenta after birth31,50. The abdominal wall closure defect was also found in mice that lack the expression of ATXN1 and ATXN1-like (ATNX1L), which bind to and stabilize CIC16,31,51 (Fig. 1b). Approximately 45% of the E18.5 Atxn1 and Atxn1l double null embryos had an omphalocele31. Taken together, these findings suggest that the CIC-ATXN1/ATXN1L complex is essential for normal embryogenesis and viability.
Brain development and function
CIC is highly expressed in the brain30,31. This molecule has been implicated in granule cell development based on the observation that Cic is highly expressed in immature granule cells in the cerebellum, hippocampus, and olfactory bulb12. A study of Cic mutant mice uncovered a critical role of CIC in brain development and function32. The deletion of Cic in the forebrain significantly reduced the thickness of cortical layers 2–4 and the dentate gyrus, potentially due to defects in the maintenance of postmitotic neurons32. The layer 2/3 pyramidal neurons of the forebrain-specific Cic null (Cicf/f;Emx1-Cre) mice also had defective dendritic branching32. CIC deficiency in the forebrain caused learning and memory deficits, and a loss of CIC in the hypothalamus and medial amygdala led to defects in social interactions32. Consistent with these mouse data, de novo heterozygous truncating mutations in CIC are associated with autism spectrum disorder, developmental delay/intellectual disability, seizures, and attention deficit hyperactivity disorder in humans32.
CIC is also associated with NSC maintenance and differentiation. Cic null NSCs presented EGF-independent hyperproliferative characteristics27. Hyperproliferation of NSCs by the loss of Cic was also confirmed in E13 embryos by a 5-ethynyl-2′-deoxyuridine (EdU) labeling experiment29. Upon the induction of differentiation in vitro, Cic null NSCs could not differentiate into mature oligodendrocytes and instead were maintained in an oligodendrocyte progenitor cell (OPC)-like stemness state27. A similar result was obtained using another forebrain-specific Cic null (Cicf/f;Foxg1-Cre) mouse model, in which Olig2+Sox2+ cells and Olig2+Pdgfra+ OPCs are increased and CNPase+ immature oligodendrocytes are decreased in the cortex29. Moreover, CIC deficiency enhanced the self-renewal capacity and promoted the symmetric division of NSCs29. Mechanistically, the derepression of Etv5 mediated the effects of CIC deficiency in NSCs29. Thus, CIC is a key transcription factor that controls brain development and function as well as the pathogenesis of neurological disorders.
Immune cell development and function
Park et al. investigated the role of CIC in the immune system by generating and characterizing hematopoietic lineage cell-specific Cic null (Cicf/f;Vav1-Cre) mice33. These mice had lymphoproliferative disorder-like symptoms at 9 weeks of age, as evidenced by an increased splenocyte count mainly due to the expansion of the B220+ B cell population and hyperglobulinemia. Cicf/f;Vav1-Cre mice eventually developed systemic autoimmune-like phenotypes, including the enlargement of secondary lymphoid organs; increased anti-dsDNA antibody serum levels; immune cell infiltration into various organs, including the liver, lung, and kidney; and IgG deposition at the glomeruli of the kidney. T cell-specific Cic null (Cicf/f;Cd4-Cre) mice also exhibited similar phenotypes to Cicf/f;Vav1-Cre mice, suggesting that CIC deficiency in T cells is critical for the induction of autoimmune-like symptoms33. CIC deficiency promotes the differentiation of follicular helper T (Tfh) cells33, which play a pivotal role in the germinal center reaction to produce isotype class switched high affinity antibodies against specific antigens52. At the molecular level, Etv5 is a critical target gene of CIC for the regulation of Tfh cell differentiation33. ETV5 levels were significantly upregulated in Cic null Tfh cells compared with WT cells. Adoptive transfer experiments using OT-II cells, ovalbumin-specific T cell receptor-expressing CD4+ T cells, revealed that ETV5 overexpression promotes Tfh cell development and that the knockdown of ETV5 substantially rescues the enhanced Tfh cell differentiation of Cic null OT-II cells33. These results indicate that the CIC-ETV5 axis controls Tfh cell development. Park et al. also proposed that Maf, which encodes a transcription factor that promotes Tfh cell differentiation53, is a target of ETV5 in CD4+ T cells under STAT3 activation33.
CIC is also involved in maintaining homeostasis of bone marrow hematopoietic stem and progenitor cells (HSPCs) and early T cell development26. Analyses of bone marrow and thymic cells in adult stage-specific (Cicf/f;UBC-Cre/ERT2) and endothelial and hematopoietic lineage cell-specific (Cicf/f;Tek-Cre) Cic null mice have shown that the number of HSPCs, including hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs), is reduced, whereas the frequency of thymic double negative 1 (DN1) cells is significantly increased26. The frequency of early T cell precursors (ETPs), a subset of DN1 cells from the bone marrow that remain pluripotent, is also elevated in the thymus of Cicf/f;UBC-Cre/ERT2 mice, suggesting that CIC regulates the self-renewal capacity of stem-like cells26.
CIC has been implicated in the development of CD8+ resident memory T (Trm) cells in the liver34. Cic-L−/− mice exhibit liver damage, as evidenced by increases in serum alanine transaminase (ALT) and hepatic proinflammatory cytokine expression levels30. These mice also have defects in the enterohepatic circulation of bile acids accompanied by the downregulation of several key genes involved in bile acid biosynthesis and transport in the liver30. These liver dysfunctions are not due to a CIC deficiency in hepatocytes because liver-specific Cic null (Cicf/f;Alb-Cre) mice do not recapitulate these phenotypes34. Cicf/f;Cd4-Cre mice have increased serum ALT and hepatic proinflammatory cytokine expression levels, indicating that CIC-deficient T cells cause inflammatory liver injury34. CIC deficiency promotes the formation of liver CD8+ Trm-like cells expressing surface markers, such as CD69+, CD49a+, CXCR6+, CXCR3+, and CD103-, in a cell intrinsic manner34. Moreover, the suppression of liver CD8+ Trm-like cell formation dramatically mitigated liver injury phenotypes in Cicf/f;Cd4-Cre mice treated with acetaminophen, which induces acute liver injury, suggesting that the increased CD8+ Trm-like cell population in the liver is responsible for the CIC deficiency-induced liver injury34. Mechanistically, the CIC–ETV5 axis controls liver CD8+ Trm-like cell differentiation. The derepression of ETV5 induces the expression of HOBIT, a transcription factor required for Trm cell development54, in Cic null CD8+ T cells, thereby promoting Trm cell differentiation34.
Regulation of CIC
RTK-RAS-MAPK pathways suppress CIC activity via the cytoplasmic translocation and/or degradation of CIC (Fig. 1b). This regulatory mechanism was originally discovered in studies of CIC expression patterns in Drosophila embryos. Torso RTK signaling in the early embryo leads to the degradation of CIC, whereas EGFR signaling in the ovarian follicle induces the partial relocalization of CIC to the cytoplasm10,55. EGF treatment resulted in the phosphorylation of human CIC-S at 20 different serine/threonine residues, presumably by ERK and p90RSK, a kinase activated by ERK56. In particular, p90RSK-mediated phosphorylation of S173 is critical for 14–3–3 binding (Fig. 1a), which inhibits CIC binding to target DNA sequences56. S1409 phosphorylation prevents the binding of importin α4/KPNA3 to the nuclear localization signal of CIC56. However, the disruption of the CIC-KPNA3 interaction does not affect the nuclear localization of CIC-S56, suggesting that other transport-related factors might be required for the cytoplasmic translocation of CIC in mammalian cells. ERK binds to the C-terminal region of human CIC-S containing residues 1335–1359 (prior to the C1 domain; Fig. 1a)57. EGFR stimulation decreased CIC levels in mammalian cells15,19,22. The inhibition of ERK by treatment with MEK1/2 inhibitors increased the levels of nuclear CIC-S at the expense of cytoplasmic CIC expression in pancreatic cancer cells58, suggesting that ERK regulates the subcellular localization of CIC (Fig. 1b). Moreover, EGFR-activated c-Src tyrosine kinase mediates cytoplasmic translocation of CIC-S via phosphorylation of the Y1455 residue59 (Fig. 1a, b). CIC is degraded in the nucleus upon EGFR-ERK activation19. In this process, the nuclear E3 ligase PRAJA1 (PJA1) polyubiquitylates CIC, leading to the proteasomal degradation of CIC in the nucleus19 (Fig. 1b). DNA binding of CIC is a prerequisite for the PJA1-mediated polyubiquitylation of CIC19. In addition, PJA1 recognizes the S173 residue of CIC-S to interact with CIC19. Since 14-3-3 also binds to S173-phosphorylated CIC-S to control the transcriptional repressor activity of CIC56, crosstalk between 14-3-3 and PJA1 might be involved in the regulation of CIC activity and/or stability.
Another regulatory mechanism underlying CIC activity is the ATXN1/ATXN1L interaction-mediated stabilization of CIC (Fig. 1b). Both ATXN1 and its homolog ATXN1L interact with and stabilize CIC31. The AXH domain of ATXN1/ATXN1L and the highly conserved N-terminal region of CIC-S, including amino acid residues 28–48, mediate their interaction60 (Fig. 1a). ATXN1L plays a more pivotal role in the stabilization of CIC than ATXN1; CIC levels decreased more substantially in response to the loss of ATXN1L than to the loss of ATXN1, leading to substantial derepression of CIC target gene expression31,61. In the absence of ATXN1L, CIC becomes unstable, resulting in proteasomal degradation61. ATXN1L also promotes CIC binding to the target gene promoter regions61. However, the reason for the relative importance of ATXN1L for CIC stabilization and function is unclear.
Long noncoding RNA (lncRNA)-mediated regulation of CIC expression has been reported62. The levels of CIC and lncRNA-AC006129.1, of which genomic locus is close to CIC in chromosome 19, were significantly decreased and increased in samples from schizophrenia patients, respectively62. AC006129.1 transgenic mice exhibited social interaction deficits, spatial working memory impairments, and sensorimotor gating disruption accompanied by upregulation of inflammatory response genes, including SOCS3 and CASP1, which are CIC target genes62. The overexpression of AC006129.1 downregulated CIC levels in both mouse and human cells62, suggesting that this lncRNA-mediated transcriptional repression of CIC expression might be conserved in mammals. Mechanistically, AC006129.1 recruits DNA methyltransferases 1 and 3a (DNMT1 and DNMT3a) and induces DNA methylation of CIC promoter regions62. The AC006129.1-mediated suppression of CIC expression leads to derepression of SOCS3 and CASP1, potentially contributing to the pathogenesis of schizophrenia62.
Concluding remarks
CIC has multiple roles in various developmental processes and in the pathogenesis of various diseases. CIC is believed to function as a tumor suppressor in various types of cancer and is a regulator of embryogenesis, brain and immune cell development, and stem cell maintenance. Our current understanding of CIC functions in mammals is largely limited to processes regulated by the CIC-ETV1/ETV4/ETV5 axis. Many molecular studies of mammalian cells have identified additional target genes of CIC, such as Spry4, Dusp4, Dusp6, Spred1, Ccnd1, Ccne1, and Per215,17,27,39,63,64. It will be important to clarify the effects of CIC regulation of various target genes at both the cellular and organismal levels. Furthermore, the mechanism by which CIC regulates target gene expression remains largely unclear and should be a focus of future research. CIC was shown to recruit the histone deacetylase complex to repress the expression of target genes in stem cells64. Another study proposed that CIC has dual functions as a transcriptional activator as well as a repressor27. There are several unanswered questions regarding the regulation of CIC activity. For example, which factors mediate the cytoplasmic translocation of CIC upon the activation of RTK signaling? How does ATXN1L stabilize CIC at the molecular level? Which transcription factors control the expression of CIC? These unresolved issues need to be addressed for a comprehensive understanding of the CIC-mediated regulation of biological processes. Finally, CIC is emerging as a key determinant of immune responses. A few studies have recently uncovered the roles of CIC in the development of T cell subsets26,33,34. However, the function of CIC in other types of immune cells, including B cells, dendritic cells, and macrophages, has not been established. Comprehensive studies of CIC functions in various types of immune cells will improve our understanding of the pathogenesis of immune disorders, such as autoimmune diseases and lymphomas, at the molecular level.
References
Jimenez, G., Guichet, A., Ephrussi, A. & Casanova, J. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev. 14, 224–231 (2000).
Andreu, M. J. et al. EGFR-dependent downregulation of Capicua and the establishment of Drosophila dorsoventral polarity. Fly 6, 234–239 (2012).
Andreu, M. J. et al. Mirror represses pipe expression in follicle cells to initiate dorsoventral axis formation in Drosophila. Development 139, 1110–1114 (2012).
Atkey, M. R., Lachance, J. F., Walczak, M., Rebello, T. & Nilson, L. A. Capicua regulates follicle cell fate in the Drosophila ovary through repression of mirror. Development 133, 2115–2123 (2006).
Cinnamon, E. et al. Capicua integrates input from two maternal systems in Drosophila terminal patterning. EMBO J. 23, 4571–4582 (2004).
Goff, D. J., Nilson, L. A. & Morisato, D. Establishment of dorsal-ventral polarity of the Drosophila egg requires capicua action in ovarian follicle cells. Development 128, 4553–4562 (2001).
Herranz, H., Hong, X. & Cohen, S. M. Mutual repression by bantam miRNA and Capicua links the EGFR/MAPK and Hippo pathways in growth control. Curr. Biol. 22, 651–657 (2012).
Roch, F., Jimenez, G. & Casanova, J. EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129, 993–1002 (2002).
Yang, L. et al. Minibrain and Wings apart control organ growth and tissue patterning through down-regulation of Capicua. Proc. Natl Acad. Sci. USA 113, 10583–10588 (2016).
Astigarraga, S. et al. A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J. 26, 668–677 (2007).
Jimenez, G., Shvartsman, S. Y. & Paroush, Z. The Capicua repressor—a general sensor of RTK signaling in development and disease. J. Cell Sci. 125, 1383–1391 (2012).
Lee, C. J. et al. CIC, a member of a novel subfamily of the HMG-box superfamily, is transiently expressed in developing granule neurons. Mol. Brain Res. 106, 151–156 (2002).
Fores, M. et al. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its mutation patterns in cancer. PLoS Genet. 13, e1006622 (2017).
Lam, Y. C. et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347 (2006).
Fryer, J. D. et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 334, 690–693 (2011).
Rousseaux, M. W. C. et al. ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar Ataxia Type 1 through a gain-of-function mechanism. Neuron 97, 1235–1243 (2018).
Kawamura-Saito, M. et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137 (2006).
Oh, S., Shin, S. & Janknecht, R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta 1826, 1–12 (2012).
Bunda, S. et al. CIC protein instability contributes to tumorigenesis in glioblastoma. Nat. Commun. 10, 661 (2019).
Choi, N. et al. miR-93/miR-106b/miR-375-CIC-CRABP1: a novel regulatory axis in prostate cancer progression. Oncotarget 6, 23533–23547 (2015).
Kim, E. et al. Capicua suppresses hepatocellular carcinoma progression by controlling the ETV4-MMP1 axis. Hepatology 67, 2287–2301 (2018).
Okimoto, R. A. et al. Inactivation of Capicua drives cancer metastasis. Nat. Genet. 49, 87–96 (2017).
Bettegowda, C. et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333, 1453–1455 (2011).
Chan, A. K. et al. Loss of CIC and FUBP1 expressions are potential markers of shorter time to recurrence in oligodendroglial tumors. Mod. Pathol. 27, 332–342 (2014).
Simon-Carrasco, L. et al. Inactivation of Capicua in adult mice causes T-cell lymphoblastic lymphoma. Genes Dev. 31, 1456–1468 (2017).
Tan, Q. et al. Loss of Capicua alters early T cell development and predisposes mice to T cell lymphoblastic leukemia/lymphoma. Proc. Natl Acad. Sci. USA 115, E1511–E1519 (2018).
Yang, R. et al. Cic loss promotes gliomagenesis via aberrant neural stem cell proliferation and differentiation. Cancer Res. 77, 6097–6108 (2017).
Lee, J. S. et al. Capicua suppresses colorectal cancer progression via repression of ETV4 expression. Cancer Cell Int. 20, 42 (2020).
Ahmad, S. T. et al. Capicua regulates neural stem cell proliferation and lineage specification through control of Ets factors. Nat. Commun. 10, 2000 (2019).
Kim, E. et al. Deficiency of Capicua disrupts bile acid homeostasis. Sci. Rep. 5, 8272 (2015).
Lee, Y. et al. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev. Cell 21, 746–757 (2011).
Lu, H. C. et al. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat. Genet. 49, 527–536 (2017).
Park, S. et al. Capicua deficiency induces autoimmunity and promotes follicular helper T cell differentiation via derepression of ETV5. Nat. Commun. 8, 16037 (2017).
Park, S., Park, J., Kim, E. & Lee, Y. The Capicua/ETS translocation variant 5 axis regulates liver-resident memory CD8(+) T-cell development and the pathogenesis of liver injury. Hepatology 70, 358–371 (2019).
Kang, S. & Hong, S. Molecular pathogenesis of spinocerebellar ataxia type 1 disease. Mol. Cells 27, 621–627 (2009).
Orr, H. T. & Zoghbi, H. Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621 (2007).
Emamian, E. S. et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38, 375–387 (2003).
Perez Ortiz, J. M. & Orr, H. T. Spinocerebellar Ataxia type 1: molecular mechanisms of neurodegeneration and preclinical studies. Adv. Exp. Med. Biol. 1049, 135–145 (2018).
Okimoto, R. A. et al. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J. Clin. Invest. 129, 3401–3406 (2019).
Graham, C., Chilton-MacNeill, S., Zielenska, M. & Somers, G. R. The CIC-DUX4 fusion transcript is present in a subgroup of pediatric primitive round cell sarcomas. Hum. Pathol. 43, 180–189 (2012).
Italiano, A. et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 51, 207–218 (2012).
Kajtar, B. et al. CD99-positive undifferentiated round cell sarcoma diagnosed on fine needle aspiration cytology, later found to harbour a CIC-DUX4 translocation: a recently described entity. Cytopathology 25, 129–132 (2014).
Mangray, S. et al. Primary undifferentiated sarcoma of the kidney harboring a novel variant of CIC-DUX4 gene fusion. Am. J. Surg. Pathol. 40, 1298–1301 (2016).
Tsukamoto, Y. et al. Primary undifferentiated small round cell sarcoma of the deep abdominal wall with a novel variant of t(10;19) CIC-DUX4 gene fusion. Pathol. Res Pract. 213, 1315–1321 (2017).
Yoshimoto, T. et al. CIC-DUX4 induces small round cell sarcomas distinct from Ewing sarcoma. Cancer Res. 77, 2927–2937 (2017).
Gleize, V. et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 78, 355–374 (2015).
Ginestier, C. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007).
Ponti, D., Zaffaroni, N., Capelli, C. & Daidone, M. G. Breast cancer stem cells: an overview. Eur. J. Cancer 42, 1219–1224 (2006).
Yoe, J., Kim, D., Kim, S. & Lee, Y. Capicua restricts cancer stem cell-like properties in breast cancer cells. Oncogene https://doi.org/10.1038/s41388-020-1230-7 (2020).
Thumkeo, D., Shimizu, Y., Sakamoto, S., Yamada, S. & Narumiya, S. ROCK-I and ROCK-II cooperatively regulate closure of eyelid and ventral body wall in mouse embryo. Genes Cells 10, 825–834 (2005).
Bowman, A. B. et al. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat. Genet. 39, 373–379 (2007).
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Bauquet, A. T. et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 10, 167–175 (2009).
Mackay, L. K. et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016).
Ajuria, L. et al. Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 138, 915–924 (2011).
Dissanayake, K. et al. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem. J. 433, 515–525 (2011).
Futran, A. S., Kyin, S., Shvartsman, S. Y. & Link, A. J. Mapping the binding interface of ERK and transcriptional repressor Capicua using photocrosslinking. Proc. Natl Acad. Sci. USA 112, 8590–8595 (2015).
Wang, B. et al. ATXN1L, CIC, and ETS transcription factors modulate sensitivity to MAPK pathway inhibition. Cell Rep. 18, 1543–1557 (2017).
Bunda, S. et al. c-Src phosphorylates and inhibits the function of the CIC tumor suppressor protein. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.MCR-18-1370 (2020).
Kim, E., Lu, H. C., Zoghbi, H. Y. & Song, J. J. Structural basis of protein complex formation and reconfiguration by polyglutamine disease protein Ataxin-1 and Capicua. Genes Dev. 27, 590–595 (2013).
Wong, D. et al. Transcriptomic analysis of CIC and ATXN1L reveal a functional relationship exploited by cancer. Oncogene 38, 273–290 (2019).
Ni, C. et al. LncRNA-AC006129.1 reactivates a SOCS3-mediated anti-inflammatory response through DNA methylation-mediated CIC downregulation in schizophrenia. Mol Psychiatry https://doi.org/10.1038/s41380-020-0662-3 (2020).
Crespo-Barreto, J., Fryer, J. D., Shaw, C. A., Orr, H. T. & Zoghbi, H. Y. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 6, e1001021 (2010).
Weissmann, S. et al. The tumor suppressor CIC directly regulates MAPK pathway genes via histone deacetylation. Cancer Res. 78, 4114–4125 (2018).
Acknowledgements
This work was supported by grants from the National Research Foundation (NRF) of Korea (2017R1A5A1015366 and 2018R1A2B2004416).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declars no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, Y. Regulation and function of capicua in mammals. Exp Mol Med 52, 531–537 (2020). https://doi.org/10.1038/s12276-020-0411-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0411-3
- Springer Nature Limited
This article is cited by
-
Molecular Determinants of Neurocognitive Deficits in Glioma: Based on 2021 WHO Classification
Journal of Molecular Neuroscience (2024)
-
Capicua regulates the development of adult-born neurons in the hippocampus
Scientific Reports (2021)