
Abstract
Background:
We aimed to investigate whether neonatal hypoxic–ischemic (HI) brain injury induces inflammatory lung damage.
Methods:
Thus, hypoxic (HYP, FiO2 10% for 30 min), ischemic (ISC, bilateral carotid flow interruption for 30 min), or HI event was performed in 1–2-d-old piglets. Dynamic compliance (Cdyn), oxygenation index (OI), and extravascular lung water (EVLW) were monitored for 6 h. Then, histologic damage was assessed in brain and lung (lung injury severity score). Total protein content (TPC) was determined in broncoalveolar lavage fluid (BALF), and IL-1β concentration was measured in lung and brain tissues and blood.
Results:
Piglets without hypoxia or ischemia served as controls (SHM). HI-induced brain damage was associated with decreased Cdyn, increased OI and EVLW, and histologic lung damage (interstitial leukocyte infiltration, congestive hyperemia, and interstitial edema). BALF TPC was increased, suggesting inflammatory damage. In agreement, tissue IL-1β concentration increased in the brain and lung, in correspondence with increased IL-1β serum concentration. Neither HYP nor ISC alone led to brain or lung damage.
Conclusion:
HI brain damage in newborn piglets led to inflammatory lung damage, suggesting an additional mechanism accounting for the development of lung dysfunction after neonatal HI encephalopathy.
Similar content being viewed by others


Main
Damage of organs other than the brain often complicates neonatal hypoxic–ischemic (HI) encephalopathy (NHIE) after perinatal asphyxia (1,2). There is a stepwise increase in the rate of adverse outcomes as the number of additional organs involved increase in newborns with NHIE (2). Lungs are often affected: depending on the definition criteria, 25 to 86% of HIE infants develop signs of respiratory dysfunction (1,2). Extracerebral impact of perinatal asphyxia has been uniformly attributed to the redistribution of blood flow and/or the effects of global hypoxia–ischemia (2,3). Observational studies from adults indicate that lung injury may develop after local brain damage such as intracranial hemorrhage, traumatic brain injury, or ischemic cerebral stroke (4,5,6). For these conditions, the incidence of lung injury is related to the severity of brain damage and represents an independent factor associated with poor clinical outcome (7).
Although the etiology of respiratory dysfunction after brain damage in adults is not completely understood, inflammation is thought to play a key role, together with other mechanisms such as neurogenic pulmonary edema, left ventricular dysfunction, or infection (8,9). In adult animal models and in patients suffering from subarachnoid hemorrhage, brain injury leads to a systemic inflammatory response with upregulation of inflammatory mediators such as TNF-α, intracellular adhesion molecule-1, IL-1β, and NF-kB and reactive oxygen species, microglial activation, and neutrophil accumulation (5,9,10). It has been hypothesized that lung injury might be triggered by the spread of that (such a systemic) response, favored by the disruption of the blood–brain barrier in the context of this inflammatory process (10). Such a cascade of events is not privative for the brain. For instance, bilateral renal ischemia is followed by an increase in serum IL-1, IL-6, and IL-12 and lung injury in mice (11). Similarly, intestinal ischemia–reperfusion injury induces acute lung injury in rats, with cellular infiltration, microvascular dysfunction, and interstitial edema (12).
Since systemic inflammation and oxidative stress, together with blood–brain barrier disruption, are events typically taking place in NHIE pathophysiology (13,14), it is attractive to hypothesize that the aforementioned mechanisms for distant lung injury after brain damage might be involved in the lung dysfunction accompanying newborn HIE. Previous studies reported that lung compliance and gas interchange are impaired in newborn piglets after brain HI insults (15,16), pointing to the existence of lung injury in that scenario.
The aim of this work was to demonstrate specifically the existence of brain HI-induced lung injury and to study the underlying mechanisms using a newborn piglet model of HI brain damage.
Results
Homeostatic Parameters
Table 1 shows the homeostatic parameters of the four different groups. No statistically significant differences were found between groups in cardiac output at all time points. However, HI led to a significant drop in mean arterial blood pressure that persisted throughout the experimental period, so that half the number of HI piglets required substantial inotropic support (dopamine, mean dose: 13 ± 4 μg/kg/min) to maintain appropriate mean arterial blood pressure values. Interestingly, such a drop in mean arterial blood pressure was not observed in the hypoxic (HYP) or ischemic (ISC) groups, with only one piglet from each of those groups receiving inotropic support (dopamine 7.5 μg/kg/min) by the end of the experiment. No control (SHM) piglet needed inotropic support. Whereas no variation in pCO2 was observed in piglets throughout the experiment, a transient but significant decrease in blood pH and in cerebral and somatic rSO2 was observed during the hypoxemic period in HI and HYP groups. Brain ischemia alone, as in ISC, led to a milder but significant drop in cerebral rSO2 with no changes in somatic rSO2. Hematocrit remained stable throughout the experiment for all groups.
Brain Damage
Assessment of brain damage. Continued sedoanalgesia determined that amplitude-integrated EEG (aEEG) amplitude decreased slightly in SHM animals throughout the experiment ( Figure 1 ), although this effect was not associated with an impairment of background EEG activity and/or the EEG pattern (data not shown). HI led to a dramatic decrease in brain activity that did not recover during the following 6 h ( Figure 1 ). Hypoxia and ischemia alone led to a slight decrease in aEEG amplitude during the insult, but in this case, aEEG regained normal amplitude to the end of the experiment. Throughout the experiment, aEEG amplitude in HYP or ISC was statistically significantly higher than that in HI ( Figure 1 ).

Brain activity studied by changes of mean amplitude of continuous aEEG recording in 1–2-d-old piglets after sham operation (SHM, open circle) or after hypoxic (HYP, open square), cerebral ischemic (ISC, filled square), or cerebral hypoxic–ischemic (HI, filled circle) insult. Results are expressed as means ± SEM of 5–8 animals. B, basal. I, insult. * P < 0.05 vs. SHM. §P < 0.05 vs. HI. See Methods for details.
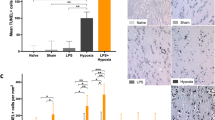
Analysis of Nissl-stained brain tissue revealed a reduced number of normal neurons in HI as compared with SHM with a 5-fold increase in the percentage of nonviable neurons in the frontoparietal cortex and a 10-fold increase in the hippocampus ( Figure 2a ). Brain histology was similar in HYP and ISC to SHM.

Brain and lung damage. Left: Representative light microphotographs of (a) Nissl staining of parietotemporal cortex (CX) and hippocampus (HPC) from brain tissue (original magnification ×200, bar = 100 µm) and (b) hematoxylin–eosin staining of lung tissue sections (top: original magnification ×80, bar = 100 µm; bottom: original magnification ×800, bar = 50 µm), obtained from 1–2-d-old piglets after sham operation (SHM, white bars) or after hypoxic (HYP, gray bars), cerebral ischemic (ISC, stripped bars), or cerebral hypoxic–ischemic (HI, black bars) insult. In brain from HI, there are an increased number of pyknotic cells (arrows). In lung from HI, there is interstitial leukocyte infiltration, congestive hyperemia, and interstitial edema. Right: (a) quantification of the number of necrotic neurons in CX and HPC; (b) quantification of lung damage by a severity score. Results are expressed as means ± SEM of 5–8 animals. * P < 0.05 vs. SHM. See Methods for details.
HI-induced neuroinflammation. Cerebral HI induced a local inflammatory response as evidenced by an increase in IL-1β concentration in brain tissue as compared with SHM ( Figure 3 ). Brain IL-1β concentration was similar in HYP or ISC to SHM.

Inflammatory response. Determination of interleukin-1α (IL-1α) concentration in (a) brain tissue, (b) plasma, and (c) lung tissue obtained from 1–2-d-old piglets 6 h after sham operation (SHM, white bars) or after hypoxic (HYP, gray bars), cerebral ischemic (ISC, stripped bars), or cerebral hypoxic–ischemic (HI, black bars) insult. Results are expressed as means ± SEM of 5–8 animals. * P < 0.05 vs. SHM. (d–f) lineal correlation between IL-1ß concentration in (d) brain tissue and serum, (e) serum and lung tissue, and (f) brain and lung tissues. Brain and lung studies were performed using microarrays after adjusting the protein concentration per well to 500 µg/ml. Plasma studies were performed by ELISA (absorbance 450 nm). See Methods for details.
Blood Inflammatory Markers
HI led to an increase in IL-1β serum levels as compared with SHM–an increase not observed in HYP or ISC ( Figure 3 ). Interestingly, a positive correlation was found between brain and serum IL-1β concentrations (R2 = 0.75; P < 0.05).
Lung Damage
Assessment of lung damage. Dynamic compliance (Cdyn) remained unchanged in the SHM group over the study period. By contrast, HI led to a fall in Cdyn as of 6 h after HI ( Figure 4 ). Cdyn remained similar to SHM in HYP or ISC. Oxygenation index (OI) also remained stable during the experiment in SHM but showed a continued increase after HI, so that by the end of the experiment, the OI was statistically significantly greater in HI than that in SHM ( Figure 4 ). In HYP or ISC, the OI remained similar to SHM throughout the experiment.

Lung dynamics. Changes in (a) dynamic lung compliance (Cdyn), (b) oxygenation index (OI), and (c) extravascular lung water (EVLW) content in 1–2-d-old piglets after sham operation (SHM, open circle) or after hypoxic (HYP, open square), cerebral ischemic (ISC, filled square), or cerebral hypoxic–ischemic (HI, filled circle) insult. Results are expressed as means ± SEM of 5–8 animals. B, basal; I, insult. *P < 0.05 vs. SHM. See Methods for details.
Hypoxia and/or ischemia were associated with an increase in extravascular lung water (EVLW) as compared with SHM during the insult ( Figure 4 ). Subsequently, ELVW tended to normalize but further increased in HI, so that by the end of the experiment, ELVW was significantly greater in HI than that in SHM. Such a final increase in ELVW was not observed in HYP or ISC, so that by the end of the experiment, ELVW was similar in those groups and in SHM.
HI led to histologic lung damage evident 6 h after the insult. Thus, HI piglets showed higher severity scores as compared with the SHM group, mainly because of the existence of interstitial leukocyte infiltration, congestive hyperemia, and interstitial edema ( Figure 2b ). Severity score was not statistically different in HYP or ISC compared with SHM.
Assessment of lung inflammation. HI was associated with an increase of IL-1β content in lung tissue, as compared with SHM ( Figure 3 ). IL-1β content was similar in HYP or ISC to SHM.
Remarkably, IL-1β concentrations in lung tissue correlated with that in brain tissue (R2 = 0.45; P < 0.05) and plasma (R2 = 0.61; P < 0.05) ( Figure 3 ).
The HI insult led to a 250% increase in broncoalveolar lavage fluid (BALF) total protein concentration from the basal condition to the end of the experiment ( Figure 5 ). No statistically significant increase in BALF protein content was observed in the other groups (P < 0.05).

Changes in bronchoalveolar lavage fluid (BALF) protein content in 1–2-d-old piglets in basal conditions or 6 h after sham operation (SHM, open circle) or after hypoxic (HYP, open square), cerebral ischemic (ISC, filled square), or cerebral hypoxic–ischemic (HI, filled circle) insult. Results are expressed as means ± SEM of 5–8 animals. *P < 0.05 vs. SHM. See Methods for details.
Discussion
In this work, we report for the first time that HI brain insult was associated with inflammatory lung damage in newborn pigs. This was demonstrated by functional, histologic, and biochemical studies. Animals subjected to HI showed signs of pulmonary dysfunction with increased ventilatory needs because of decreased lung compliance and impaired gas exchange. The lung is one of the most commonly affected extracerebral organs in NHIE, and ventilatory support and high oxygen requirements are frequently needed in these patients (1,2). We observed increased EVLW in HI piglets throughout the experimental period. In the absence of cardiac dysfunction, EVLW is a good indicator of noncardiogenic pulmonary edema earlier and more sensitive than other parameters such as chest x-ray or OI (17). It has been validated against the gold-standard gravimetric method in animal models and the in vivo double indicator technique (18,19). Interestingly, EVLW is increased in patients suffering from stroke (6) or intracranial trauma (20). The relationship of EVLW with lung edema agrees with our finding of histologic lung damage in HI piglets, which mainly consists of interstitial leukocyte infiltration, congestive hyperemia, and interstitial edema. To the best of our knowledge, this is the first study to assess lung histology in a neonatal animal model of cerebral HI. Altogether, these findings pointed to lung inflammatory damage. In support, we observed an increase in total protein concentration in BALF from HI piglets. Total protein concentration in BALF is a direct indicator of vascular permeability and alveolar epithelium integrity (10). Increased vascular permeability can be a consequence of vascular damage or the result of a cell-mediated inflammatory response (21). In agreement, we found an increased concentration of IL-1β in lung tissue from HI piglets. It has been reported that IL-1, IL-6, and other inflammatory mediators such as COX-2, LTB4, or intracellular adhesion molecule-1 are increased in lung tissue after traumatic brain injury (9,10). All changes observed in the lung in our experiments were linked to the existence of an HI insult, since neither hypoxia nor brain ischemia alone led to significant lung damage in our experiments. However, due to the design of our work, we cannot rule out entirely that hypoxemia could affect lungs to some extent. Sustained hypoxemia leads to pulmonary hypertension because of lung artery vasoconstriction in ~10 to 25% of infants with NHIE (2). Transient pulmonary hypertension is observed during and after 60 min of hypoxemia in piglets subsequently resuscitated with 21% oxygen (22,23). In agreement, in our experiments, a shorter duration of the hypoxemic episode was not sufficient to induce significant and sustained pulmonary hypertension, as reflected by the prompt normalization of the OI in the HYP group.
It is well known that a combination of hypoxia and ischemia is needed to induce brain damage in newborns (13). In fact, in experiments in which only hypoxemia was induced in newborn animals, brain damage only appears when hypoxemia is long enough to induce hypotension and thus ischemia (24). In agreement, in our experiments, the hypoxic episode lasted for just 30 min; it was not associated with hypotension and thus did not lead to either histologic brain damage or sustained impairment in aEEG activity. Prolonged ischemia, for instance, because of sustained hypotension, may lead to brain damage in newborn pigs (24). In our experiments, however, the ischemic episode did not last long enough to induce histologic or functional (aEEG) brain damage. This may, at least in part, be because of the fact that during such a short ischemic period, some remaining cerebral blood flow may exist despite the bilateral carotid compression thanks to the vertebral circulation, as suggested by the modest decline in crSO2 in ISC piglets during the ischemic episode. HI events trigger a cascade of events leading to brain damage. Among these, three variables constitute the “deadly triad”: excitotoxicity, oxidative stress, and inflammation. HI, traumatic, or hemorrhagic brain injury induces an inflammatory response characterized by activation of microglia, release of excitatory amino acids, reactive oxygen species, nitric oxide, proteases, and proinflammatory cytokines (13,25,26). Neutrophil infiltration further increases local accumulation of reactive oxygen species and cytokines (13,25). The importance paid to inflammation in NHIE pathophysiology has been on the rise recently (26). Among the different proinflammatory cytokines, IL-1 is especially important in the context of HI-induced brain damage (25), with IL-1 levels increasing in the cerebrospinal fluid of HI infants in parallel with the severity of encephalopathy. Indeed, this cytokine predicts HI brain injury better than TNFα (27). In this work, we confirmed that hypoxia and ischemia together are needed for IL-1β levels to be increased in the brain since IL-1β concentration was similar in HYP or ISC to SHM.
NHIE is often followed by distant organ injury (1,2). This complication has been classically attributed to a compensatory mechanism of redistribution of blood flow (diving reflex) to organs such as the brain, myocardium, or adrenal glands (2,3). Alvarez et al. (15) reported decreased lung compliance and increased OI after brain HI in piglets. These findings were attributed to ventilator-induced lung injury. However, in our study, the same ventilation protocol was applied in all groups, but lung injury was observed only in HI piglets. Thus, we can rule out a ventilator-induced lung injury. Interestingly, in our study, we found an increase in plasma IL-1β levels following HI brain injury. Proinflammatory cytokines such as IL-6, IL-10, IL-1β, or TNF-α are elevated in the blood of infants with HIE (14). Studies in animal models of brain injury suggest that the aforementioned inflammatory response is not limited to the brain and may spread to distant organs (9,10). In fact, severe brain injury induces release of inflammatory mediators such as TNF-α, IL, and intracellular adhesion molecule-1 into the systemic circulation (28) thanks to the increase in blood–brain barrier permeability that follows inflammatory brain injury (10,29). In our study, the linear relationship between IL-1β concentration in brain tissue and plasma, and between IL-1β concentration in plasma and the lung, supports a pathophysiologic link between brain and lung damage after a brain HI insult in piglets. Wu et al. (9) report a transient elevation in IL-1β in the lung of rats exposed to hemorrhagic brain injury, which is parallel to an increase of this cytokine in the brain. In this regard, several clinical studies in stroke patients demonstrate a correlation between the severity and extent of brain injury and the degree of systemic inflammatory response (30,31). Furthermore, thrombolysis in this setting, and thus reduction of the ischemic brain area, is followed by decreased plasma levels of inflammatory parameters (30).
To the best of our knowledge, the lung injury mechanism we propose in our study, linking cerebral HI and inflammatory lung damage, has never been specifically investigated in newborns. However, several studies have investigated the effects on the lung of ischemia–reperfusion injury in other organs. Renal ischemia–reperfusion, for instance, induces histologic lung damage and increases pulmonary vascular permeability and expression of proinflammatory cytokines in lung tissue (11,32). Some studies also demonstrate a link between intestinal ischemia–reperfusion injury and lung damage (12,33). Although much less studied, lung injury can also induce inflammatory brain damage. In this regard, a recent experimental study demonstrates that ventilation with excessive tidal volume during resuscitation at birth induces lung injury and triggers brain inflammation (34). Characteristically, the lung, whose epithelial surface area is so vast that its cross-sectional area is approximately the size of a tennis court, receives most of the systemic blood flow (35). These factors make the lung both a potential source and a target organ for a systemic inflammatory response, as reported in acute respiratory distress syndrome (36) and traumatic brain injury (10). Altogether, those data suggest that ischemia–reperfusion injury and/or inflammation may establish a crosstalk between organs spreading the inflammatory process to induce distant damage.
This study has some limitations. Firstly, the animal model we used, although widely accepted, is based on the combination of local cerebral ischemia and systemic hypoxia, whereas in the clinical setting NHIE occurs in the context of global ischemia and/or hypoxia, for example, after interruption of placental blood supply (13). However, this peculiarity enabled us to isolate and independently investigate the specific role of cerebral HI and reperfusion injury in the generation of lung damage. Another limitation is the short period of study. This could determine that lung injury was significant but not very severe. Such a short period of study, however, allow us to avoid the detrimental effects of mechanical ventilation by itself, as suggested by the mild signs of lung damage appearing even in SHM animals after 6 h of ventilation. Therefore, lung damage observed in our experiments could be attributed to the pathological condition and not to iatrogenic effects. Nevertheless, further studies investigating medium- and long-term effects of cerebral HI on lungs are warranted. Finally, since in our model we avoided severe systemic hypoxemia and/or hypotension to prevent a significant effect of those conditions on lung, the resulting brain damage was not severe. This indicates that even mild brain damage was able to induce distant lung damage. Nevertheless, further studies investigating the effects of severe cerebral HI damage on the lungs would be of great interest.
We can draw the conclusions that in newborn piglets cerebral HI triggers per se inflammatory lung damage that cannot be attributed to the effects of systemic hypoxia. Our results suggest that this is because of a local inflammatory response not confined to the brain, as reflected by the elevation of inflammatory mediators in serum, spreading to a distant organ such as the lung where it induces histologic and functional damage. Thus, we propose an additional mechanism to account for the development of lung dysfunction after neonatal HIE, together with other well-known mechanisms such as redistribution of organ blood flow. Determining the relative importance of those mechanisms warrants further research.
Methods
The experimental protocol met European and Spanish regulations for protection of experimental animals (86/609/EEC and RD 53/2013) and was approved by the Ethics Committee for Animal Welfare from the University Hospital Puerta de Hierro Majadahonda (Madrid, Spain). The number of animals used was determined to be the minimum number necessary to attain statistical significance.
Animal Preparation
As previously reported (15,16), 1–2-d-old male piglets were intubated and then mechanically ventilated (Evita 4; Dräger, Lübeck, Germany). Initial ventilator settings were: PEEP 5 cmH2O, respiratory rate 30 bpm, inspiratory time 0.5 s, peak inspiratory pressure as required for a tidal volume of 6 ml/Kg, FiO2 0.21. Ventilatory parameters (by the ventilator’s computerized pneumotachography), end-tidal CO2 concentration and O2 saturation (SpO2) (Ohmeda 5250 RGM, Louisville, CO). hemodynamic parameters, EVLW content, and central temperature (PiCCO Plus, Pulsion, Feldkirchen, Germany) were continuously monitored. Arterial blood gases were obtained hourly to adjust ventilatory settings and FiO2 and to calculate OI (PMVA × FiO2/PaO2 × 100). Brain activity was continuously monitored by aEEG (BRM3; BrainZ Instruments, Auckland, New Zealand). Cerebral (crSO2) and somatic (srSO2) regional hemoglobin oxygen saturation were continuously measured using a near-infrared spectroscopy system (INVOS 5100C; Covidien, Mansfield, MA).
Experimental Protocol
Animals were then randomly assigned to 30 min hypoxia (HYP, n = 7) by lowering FiO2 to 0.10; 30 min brain ischemia (ISC, n = 5) by interrupting carotid blood flow by pulling out elastic bands; 30 min brain hypoxia–ischemia (HI, n = 8); or control group (SHM, n = 6). At the end of the period of ischemia and/or hypoxia, carotid flow was restored and/or FiO2 increased to 0.21.
Six hours after HI/HYP/ISC, or the equivalent period in SHAM, piglets were killed by i.v. potassium chloride bolus.
Brain Processing
As previously reported (16), 4-µm-width brain coronal sections were obtained to assess early neuronal necrosis by Nissl staining in the parietal cortex at 3 mm in the posterior plane, as shown in a stereotaxic atlas of pig brain (37).
Bronchoalveolar Lavage Fluid Collection and Processing
BALF was obtained at baseline and after sacrifice following a standardized procedure (38). After centrifugation (1,000 rpm for 10 min at 4 °C), the supernatant was collected and frozen (−20 °C). Total protein content in BALF was subsequently determined using a BCA Protein Assay Kit (Thermo Scientific, Waltham, MA).
Lung Processing
After sacrifice, lungs were inflated and rinsed with saline. Paraformaldehyde 4%-fixed paraffin-embedded sections from the upper, lower, middle-anterior, and middle-posterior lobes from the right lung were stained with hematoxylin–eosin to perform a four-point, semi-quantitative, severity-based scoring system (negative = 0, slight = 1, moderate = 2, and severe = 3) assessing atelectasis, edema, inflammation, and other variables (39).
Assessment of Cytokine Expression in Brain and Lung Tissue and Plasma
IL-1β concentration was determined in the Madrid Science Park (Spain) in samples of frozen brain or left lung (30 mg) using cytokine arrays (Quantibody Porcine Cytokine Array, RayBiotech, Atlanta, GA).
IL-1β concentration was determined by ELISA (IL1ß Pig ELISA Kit; Abcam, Madrid, Spain) in plasma from blood samples collected in EDTA-treated tubes and centrifuged at 1,500 rpm for 10 min at 4 °C.
Statistical Analysis
SPSS 15.0.0 software (IBM Software, Armonk, NY) was used for all statistical analyses. All data are presented as mean ± SD. Mean values were compared using the Mann–Whitney test. For multiple comparisons, mean values were compared using the Kruskall–Wallis test for range analysis with the post hoc Dunn test for multiple comparisons. The Spearman test was used to analyze correlation between variables. P < 0.05 was considered to be statistically significant.
Statement of Financial Support
This work was supported by grants from the Research Institute Carlos III (ISCiii) according to the Plan Nacional de I+D+I 2008–2011 and the Plan Estatal de Investigación Científica y Técnica y de Innovación 2013–2016, with co-funding from the European Regional Development Funds (FEDER) (FIS-PS09/01900, PS09/00434, PI12/00852, and PS12/00192) and from the Biomedicine Programme, Community of Madrid (S2010/BMD-2308). None of the funding sources were involved in the study design, collection, analysis, and interpretation of data, the drawing up of the report, or the decision to submit the article for publication.
Disclosure
J.M.-O. has a research agreement with GW Pharma, Cambridge, UK (GWCRI09119). The other authors have no competing financial interests to declare.
References
Martín-Ancel A, García-Alix A, Gayá F, Cabañas F, Burgueros M, Quero J. Multiple organ involvement in perinatal asphyxia. J Pediatr 1995;127:786–93.
Shah P, Riphagen S, Beyene J, Perlman M. Multiorgan dysfunction in infants with post-asphyxial hypoxic-ischaemic encephalopathy. Arch Dis Child Fetal Neonatal Ed 2004;89:F152–5.
Bocking AD, Gagnon R, White SE, Homan J, Milne KM, Richardson BS. Circulatory responses to prolonged hypoxemia in fetal sheep. Am J Obstet Gynecol 1988;159:1418–24.
Holland MC, Mackersie RC, Morabito D, et al. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J Trauma 2003;55:106–11.
Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010;50:456–60.
Verein M, Valiahmedov A, Churliaev Y, Sitnikov P, Redkokasha L, Lukashev K. Dynamics of extravascular pulmonary water and intracranial pressure in patients with ischemic stroke. Semin Cardiothorac Vasc Anesth 2010;14:226–30.
Gruber A, Reinprecht A, Görzer H, et al. Pulmonary function and radiographic abnormalities related to neurological outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 1998;88:28–37.
Macmillan CS, Grant IS, Andrews PJ. Pulmonary and cardiac sequelae of subarachnoid haemorrhage: time for active management? Intensive Care Med 2002;28:1012–23.
Wu S, Fang CX, Kim J, Ren J. Enhanced pulmonary inflammation following experimental intracerebral hemorrhage. Exp Neurol 2006;200:245–9.
Kalsotra A, Zhao J, Anakk S, Dash PK, Strobel HW. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab 2007;27:963–74.
Hoke TS, Douglas IS, Klein CL, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol 2007;18:155–64.
Schmeling DJ, Caty MG, Oldham KT, Guice KS, Hinshaw DB. Evidence for neutrophil-related acute lung injury after intestinal ischemia-reperfusion. Surgery 1989;106:195–201; discussion 201–2.
Volpe JJ. Hypoxic-ischemic encephalopathy: biochemical and physiological aspects. In: Volpe JJ, ed. Neurology of the Newborn, 5th edn. Philadelphia, PA, USA: Saunders-Elsevier, 2008:247–324.
Xanthou M, Fotopoulos S, Mouchtouri A, Lipsou N, Zika I, Sarafidou J. Inflammatory mediators in perinatal asphyxia and infection. Acta Paediatr Suppl 2002;91:92–7.
Alvarez FJ, Lafuente H, Rey-Santano MC, et al. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr Res 2008;64:653–8.
Pazos MR, Mohammed N, Lafuente H, et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: role of 5HT(1A) and CB2 receptors. Neuropharmacology 2013;71:282–91.
Baudendistel L, Shields JB, Kaminski DL. Comparison of double indicator thermodilution measurements of extravascular lung water (EVLW) with radiographic estimation of lung water in trauma patients. J Trauma 1982;22:983–8.
Katzenelson R, Perel A, Berkenstadt H, et al. Accuracy of transpulmonary thermodilution versus gravimetric measurement of extravascular lung water. Crit Care Med 2004;32:1550–4.
Neumann P. Extravascular lung water and intrathoracic blood volume: double versus single indicator dilution technique. Intensive Care Med 1999;25:216–9.
Mackersie RC, Christensen JM, Pitts LH, Lewis FR. Pulmonary extravascular fluid accumulation following intracranial injury. J Trauma 1983;23:968–75.
Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest 1989;84:1609–19.
Medbø S, Yu XQ, Asberg A, Saugstad OD. Pulmonary hemodynamics and plasma endothelin-1 during hypoxemia and reoxygenation with room air or 100% oxygen in a piglet model. Pediatr Res 1998;44:843–9.
Cheung PY, Johnson ST, Obaid L, Chan GS, Bigam DL. The systemic, pulmonary and regional hemodynamic recovery of asphyxiated newborn piglets resuscitated with 18%, 21% and 100% oxygen. Resuscitation 2008;76:457–64.
Bunt JE, Gavilanes AW, Reulen JP, Blanco CE, Vles JS. The influence of acute hypoxemia and hypovolemic hypotension of neuronal brain activity measured by the cerebral function monitor in newborn piglets. Neuropediatrics 1996;27:260–4.
Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci 2001;2:734–44.
Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol 2011;10:372–82.
Oygür N, Sönmez O, Saka O, Yeğin O. Predictive value of plasma and cerebrospinal fluid tumour necrosis factor-alpha and interleukin-1 beta concentrations on outcome of full term infants with hypoxic-ischaemic encephalopathy. Arch Dis Child Fetal Neonatal Ed 1998;79:F190–3.
Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res 2000;871:57–65.
Lampl Y, Shmuilovich O, Lockman J, Sadeh M, Lorberboym M. Prognostic significance of blood brain barrier permeability in acute hemorrhagic stroke. Cerebrovasc Dis 2005;20:433–7.
Audebert HJ, Rott MM, Eck T, Haberl RL. Systemic inflammatory response depends on initial stroke severity but is attenuated by successful thrombolysis. Stroke 2004;35:2128–33.
Smith CJ, Emsley HC, Gavin CM, et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol 2004;4:2.
Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H. Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int 1999;55:2362–7.
Kadesky KM, Turnage RH, Rogers TE, Inman L, Myers SI. In vitro evidence of neutrophil-mediated lung injury after intestinal reperfusion. Shock 1995;4:102–6.
Polglase GR, Miller SL, Barton SK, et al. Initiation of resuscitation with high tidal volumes causes cerebral hemodynamic disturbance, brain inflammation and injury in preterm lambs. PLoS One 2012;7:e39535.
Slutsky AS. Ventilator-induced lung injury: from barotrauma to biotrauma. Respir Care 2005;50:646–59.
Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 1999;160:109–16.
Félix B, Léger ME, Albe-Fessard D, Marcilloux JC, Rampin O, Laplace JP. Stereotaxic atlas of the pig brain. Brain Res Bull 1999;49:1–137.
de Blic J, Midulla F, Barbato A, et al. Bronchoalveolar lavage in children. ERS Task Force on bronchoalveolar lavage in children. European Respiratory Society. Eur Respir J 2000;15:217–31.
Merz U, Kellinghaus M, Häusler M, Pakrawan N, Klosterhalfen B, Hörnchen H. Partial liquid ventilation with surfactant: effects on gas exchange and lung pathology in surfactant-depleted piglets. Intensive Care Med 2000;26:109–16.
Acknowledgements
Technical and human support provided by María Dolores Molina Corzo is gratefully acknowledged. We are also indebted to Francisco Tendillo for his help in performing this experiment. We are grateful to Jason Willis-Lee for author’s editing services provided during preparation of the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Arruza, L., Pazos, M., Mohammed, N. et al. Hypoxic–ischemic brain damage induces distant inflammatory lung injury in newborn piglets. Pediatr Res 79, 401–408 (2016). https://doi.org/10.1038/pr.2015.87
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.87
- Springer Nature America, Inc.
This article is cited by
-
aEEG and neurologic exam findings correlate with hypoxic–ischemic brain damage severity in a piglet survival model
Pediatric Research (2019)
-
Inflammation markers in the saliva of infants born from Zika-infected mothers: exploring potential mechanisms of microcephaly during fetal development
Scientific Reports (2019)
-
Cannabidiol reduces lung injury induced by hypoxic–ischemic brain damage in newborn piglets
Pediatric Research (2017)
-
Sex-specific associations between cerebrovascular blood pressure autoregulation and cardiopulmonary injury in neonatal encephalopathy and therapeutic hypothermia
Pediatric Research (2017)